
Genome Assembly and Annotation of Vietnamese Rice Lines with Diverse Life-Cycle Durations

 , , , ,
, , , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Phenotypic Data
2.2. Plant Growth, Sequencing, Genome Assembly, and Annotation
2.3. Comparison between Genomes
3. Results
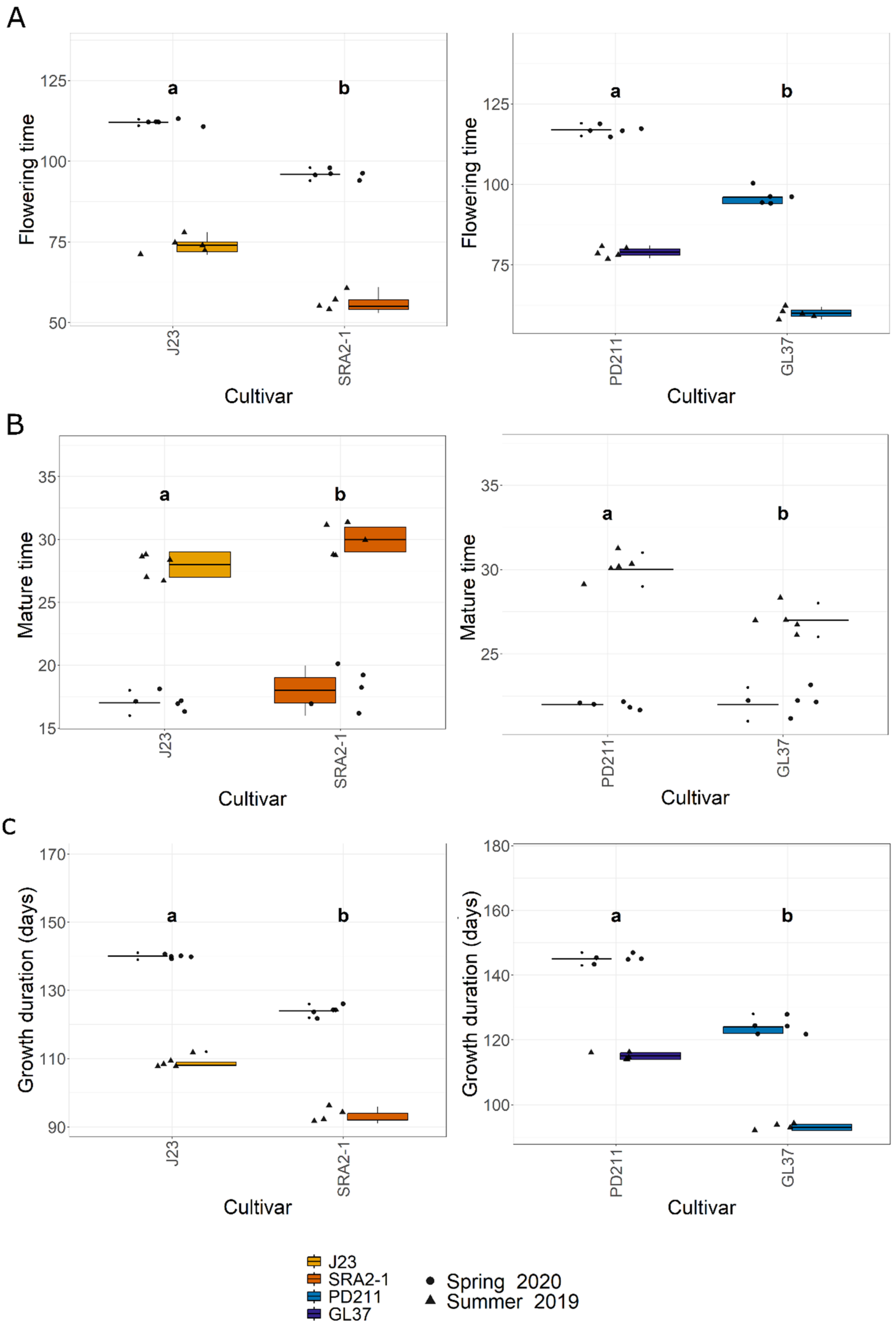
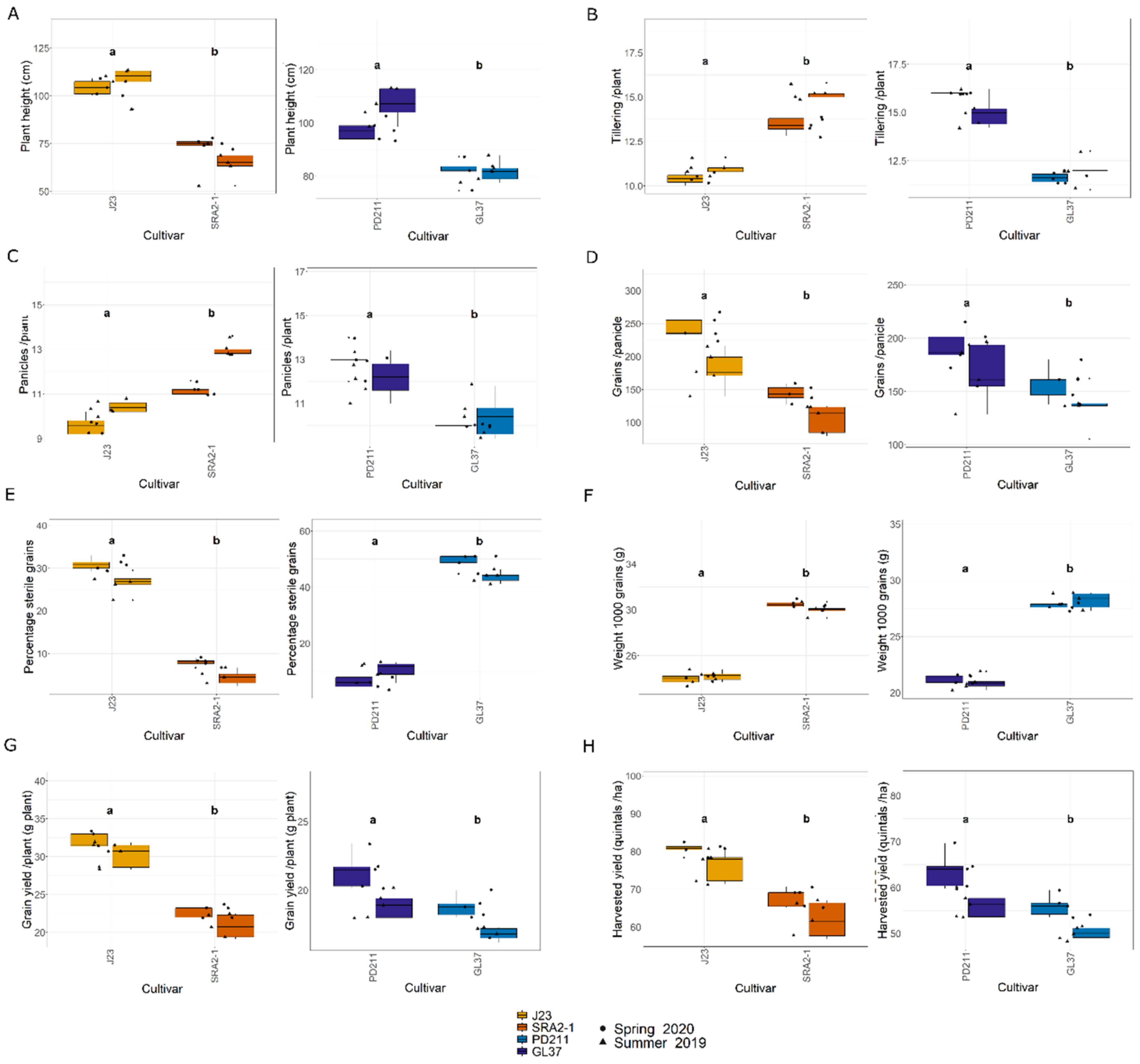
3.1. Flowering Time and Yield
3.2. Genome Statistics
3.3. Similarity between Genomes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FAOSTAT. Available online: https://www.fao.org/faostat/en/#data/QCL (accessed on 9 February 2024).
- Wang, W.; Mauleon, R.; Hu, Z.; Chebotarov, D.; Tai, S.; Wu, Z.; Li, M.; Zheng, T.; Fuentes, R.R.; Zhang, F.; et al. Genomic Variation in 3,010 Diverse Accessions of Asian Cultivated Rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Garris, A.J.; Tai, T.H.; Coburn, J.; Kresovich, S.; McCouch, S. Genetic Structure and Diversity in Oryza sativa L. Genetics 2005, 169, 1631–1638. [Google Scholar] [CrossRef]
- Glaszmann, J.C. Isozymes and Classification of Asian Rice Varieties. Theor. Appl. Genet. 1987, 74, 21–30. [Google Scholar] [CrossRef] [PubMed]
- FAO. The Contribution of Plant Genetic Resources for Food and Agriculture to Food Security and Sustainable Agricultural Development. In The Second Report on the State of the World’s Plant Genetic Resources for Food and Agriculture; FAO: Rome, Italy, 2010; pp. 182–201. [Google Scholar]
- Li, Z.; Fu, B.Y.; Gao, Y.M.; Wang, W.S.; Xu, J.L.; Zhang, F.; Zhao, X.Q.; Zheng, T.Q.; Zhou, Y.L.; Zhang, G.; et al. The 3,000 Rice Genomes Project. Gigascience 2014, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.; Santos, B.; Khanh, T.D.; Trung, K.H.; Duong, T.D.; Doai, N.T.P.; Khoa, N.T.; Ha, D.T.T.; Diep, N.T.; Dung, K.T.; et al. Resequencing of 672 Native Rice Accessions to Explore Genetic Diversity and Trait Associations in Vietnam. Rice 2021, 14, 52. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.M.; Lee, S.H.; Lee, J.Y. The Impact of Natural Disasters and Pest Infestations on Technical Efficiency in Rice Production: A Study in Vietnam. Sustainability 2023, 15, 11633. [Google Scholar] [CrossRef]
- General Statistics Office. Available online: https://www.gso.gov.vn/en/data-and-statistics/2024/07/statistical-yearbook-of-2023/ (accessed on 18 July 2024).
- Mathews, S.; Donoghue, M.J. The Root of Angiosperm Phylogeny Inferred from Duplicate Phytochrome Genes. Science 1999, 286, 947–950. [Google Scholar] [CrossRef]
- BBMap Guide—DOE Joint Genome Institute. Available online: https://jgi.doe.gov/data-and-tools/software-tools/bbtools/bb-tools-user-guide/bbmap-guide/ (accessed on 2 January 2024).
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Minkin, I.; Medvedev, P. Scalable Multiple Whole-Genome Alignment and Locally Collinear Block Construction with SibeliaZ. Nat. Commun. 2020, 11, 6327. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Raney, B.; Paten, B.; Pham, S. Ragout—A Reference-Assisted Assembly Tool for Bacterial Genomes. Bioinformatics 2014, 30, i302–i309. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov, M.; Armstrong, J.; Raney, B.J.; Streeter, I.; Dunn, M.; Yang, F.; Odom, D.; Flicek, P.; Keane, T.M.; Thybert, D.; et al. Chromosome Assembly of Large and Complex Genomes Using Multiple References. Genome Res. 2018, 28, 1720–1732. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Zdobnov, E.M. BUSCO: Assessing Genomic Data Quality and Beyond. Curr. Protoc. 2021, 1, e323. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for Automated Genomic Discovery of Transposable Element Families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef] [PubMed]
- Brůna, T.; Hoff, K.J.; Lomsadze, A.; Stanke, M.; Borodovsky, M. BRAKER2: Automatic Eukaryotic Genome Annotation with GeneMark-EP+ and AUGUSTUS Supported by a Protein Database. NAR Genom. Bioinform. 2021, 3, lqaa108. [Google Scholar] [CrossRef] [PubMed]
- Zdobnov, E.M.; Kuznetsov, D.; Tegenfeldt, F.; Manni, M.; Berkeley, M.; Kriventseva, E.V. OrthoDB in 2020: Evolutionary and Functional Annotations of Orthologs. Nucleic Acids Res. 2021, 49, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. EggNOG 5.0: A Hierarchical, Functionally and Phylogenetically Annotated Orthology Resource Based on 5090 Organisms and 2502 Viruses. Nucleic Acids Res. 2019, 47, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. EggNOG-Mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- Caballero, M.; Wegrzyn, J. GFACs: Gene Filtering, Analysis, and Conversion to Unify Genome Annotations Across Alignment and Gene Prediction Frameworks. Genom. Proteom. Bioinform. 2019, 17, 305. [Google Scholar] [CrossRef]
- Hart, A.J.; Ginzburg, S.; Xu, M.; Fisher, C.R.; Rahmatpour, N.; Mitton, J.B.; Paul, R.; Wegrzyn, J.L. EnTAP: Bringing Faster and Smarter Functional Annotation to Non-Model Eukaryotic Transcriptomes. Mol. Ecol. Resour. 2020, 20, 591–604. [Google Scholar] [CrossRef]
- Ou, S.; Su, W.; Liao, Y.; Chougule, K.; Agda, J.R.A.; Hellinga, A.J.; Lugo, C.S.B.; Elliott, T.A.; Ware, D.; Peterson, T.; et al. Benchmarking Transposable Element Annotation Methods for Creation of a Streamlined, Comprehensive Pipeline. Genome Biol. 2019, 20, 275. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast Genome and Metagenome Distance Estimation Using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. TrimAl: A Tool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hasegawa, M.; Kishino, H.; Yano, T. aki Dating of the Human-Ape Splitting by a Molecular Clock of Mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. A Space-Time Process Model for the Evolution of DNA Sequences. Genetics 1995, 139, 993–1005. [Google Scholar] [CrossRef]
- Soubrier, J.; Steel, M.; Lee, M.S.Y.; Der Sarkissian, C.; Guindon, S.; Ho, S.Y.W.; Cooper, A. The Influence of Rate Heterogeneity among Sites on the Time Dependence of Molecular Rates. Mol. Biol. Evol. 2012, 29, 3345–3358. [Google Scholar] [CrossRef]
- Won, P.L.P.; Liu, H.; Banayo, N.P.M.; Nie, L.; Peng, S.; Islam, M.R.; Cruz, P.S.; Collard, B.C.Y.; Kato, Y. Identification and Characterization of High-yielding, Short-duration Rice Genotypes for Tropical Asia. Crop. Sci. 2020, 60, 2241. [Google Scholar] [CrossRef]
- Thúy, L.T.; Vu, T.-N.; Pham, V.-T.; Nguyen, A.-D.; Nguyen, T.-K. Variability, Correlation and Path Analysis for Several Quantitative Traits Derived Multi-Parent Advanced Generation Inter-Cross (Magic) F2 Population of Rice (Oryza sativa L.). Int. J. Sci. Res. Manag. 2022, 10, 356–363. [Google Scholar] [CrossRef]
- Jing, Q.; Spiertz, J.H.J.; Hengsdijk, H.; Van Keulen, H.; Cao, W.; Dai, T. Adaptation and Performance of Rice Genotypes in Tropical and Subtropical Environments. NJAS 2010, 57, 149–157. [Google Scholar] [CrossRef]
- Li, B.; Du, X.; Fei, Y.; Wang, F.; Xu, Y.; Li, X.; Li, W.; Chen, Z.; Fan, F.; Wang, J.; et al. Efficient Breeding of Early-Maturing Rice Cultivar by Editing PHYC via CRISPR/Cas9. Rice 2021, 14, 86. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.; Santos, B.; Khanh, T.D.; Trung, K.H.; Duong, T.D.; Doai, N.T.P.; Hall, A.; Dyer, S.; Ham, L.H.; Caccamo, M.; et al. Genomic Regions and Candidate Genes Selected during the Breeding of Rice in Vietnam. Evol. Appl. 2022, 15, 1141. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Varieties | Flowering Time (Days) | Flowering Day | Maturity Time (Days) | Growth Duration (Days) | Plant Height (cm) | Tillers/Plnat | Panicles/Plant | Grains/Panicle | Sterile Grain Rate (%) | Weight of 1000 Grains (g) | Grain Yield/Plant (g/Plant) | Harvested Yield (Quintals/ha) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| J23 | 112.0 | 11.0 | 17.0 | 140.0 | 104.4 | 10.4 | 9.6 | 243.3 | 30.9 | 23.9 | 32.0 | 80.7 |
| SRA2-1 | 96.0 | 10.0 | 18.0 | 124.0 | 75.0 | 13.4 | 11.2 | 144.1 | 8.0 | 30.5 | 22.8 | 67.9 |
| PD211 | 117.0 | 6.0 | 22.0 | 145.0 | 97.2 | 16.0 | 13.0 | 191.8 | 6.3 | 21.3 | 21.4 | 63.7 |
| GL37 | 96.0 | 6.0 | 22.0 | 124.0 | 81.9 | 11.6 | 10.0 | 157.4 | 49.3 | 27.7 | 18.8 | 56.0 |
| Varieties | Flowering Time (Days) | Flowering Day | Maturity Time (Days) | Growth Duration (Days) | Plant Height (cm) | Tillers/Plnat | Panicles/Plant | Grains/Panicle | Sterile Grain Rate (%) | Weight of 1000 Grains (g) | Grain Yield/Plant (g/Plant) | Harvested Yield (Quintals/ha) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| J23 | 74.0 | 7.0 | 28.0 | 109.0 | 107.4 | 11.0 | 10.4 | 180.4 | 26.5 | 24.2 | 30.2 | 76.1 |
| SRA2-1 | 56.0 | 7.0 | 30.0 | 93.0 | 65.0 | 15.2 | 13.0 | 105.1 | 4.4 | 30.0 | 20.8 | 61.9 |
| PD211 | 79.0 | 6.0 | 30.0 | 115.0 | 107.2 | 15.0 | 12.2 | 166.8 | 10.6 | 20.9 | 18.9 | 56.3 |
| GL37 | 60.0 | 6.0 | 27.0 | 93.0 | 81.9 | 12.0 | 10.4 | 135.8 | 43.7 | 28.2 | 16.8 | 50.0 |
| Contigs | Unassembled Contigs to Nipponbare | ||||||
|---|---|---|---|---|---|---|---|
| Contigs | Total Length | Minimum Length Contigs | Average Length Contigs | Maximum Length Contigs | Number Contig | Total Length | |
| J23 | 124,180 | 333,355,875 | 66 | 2684.5 | 244,097 | 83,865 | 37,422,112 |
| SRA2-1 | 104,659 | 319,081,948 | 64 | 3048.8 | 168,733 | 66,421 | 26,594,505 |
| PD211 | 106,013 | 319,376,162 | 70 | 3012.6 | 149,118 | 72,349 | 31,732,613 |
| GL37 | 101,153 | 316,744,078 | 65 | 3131.3 | 166,595 | 68,785 | 30,724,240 |
| GC% | N50 | Total Length | %Ns | Coverage | |
|---|---|---|---|---|---|
| J23 | 42.73 | 30,217,895 | 379,887,007 | 19.7% | 131.32 |
| SRA2-1 | 42.71 | 30,335,714 | 381,425,794 | 21% | 131.821 |
| PD211 | 42.63 | 30,796,562 | 384,074,978 | 23.2% | 132.181 |
| GL37 | 42.64 | 30,961,767 | 383,427,667 | 23.5% | 127.264 |
| BUSCO % Poales Total | Complete BUSCOs (C) | Complete and Single-Copy BUSCOs (S) | Complete and Duplicated BUSCOs (D) | Fragmented BUSCOs (F) | Missing BUSCOs (M) | ||
|---|---|---|---|---|---|---|---|
| J23 | Percentage | 96.9% | 95.5% | 1.4% | 0.6% | 2.5% | |
| Genes | 4896 | 4745 | 4678 | 67 | 28 | 123 | |
| SRA2-1 | Percentage | 96.7% | 95.3% | 1.4% | 0.8% | 2.5% | |
| Genes | 4896 | 4735 | 4668 | 67 | 38 | 123 | |
| PD211 | Percentage | 96.7% | 95.4% | 1.3% | 0.8% | 2.8% | |
| Genes | 4896 | 4737 | 4671 | 66 | 38 | 121 | |
| GL37 | Percentage | 90.5% | 89.1% | 1.4% | 0.0% | 9.5% | |
| Genes | 4896 | 4432 | 4363 | 69 | 0 | 464 |
| Number of Genes BRAKER | Functional Annotation Total Genes eggNOG | Retroelements (LTR) | DNA Transposons | Unclassified TE Elements | Total TEs (%) | |
|---|---|---|---|---|---|---|
| J23 | 48,190 | 45,798 (95.0%) | 5.27% | 25.48% | 0.21% | 30.96% |
| SRA2-1 | 48,704 | 46,487 (95.4%) | 4.43% | 25.66% | 0.16% | 30.26% |
| PD211 | 47,411 | 45,160 (95.3%) | 3.62% | 24.65% | 0.10% | 28.37% |
| GL37 | 44,427 | 43,000 (96.8%) | 3.31% | 24.89% | 0.10% | 28.3% |
| Mash Distance | p-Value | Shared-Hashes | ||
|---|---|---|---|---|
| J23 (Japonica) | GL37 (Indica) | 0.0118 | 0 | 640/1000 |
| PD211 (Indica) | GL37 (Indica) | 0.0051 | 0 | 817/1000 |
| PD211 (Indica) | J23 (Japonica) | 0.01111 | 0 | 654/1000 |
| SRA2-1 (Japonica) | GL37 (Indica) | 0.0117 | 0 | 641/1000 |
| SRA2-1 (Japonica) | J23 (Japonica) | 0.0029 | 0 | 890/1000 |
| SRA2-1 (Japonica) | PD211 (Indica) | 0.0110 | 0 | 659/1000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco Ortega, S.; Thi Thuy, L.; Trong Khanh, N.; Thu Hang, L.; Thi Yen, T.; Thi Ngoan, L.; Thi Thanh, L.; Thien Thanh, P.; Ouyang, X.; Tao, W.; et al. Genome Assembly and Annotation of Vietnamese Rice Lines with Diverse Life-Cycle Durations. DNA 2024, 4, 239-251. https://doi.org/10.3390/dna4030016
Franco Ortega S, Thi Thuy L, Trong Khanh N, Thu Hang L, Thi Yen T, Thi Ngoan L, Thi Thanh L, Thien Thanh P, Ouyang X, Tao W, et al. Genome Assembly and Annotation of Vietnamese Rice Lines with Diverse Life-Cycle Durations. DNA. 2024; 4(3):239-251. https://doi.org/10.3390/dna4030016
Chicago/Turabian StyleFranco Ortega, Sara, Luu Thi Thuy, Nguyen Trong Khanh, Le Thu Hang, Tran Thi Yen, Le Thi Ngoan, Le Thi Thanh, Pham Thien Thanh, Xinhao Ouyang, Wenjing Tao, and et al. 2024. "Genome Assembly and Annotation of Vietnamese Rice Lines with Diverse Life-Cycle Durations" DNA 4, no. 3: 239-251. https://doi.org/10.3390/dna4030016
APA StyleFranco Ortega, S., Thi Thuy, L., Trong Khanh, N., Thu Hang, L., Thi Yen, T., Thi Ngoan, L., Thi Thanh, L., Thien Thanh, P., Ouyang, X., Tao, W., James, S., Gilbert, L., Davis, A. M., Gomez, L. D., Harper, A. L., McQueen-Mason, S. J., Xuan Tu, D., & Davis, S. J. (2024). Genome Assembly and Annotation of Vietnamese Rice Lines with Diverse Life-Cycle Durations. DNA, 4(3), 239-251. https://doi.org/10.3390/dna4030016