

Complex Roles of NEIL1 and OGG1: Insights Gained from Murine Knockouts and Human Polymorphic Variants


Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
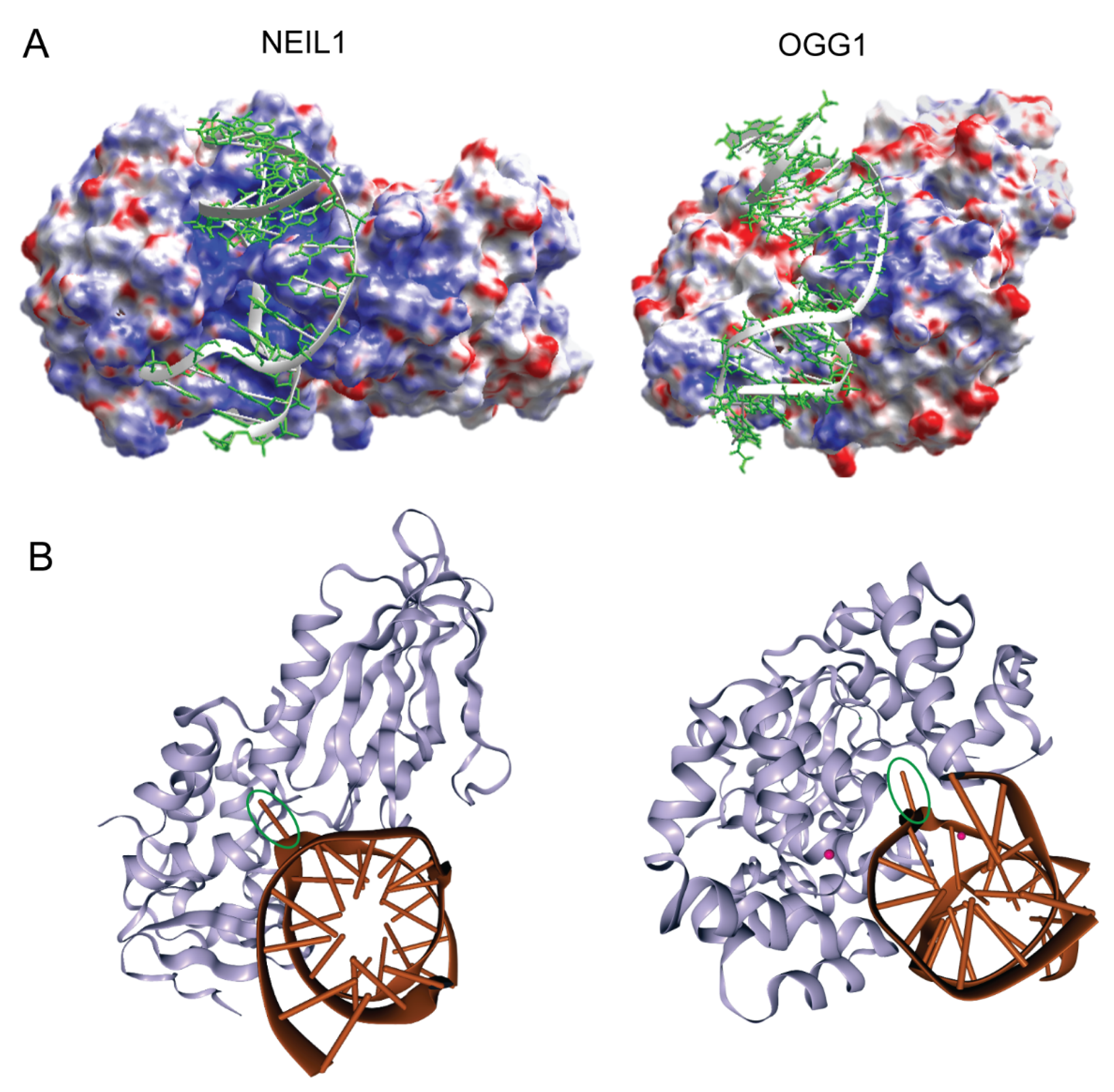
2. Target Site Location and Substrate Recognition
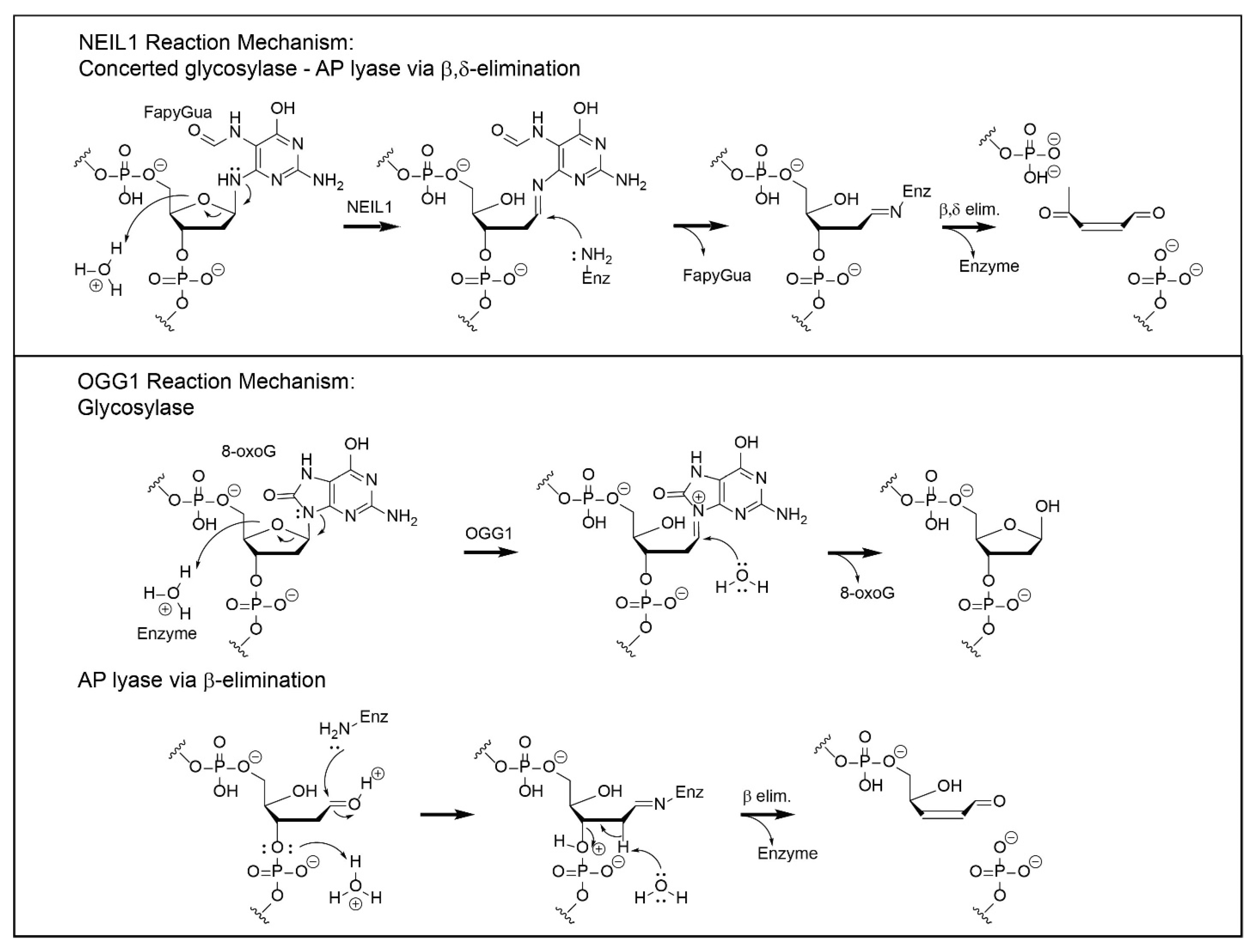
3. Mechanisms of Chemical Catalysis with Implications for the Completion of the BER Pathway
4. Cell-Cycle Specificity and Protein-Binding Partners
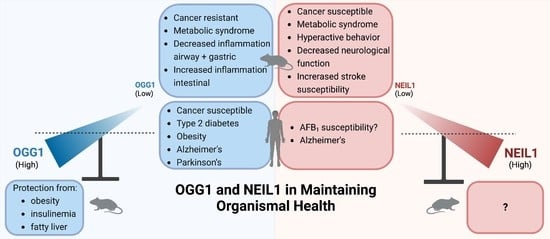
5. Phenotypes in Knockout Mice
5.1. Cancers
5.2. Metabolic Syndrome
5.3. Inflammation: The Role of OGG1 as a Regulator of Transcriptional Responses to Oxidatively Induced Base Damage
5.4. Roles of OGG1 and NEIL1 in Huntington’s Disease
5.5. Neurodegeneration in Neil1 Deficiency
6. Extrapolation to Human Investigations
6.1. OGG1-Associated Cancer Variants
6.2. Low-Level Expression of OGG1 in Therapeutically Responsive Subtypes of Human Acute Myeloid Leukemia (AML)
6.3. NEIL1-Associated Cancers
6.4. Potential Applications of Agonists of OGG1 in Disease Prevention
7. Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Kow, Y.W. Base excision repair in E. coli—An overview. Ann. N. Y. Acad. Sci. 1994, 726, 178–180. [Google Scholar] [CrossRef] [PubMed]
- Laval, J. Role of DNA repair enzymes in the cellular resistance to oxidative stress. Pathol. Biol. 1996, 44, 14–24. [Google Scholar] [PubMed]
- Beard, W.A.; Horton, J.K.; Prasad, R.; Wilson, S.H. Eukaryotic base excision repair: New approaches shine light on mechanism. Annu. Rev. Biochem. 2019, 88, 137–162. [Google Scholar] [CrossRef] [PubMed]
- Bandaru, V.; Sunkara, S.; Wallace, S.S.; Bond, J.P. A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair 2002, 1, 517–529. [Google Scholar] [CrossRef]
- Hazra, T.K.; Izumi, T.; Boldogh, I.; Imhoff, B.; Kow, Y.W.; Jaruga, P.; Dizdaroglu, M.; Mitra, S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. USA 2002, 99, 3523–3528. [Google Scholar] [CrossRef]
- Morland, I.; Rolseth, V.; Luna, L.; Rognes, T.; Bjoras, M.; Seeberg, E. Human DNA glycosylases of the bacterial Fpg/MutM superfamily: An alternative pathway for the repair of 8-oxoguanine and other oxidation products in DNA. Nucleic Acids Res. 2002, 30, 4926–4936. [Google Scholar] [CrossRef]
- Rosenquist, T.A.; Zaika, E.; Fernandes, A.S.; Zharkov, D.O.; Miller, H.; Grollman, A.P. The novel DNA glycosylase, NEIL1, protects mammalian cells from radiation-mediated cell death. DNA Repair 2003, 2, 581–591. [Google Scholar] [CrossRef]
- van der Kemp, P.A.; Thomas, D.; Barbey, R.; de Oliveira, R.; Boiteux, S. Cloning and expression in Escherichia coli of the OGG1 gene of Saccharomyces cerevisiae, which codes for a DNA glycosylase that excises 7,8-dihydro-8-oxoguanine and 2,6-diamino-4-hydroxy-5-N-methylformamidopyrimidine. Proc. Natl. Acad. Sci. USA 1996, 93, 5197–5202. [Google Scholar] [CrossRef]
- Aburatani, H.; Hippo, Y.; Ishida, T.; Takashima, R.; Matsuba, C.; Kodama, T.; Takao, M.; Yasui, A.; Yamamoto, K.; Asano, M. Cloning and characterization of mammalian 8-hydroxyguanine-specific DNA glycosylase/apurinic, apyrimidinic lyase, a functional mutM homologue. Cancer Res. 1997, 57, 2151–2156. [Google Scholar]
- Arai, K.; Morishita, K.; Shinmura, K.; Kohno, T.; Kim, S.R.; Nohmi, T.; Taniwaki, M.; Ohwada, S.; Yokota, J. Cloning of a human homolog of the yeast OGG1 gene that is involved in the repair of oxidative DNA damage. Oncogene 1997, 14, 2857–2861. [Google Scholar] [CrossRef]
- Bjoras, M.; Luna, L.; Johnsen, B.; Hoff, E.; Haug, T.; Rognes, T.; Seeberg, E. Opposite base-dependent reactions of a human base excision repair enzyme on DNA containing 7,8-dihydro-8-oxoguanine and abasic sites. EMBO J. 1997, 16, 6314–6322. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Nash, H.M.; Verdine, G.L. A mammalian DNA repair enzyme that excises oxidatively damaged guanines maps to a locus frequently lost in lung cancer. Curr. Biol. 1997, 7, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Radicella, J.P.; Dherin, C.; Desmaze, C.; Fox, M.S.; Boiteux, S. Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1997, 94, 8010–8015. [Google Scholar] [CrossRef] [PubMed]
- Roldan-Arjona, T.; Wei, Y.F.; Carter, K.C.; Klungland, A.; Anselmino, C.; Wang, R.P.; Augustus, M.; Lindahl, T. Molecular cloning and functional expression of a human cDNA encoding the antimutator enzyme 8-hydroxyguanine-DNA glycosylase. Proc. Natl. Acad. Sci. USA 1997, 94, 8016–8020. [Google Scholar] [CrossRef] [PubMed]
- Rosenquist, T.A.; Zharkov, D.O.; Grollman, A.P. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proc. Natl. Acad. Sci. USA 1997, 94, 7429–7434. [Google Scholar] [CrossRef] [PubMed]
- Doublie, S.; Bandaru, V.; Bond, J.P.; Wallace, S.S. The crystal structure of human endonuclease VIII-like 1 (NEIL1) reveals a zincless finger motif required for glycosylase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 10284–10289. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Yang, W.; Karplus, M.; Verdine, G.L. Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damaged DNA. Nature 2005, 434, 612–618. [Google Scholar] [CrossRef]
- Imamura, K.; Wallace, S.S.; Doublie, S. Structural characterization of a viral NEIL1 ortholog unliganded and bound to abasic site-containing DNA. J. Biol. Chem. 2009, 284, 26174–26183. [Google Scholar] [CrossRef]
- Imamura, K.; Averill, A.; Wallace, S.S.; Doublie, S. Structural characterization of viral ortholog of human DNA glycosylase NEIL1 bound to thymine glycol or 5-hydroxyuracil-containing DNA. J. Biol. Chem. 2012, 287, 4288–4298. [Google Scholar] [CrossRef]
- Zhu, C.; Lu, L.; Zhang, J.; Yue, Z.; Song, J.; Zong, S.; Liu, M.; Stovicek, O.; Gao, Y.Q.; Yi, C. Tautomerization-dependent recognition and excision of oxidation damage in base-excision DNA repair. Proc. Natl. Acad. Sci. USA 2016, 113, 7792–7797. [Google Scholar] [CrossRef]
- Li, H.; Endutkin, A.V.; Bergonzo, C.; Fu, L.; Grollman, A.; Zharkov, D.O.; Simmerling, C. DNA deformation-coupled recognition of 8-oxoguanine: Conformational kinetic gating in human DNA glycosylase. J. Am. Chem. Soc. 2017, 139, 2682–2692. [Google Scholar] [CrossRef]
- Shigdel, U.K.; Ovchinnikov, V.; Lee, S.J.; Shih, J.A.; Karplus, M.; Nam, K.; Verdine, G.L. The trajectory of intrahelical lesion recognition and extrusion by the human 8-oxoguanine DNA glycosylase. Nat. Commun. 2020, 11, 4437. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, J.; Zhu, C.; Zhang, X.; Xiao, W.; Yan, Y.; Liu, L.; Zeng, H.; Gao, Y.Q.; Yi, C. DNA repair glycosylase hNEIL1 triages damaged bases via competing interaction modes. Nat. Commun. 2021, 12, 4108. [Google Scholar] [CrossRef]
- Lloyd, R.S.; Hanawalt, P.C.; Dodson, M.L. Processive action of T4 endonuclease V on ultraviolet-irradiated DNA. Nucleic Acids Res. 1980, 8, 5113–5127. [Google Scholar] [CrossRef]
- Gruskin, E.A.; Lloyd, R.S. The DNA scanning mechanism of T4 endonuclease V. Effect of NaCl concentration on processive nicking activity. J. Biol. Chem. 1986, 261, 9607–9613. [Google Scholar] [CrossRef]
- Dowd, D.R.; Lloyd, R.S. Site-directed mutagenesis of the T4 endonuclease V gene: The role of arginine-3 in the target search. Biochemistry 1989, 28, 8699–8705. [Google Scholar] [CrossRef]
- Dowd, D.R.; Lloyd, R.S. Biological significance of facilitated diffusion in protein-DNA interactions. Applications to T4 endonuclease V-initiated DNA repair. J. Biol. Chem. 1990, 265, 3424–3431. [Google Scholar] [CrossRef]
- Zharkov, D.O.; Grollman, A.P. The DNA trackwalkers: Principles of lesion search and recognition by DNA glycosylases. Mutat. Res. 2005, 577, 24–54. [Google Scholar] [CrossRef]
- Prakash, A.; Doublie, S.; Wallace, S.S. The Fpg/Nei family of DNA glycosylases: Substrates, structures, and search for damage. Prog. Mol. Biol. Transl. Sci. 2012, 110, 71–91. [Google Scholar] [CrossRef]
- Lee, A.J.; Wallace, S.S. Hide and seek: How do DNA glycosylases locate oxidatively damaged DNA bases amidst a sea of undamaged bases? Free Radic. Biol. Med. 2017, 107, 170–178. [Google Scholar] [CrossRef]
- D’Augustin, O.; Huet, S.; Campalans, A.; Radicella, J.P. Lost in the crowd: How does human 8-oxoguanine DNA glycosylase 1 (OGG1) find 8-oxoguanine in the genome? Int. J. Mol. Sci. 2020, 21, 8360. [Google Scholar] [CrossRef]
- Kong, M.; Beckwitt, E.C.; Van Houten, B. Dynamic action of DNA repair proteins as revealed by single molecule techniques: Seeing is believing. DNA Repair 2020, 93, 102909. [Google Scholar] [CrossRef]
- Rose, A.S.; Bradley, A.R.; Valasatava, Y.; Duarte, J.M.; Prlic, A.; Rose, P.W. NGL viewer: Web-based molecular graphics for large complexes. Bioinformatics 2018, 34, 3755–3758. [Google Scholar] [CrossRef]
- McCullough, A.K.; Minko, I.G.; Nilsen, A.; Nagarajan, S.; Lloyd, R.S. Modulation of DNA glycosylase activities via small molecules. In DNA Damage, DNA Repair and Disease; Dizdaroglu, M., Lloyd, R.S., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2021; Volume 1, pp. 323–347. [Google Scholar]
- Hegde, M.L.; Hegde, P.M.; Bellot, L.J.; Mandal, S.M.; Hazra, T.K.; Li, G.M.; Boldogh, I.; Tomkinson, A.E.; Mitra, S. Prereplicative repair of oxidized bases in the human genome is mediated by NEIL1 DNA glycosylase together with replication proteins. Proc. Natl. Acad. Sci. USA 2013, 110, E3090–E3099. [Google Scholar] [CrossRef]
- Hegde, P.M.; Dutta, A.; Sengupta, S.; Mitra, J.; Adhikari, S.; Tomkinson, A.E.; Li, G.M.; Boldogh, I.; Hazra, T.K.; Mitra, S.; et al. The C-terminal domain (CTD) of human DNA glycosylase NEIL1 is required for forming BERosome repair complex with DNA replication proteins at the replicating genome: Dominant negative function of the CTD. J. Biol. Chem. 2015, 290, 20919–20933. [Google Scholar] [CrossRef]
- Dou, H.; Mitra, S.; Hazra, T.K. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J. Biol. Chem. 2003, 278, 49679–49684. [Google Scholar] [CrossRef]
- Das, A.; Boldogh, I.; Lee, J.W.; Harrigan, J.A.; Hegde, M.L.; Piotrowski, J.; de Souza Pinto, N.; Ramos, W.; Greenberg, M.M.; Hazra, T.K.; et al. The human Werner syndrome protein stimulates repair of oxidative DNA base damage by the DNA glycosylase NEIL1. J. Biol. Chem. 2007, 282, 26591–26602. [Google Scholar] [CrossRef]
- Hegde, M.L.; Theriot, C.A.; Das, A.; Hegde, P.M.; Guo, Z.; Gary, R.K.; Hazra, T.K.; Shen, B.; Mitra, S. Physical and functional interaction between human oxidized base-specific DNA glycosylase NEIL1 and flap endonuclease 1. J. Biol. Chem. 2008, 283, 27028–27037. [Google Scholar] [CrossRef]
- Yeo, J.; Goodman, R.A.; Schirle, N.T.; David, S.S.; Beal, P.A. RNA editing changes the lesion specificity for the DNA repair enzyme NEIL1. Proc. Natl. Acad. Sci. USA 2010, 107, 20715–20719. [Google Scholar] [CrossRef]
- Zhao, X.; Krishnamurthy, N.; Burrows, C.J.; David, S.S. Mutation versus repair: NEIL1 removal of hydantoin lesions in single-stranded, bulge, bubble, and duplex DNA contexts. Biochemistry 2010, 49, 1658–1666. [Google Scholar] [CrossRef]
- Hegde, M.L.; Banerjee, S.; Hegde, P.M.; Bellot, L.J.; Hazra, T.K.; Boldogh, I.; Mitra, S. Enhancement of NEIL1 protein-initiated oxidized DNA base excision repair by heterogeneous nuclear ribonucleoprotein U (hnRNP-U) via direct interaction. J. Biol. Chem. 2012, 287, 34202–34211. [Google Scholar] [CrossRef]
- Vartanian, V.; Minko, I.G.; Chawanthayatham, S.; Egner, P.A.; Lin, Y.C.; Earley, L.F.; Makar, R.; Eng, J.R.; Camp, M.T.; Li, L.; et al. NEIL1 protects against aflatoxin-induced hepatocellular carcinoma in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 4207–4212. [Google Scholar] [CrossRef]
- Roy, L.M.; Jaruga, P.; Wood, T.G.; McCullough, A.K.; Dizdaroglu, M.; Lloyd, R.S. Human polymorphic variants of the NEIL1 DNA glycosylase. J. Biol. Chem. 2007, 282, 15790–15798. [Google Scholar] [CrossRef]
- Dizdaroglu, M.; Coskun, E.; Jaruga, P. Repair of oxidatively induced DNA damage by DNA glycosylases: Mechanisms of action, substrate specificities and excision kinetics. Mutat. Res. Rev. Mutat. Res. 2017, 771, 99–127. [Google Scholar] [CrossRef]
- Minko, I.G.; Vartanian, V.L.; Tozaki, N.N.; Coskun, E.; Coskun, S.H.; Jaruga, P.; Yeo, J.; David, S.S.; Stone, M.P.; Egli, M.; et al. Recognition of DNA adducts by edited and unedited forms of DNA glycosylase NEIL1. DNA Repair 2020, 85, 102741. [Google Scholar] [CrossRef]
- Prakash, A.; Carroll, B.L.; Sweasy, J.B.; Wallace, S.S.; Doublie, S. Genome and cancer single nucleotide polymorphisms of the human NEIL1 DNA glycosylase: Activity, structure, and the effect of editing. DNA Repair 2014, 14, 17–26. [Google Scholar] [CrossRef]
- Minko, I.G.; Christov, P.P.; Li, L.; Stone, M.P.; McCullough, A.K.; Lloyd, R.S. Processing of N5-substituted formamidopyrimidine DNA adducts by DNA glycosylases NEIL1 and NEIL3. DNA Repair 2019, 73, 49–54. [Google Scholar] [CrossRef]
- Tomar, R.; Minko, I.G.; Kellum, A.H., Jr.; Voehler, M.W.; Stone, M.P.; McCullough, A.K.; Lloyd, R.S. DNA sequence modulates the efficiency of NEIL1-catalyzed excision of the aflatoxin B1-induced formamidopyrimidine guanine adduct. Chem. Res. Toxicol. 2021, 34, 901–911. [Google Scholar] [CrossRef]
- Hailer, M.K.; Slade, P.G.; Martin, B.D.; Rosenquist, T.A.; Sugden, K.D. Recognition of the oxidized lesions spiroiminodihydantoin and guanidinohydantoin in DNA by the mammalian base excision repair glycosylases NEIL1 and NEIL2. DNA Repair 2005, 4, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, N.; Zhao, X.; Burrows, C.J.; David, S.S. Superior removal of hydantoin lesions relative to other oxidized bases by the human DNA glycosylase hNEIL1. Biochemistry 2008, 47, 7137–7146. [Google Scholar] [CrossRef]
- McKibbin, P.L.; Fleming, A.M.; Towheed, M.A.; Van Houten, B.; Burrows, C.J.; David, S.S. Repair of hydantoin lesions and their amine adducts in DNA by base and nucleotide excision repair. J. Am. Chem. Soc. 2013, 135, 13851–13861. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liu, M.; Fleming, A.M.; Burrows, C.J.; Wallace, S.S. Neil3 and NEIL1 DNA glycosylases remove oxidative damages from quadruplex DNA and exhibit preferences for lesions in the telomeric sequence context. J. Biol. Chem. 2013, 288, 27263–27272. [Google Scholar] [CrossRef] [PubMed]
- Slyvka, A.; Mierzejewska, K.; Bochtler, M. Nei-like 1 (NEIL1) excises 5-carboxylcytosine directly and stimulates TDG-mediated 5-formyl and 5-carboxylcytosine excision. Sci. Rep. 2017, 7, 9001. [Google Scholar] [CrossRef] [PubMed]
- Couve-Privat, S.; Mace, G.; Rosselli, F.; Saparbaev, M.K. Psoralen-induced DNA adducts are substrates for the base excision repair pathway in human cells. Nucleic Acids Res. 2007, 35, 5672–5682. [Google Scholar] [CrossRef] [PubMed]
- Couve, S.; Mace-Aime, G.; Rosselli, F.; Saparbaev, M.K. The human oxidative DNA glycosylase NEIL1 excises psoralen-induced interstrand DNA cross-links in a three-stranded DNA structure. J. Biol. Chem. 2009, 284, 11963–11970. [Google Scholar] [CrossRef]
- Martin, P.R.; Couve, S.; Zutterling, C.; Albelazi, M.S.; Groisman, R.; Matkarimov, B.T.; Parsons, J.L.; Elder, R.H.; Saparbaev, M.K. The human DNA glycosylases NEIL1 and NEIL3 excise psoralen-induced DNA-DNA cross-links in a four-stranded DNA structure. Sci. Rep. 2017, 7, 17438. [Google Scholar] [CrossRef]
- Hu, J.; de Souza-Pinto, N.C.; Haraguchi, K.; Hogue, B.A.; Jaruga, P.; Greenberg, M.M.; Dizdaroglu, M.; Bohr, V.A. Repair of formamidopyrimidines in DNA involves different glycosylases: Role of the OGG1, NTH1, and NEIL1 enzymes. J. Biol. Chem. 2005, 280, 40544–40551. [Google Scholar] [CrossRef]
- Sidorenko, V.S.; Grollman, A.P.; Jaruga, P.; Dizdaroglu, M.; Zharkov, D.O. Substrate specificity and excision kinetics of natural polymorphic variants and phosphomimetic mutants of human 8-oxoguanine-DNA glycosylase. FEBS J. 2009, 276, 5149–5162. [Google Scholar] [CrossRef]
- Sassa, A.; Beard, W.A.; Prasad, R.; Wilson, S.H. DNA sequence context effects on the glycosylase activity of human 8-oxoguanine DNA glycosylase. J. Biol. Chem. 2012, 287, 36702–36710. [Google Scholar] [CrossRef]
- Sidorenko, V.S.; Nevinsky, G.A.; Zharkov, D.O. Specificity of stimulation of human 8-oxoguanine-DNA glycosylase by AP endonuclease. Biochem. Biophys. Res. Commun. 2008, 368, 175–179. [Google Scholar] [CrossRef]
- Dherin, C.; Radicella, J.P.; Dizdaroglu, M.; Boiteux, S. Excision of oxidatively damaged DNA bases by the human a-hOgg1 protein and the polymorphic a-hOgg1(Ser326Cys) protein which is frequently found in human populations. Nucleic Acids Res. 1999, 27, 4001–4007. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M. Base-excision repair of oxidative DNA damage by DNA glycosylases. Mutat. Res. 2005, 591, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Schrock, R.D., 3rd; Lloyd, R.S. Reductive methylation of the amino terminus of endonuclease V eradicates catalytic activities. Evidence for an essential role of the amino terminus in the chemical mechanisms of catalysis. J. Biol. Chem. 1991, 266, 17631–17639. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.L.; Schrock, R.D., 3rd; Lloyd, R.S. Evidence for an imino intermediate in the T4 endonuclease V reaction. Biochemistry 1993, 32, 8284–8290. [Google Scholar] [CrossRef]
- Schrock, R.D., 3rd; Lloyd, R.S. Site-directed mutagenesis of the NH2 terminus of T4 endonuclease V. The position of the aNH2 moiety affects catalytic activity. J. Biol. Chem. 1993, 268, 880–886. [Google Scholar] [CrossRef]
- Golan, G.; Zharkov, D.O.; Grollman, A.P.; Dodson, M.L.; McCullough, A.K.; Lloyd, R.S.; Shoham, G. Structure of T4 pyrimidine dimer glycosylase in a reduced imine covalent complex with abasic site-containing DNA. J. Mol. Biol. 2006, 362, 241–258. [Google Scholar] [CrossRef]
- Dodson, M.L.; Michaels, M.L.; Lloyd, R.S. Unified catalytic mechanism for DNA glycosylases. J. Biol. Chem. 1994, 269, 32709–32712. [Google Scholar] [CrossRef]
- Sun, B.; Latham, K.A.; Dodson, M.L.; Lloyd, R.S. Studies on the catalytic mechanism of five DNA glycosylases. Probing for enzyme-DNA imino intermediates. J. Biol. Chem. 1995, 270, 19501–19508. [Google Scholar] [CrossRef]
- Vik, E.S.; Alseth, I.; Forsbring, M.; Helle, I.H.; Morland, I.; Luna, L.; Bjoras, M.; Dalhus, B. Biochemical mapping of human NEIL1 DNA glycosylase and AP lyase activities. DNA Repair 2012, 11, 766–773. [Google Scholar] [CrossRef]
- Forsbring, M.; Vik, E.S.; Dalhus, B.; Karlsen, T.H.; Bergquist, A.; Schrumpf, E.; Bjoras, M.; Boberg, K.M.; Alseth, I. Catalytically impaired hMYH and NEIL1 mutant proteins identified in patients with primary sclerosing cholangitis and cholangiocarcinoma. Carcinogenesis 2009, 30, 1147–1154. [Google Scholar] [CrossRef]
- Dalhus, B.; Forsbring, M.; Helle, I.H.; Vik, E.S.; Forstrom, R.J.; Backe, P.H.; Alseth, I.; Bjoras, M. Separation-of-function mutants unravel the dual-reaction mode of human 8-oxoguanine DNA glycosylase. Structure 2011, 19, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Allgayer, J.; Kitsera, N.; Bartelt, S.; Epe, B.; Khobta, A. Widespread transcriptional gene inactivation initiated by a repair intermediate of 8-oxoguanine. Nucleic Acids Res. 2016, 44, 7267–7280. [Google Scholar] [CrossRef] [PubMed]
- Morland, I.; Luna, L.; Gustad, E.; Seeberg, E.; Bjoras, M. Product inhibition and magnesium modulate the dual reaction mode of hOgg1. DNA Repair 2005, 4, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Kidane, D.; Murphy, D.L.; Sweasy, J.B. Accumulation of abasic sites induces genomic instability in normal human gastric epithelial cells during Helicobacter pylori infection. Oncogenesis 2014, 3, e128. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Maayah, M.; Sweasy, J.B.; Alnajjar, K.S. The role of cysteines in the structure and function of OGG1. J. Biol. Chem. 2021, 296, 100093. [Google Scholar] [CrossRef]
- Popov, A.V.; Endutkin, A.V.; Yatsenko, D.D.; Yudkina, A.V.; Barmatov, A.E.; Makasheva, K.A.; Raspopova, D.Y.; Diatlova, E.A.; Zharkov, D.O. Molecular dynamics approach to identification of new OGG1 cancer-associated somatic variants with impaired activity. J. Biol. Chem. 2021, 296, 100229. [Google Scholar] [CrossRef]
- Wiederhold, L.; Leppard, J.B.; Kedar, P.; Karimi-Busheri, F.; Rasouli-Nia, A.; Weinfeld, M.; Tomkinson, A.E.; Izumi, T.; Prasad, R.; Wilson, S.H.; et al. AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell 2004, 15, 209–220. [Google Scholar] [CrossRef]
- Whitehouse, C.J.; Taylor, R.M.; Thistlethwaite, A.; Zhang, H.; Karimi-Busheri, F.; Lasko, D.D.; Weinfeld, M.; Caldecott, K.W. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell 2001, 104, 107–117. [Google Scholar] [CrossRef]
- Guan, X.; Bai, H.; Shi, G.; Theriot, C.A.; Hazra, T.K.; Mitra, S.; Lu, A.L. The human checkpoint sensor Rad9-Rad1-Hus1 interacts with and stimulates NEIL1 glycosylase. Nucleic Acids Res. 2007, 35, 2463–2472. [Google Scholar] [CrossRef]
- Dou, H.; Theriot, C.A.; Das, A.; Hegde, M.L.; Matsumoto, Y.; Boldogh, I.; Hazra, T.K.; Bhakat, K.K.; Mitra, S. Interaction of the human DNA glycosylase NEIL1 with proliferating cell nuclear antigen. The potential for replication-associated repair of oxidized bases in mammalian genomes. J. Biol. Chem. 2008, 283, 3130–3140. [Google Scholar] [CrossRef]
- Theriot, C.A.; Hegde, M.L.; Hazra, T.K.; Mitra, S. RPA physically interacts with the human DNA glycosylase NEIL1 to regulate excision of oxidative DNA base damage in primer-template structures. DNA Repair 2010, 9, 643–652. [Google Scholar] [CrossRef]
- Noren Hooten, N.; Fitzpatrick, M.; Kompaniez, K.; Jacob, K.D.; Moore, B.R.; Nagle, J.; Barnes, J.; Lohani, A.; Evans, M.K. Coordination of DNA repair by NEIL1 and PARP-1: A possible link to aging. Aging 2012, 4, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Dhenaut, A.; Boiteux, S.; Radicella, J.P. Characterization of the hOGG1 promoter and its expression during the cell cycle. Mutat. Res. 2000, 461, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Boldogh, I.; Milligan, D.; Lee, M.S.; Bassett, H.; Lloyd, R.S.; McCullough, A.K. hMYH cell cycle-dependent expression, subcellular localization and association with replication foci: Evidence suggesting replication-coupled repair of adenine:8-oxoguanine mispairs. Nucleic Acids Res. 2001, 29, 2802–2809. [Google Scholar] [CrossRef] [PubMed]
- Luna, L.; Rolseth, V.; Hildrestrand, G.A.; Otterlei, M.; Dantzer, F.; Bjoras, M.; Seeberg, E. Dynamic relocalization of hOGG1 during the cell cycle is disrupted in cells harbouring the hOGG1-Cys326 polymorphic variant. Nucleic Acids Res. 2005, 33, 1813–1824. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.W.; Hazra, T.K.; Izumi, T.; Mitra, S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: Potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 2001, 29, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.E.; Hickson, I.D.; Boiteux, S.; Radicella, J.P. Mechanism of stimulation of the DNA glycosylase activity of hOGG1 by the major human AP endonuclease: Bypass of the AP lyase activity step. Nucleic Acids Res. 2001, 29, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Marsin, S.; Vidal, A.E.; Sossou, M.; Menissier-de Murcia, J.; Le Page, F.; Boiteux, S.; de Murcia, G.; Radicella, J.P. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. J. Biol. Chem. 2003, 278, 44068–44074. [Google Scholar] [CrossRef] [PubMed]
- Mokkapati, S.K.; Wiederhold, L.; Hazra, T.K.; Mitra, S. Stimulation of DNA glycosylase activity of OGG1 by NEIL1: Functional collaboration between two human DNA glycosylases. Biochemistry 2004, 43, 11596–11604. [Google Scholar] [CrossRef] [PubMed]
- Park, M.J.; Park, J.H.; Hahm, S.H.; Ko, S.I.; Lee, Y.R.; Chung, J.H.; Sohn, S.Y.; Cho, Y.; Kang, L.W.; Han, Y.S. Repair activities of human 8-oxoguanine DNA glycosylase are stimulated by the interaction with human checkpoint sensor Rad9-Rad1-Hus1 complex. DNA Repair 2009, 8, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Noren Hooten, N.; Kompaniez, K.; Barnes, J.; Lohani, A.; Evans, M.K. Poly(ADP-ribose) polymerase 1 (PARP-1) binds to 8-oxoguanine-DNA glycosylase (OGG1). J. Biol. Chem. 2011, 286, 44679–44690. [Google Scholar] [CrossRef] [PubMed]
- Ramdzan, Z.M.; Vadnais, C.; Pal, R.; Vandal, G.; Cadieux, C.; Leduy, L.; Davoudi, S.; Hulea, L.; Yao, L.; Karnezis, A.N.; et al. RAS transformation requires CUX1-dependent repair of oxidative DNA damage. PLoS Biol. 2014, 12, e1001807. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Ramdzan, Z.M.; Kaur, S.; Duquette, P.M.; Marcotte, R.; Leduy, L.; Davoudi, S.; Lamarche-Vane, N.; Iulianella, A.; Nepveu, A. CUX2 protein functions as an accessory factor in the repair of oxidative DNA damage. J. Biol. Chem. 2015, 290, 22520–22531. [Google Scholar] [CrossRef]
- Ramdzan, Z.M.; Pal, R.; Kaur, S.; Leduy, L.; Berube, G.; Davoudi, S.; Vadnais, C.; Nepveu, A. The function of CUX1 in oxidative DNA damage repair is needed to prevent premature senescence of mouse embryo fibroblasts. Oncotarget 2015, 6, 3613–3626. [Google Scholar] [CrossRef]
- Kaur, S.; Coulombe, Y.; Ramdzan, Z.M.; Leduy, L.; Masson, J.Y.; Nepveu, A. Special AT-rich sequence-binding protein 1 (SATB1) functions as an accessory factor in base excision repair. J. Biol. Chem. 2016, 291, 22769–22780. [Google Scholar] [CrossRef]
- Jang, S.; Kumar, N.; Beckwitt, E.C.; Kong, M.; Fouquerel, E.; Rapic-Otrin, V.; Prasad, R.; Watkins, S.C.; Khuu, C.; Majumdar, C.; et al. Damage sensor role of UV-DDB during base excision repair. Nat. Struct. Mol. Biol. 2019, 26, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Han, X.; Akbari, M.; Croteau, D.L.; Rasmussen, L.J.; Bohr, V.A. Interaction between RECQL4 and OGG1 promotes repair of oxidative base lesion 8-oxoG and is regulated by SIRT1 deacetylase. Nucleic Acids Res. 2020, 48, 6530–6546. [Google Scholar] [CrossRef]
- Kumar, N.; Theil, A.F.; Roginskaya, V.; Ali, Y.; Calderon, M.; Watkins, S.C.; Barnes, R.P.; Opresko, P.L.; Pines, A.; Lans, H.; et al. Global and transcription-coupled repair of 8-oxoG is initiated by nucleotide excision repair proteins. Nat. Commun. 2022, 13, 974. [Google Scholar] [CrossRef]
- Ba, X.; Boldogh, I. 8-Oxoguanine DNA glycosylase 1: Beyond repair of the oxidatively modified base lesions. Redox Biol. 2018, 14, 669–678. [Google Scholar] [CrossRef]
- Bangalore, D.M.; Tessmer, I. Direct hOGG1-Myc interactions inhibit hOGG1 catalytic activity and recruit Myc to its promoters under oxidative stress. Nucleic Acids Res. 2022, 50, 10385–10398. [Google Scholar] [CrossRef]
- Sharma, N.; Chakravarthy, S.; Longley, M.J.; Copeland, W.C.; Prakash, A. The C-terminal tail of the NEIL1 DNA glycosylase interacts with the human mitochondrial single-stranded DNA binding protein. DNA Repair 2018, 65, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Thompson, M.K.; Arrington, J.F.; Terry, D.M.; Chakravarthy, S.; Prevelige, P.E.; Prakash, A. Novel interaction interfaces mediate the interaction between the NEIL1 DNA glycosylase and mitochondrial transcription factor A. Front. Cell Dev. Biol. 2022, 10, 893806. [Google Scholar] [CrossRef] [PubMed]
- Kamenisch, Y.; Fousteri, M.; Knoch, J.; von Thaler, A.K.; Fehrenbacher, B.; Kato, H.; Becker, T.; Dolle, M.E.; Kuiper, R.; Majora, M.; et al. Proteins of nucleotide and base excision repair pathways interact in mitochondria to protect from loss of subcutaneous fat, a hallmark of aging. J. Exp. Med. 2010, 207, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.T.; De Luca, G.; Degan, P.; Parlanti, E.; Dogliotti, E.; Barnes, D.E.; Lindahl, T.; Yang, H.; Miller, J.H.; Bignami, M. Accumulation of the oxidative base lesion 8-hydroxyguanine in DNA of tumor-prone mice defective in both the Myh and Ogg1 DNA glycosylases. Cancer Res. 2004, 64, 4411–4414. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Yang, H.; Cunanan, C.; Okamoto, K.; Shibata, D.; Pan, J.; Barnes, D.E.; Lindahl, T.; McIlhatton, M.; Fishel, R.; et al. Deficiencies in mouse Myh and Ogg1 result in tumor predisposition and G to T mutations in codon 12 of the K-ras oncogene in lung tumors. Cancer Res. 2004, 64, 3096–3102. [Google Scholar] [CrossRef]
- Arai, T.; Kelly, V.P.; Minowa, O.; Noda, T.; Nishimura, S. The study using wild-type and Ogg1 knockout mice exposed to potassium bromate shows no tumor induction despite an extensive accumulation of 8-hydroxyguanine in kidney DNA. Toxicology 2006, 221, 179–186. [Google Scholar] [CrossRef]
- Kakehashi, A.; Ishii, N.; Okuno, T.; Fujioka, M.; Gi, M.; Wanibuchi, H. Enhanced susceptibility of Ogg1 mutant mice to multiorgan carcinogenesis. Int. J. Mol. Sci. 2017, 18, 1801. [Google Scholar] [CrossRef]
- Kunisada, M.; Sakumi, K.; Tominaga, Y.; Budiyanto, A.; Ueda, M.; Ichihashi, M.; Nakabeppu, Y.; Nishigori, C. 8-Oxoguanine formation induced by chronic UVB exposure makes Ogg1 knockout mice susceptible to skin carcinogenesis. Cancer Res. 2005, 65, 6006–6010. [Google Scholar] [CrossRef]
- Kakehashi, A.; Ishii, N.; Okuno, T.; Fujioka, M.; Gi, M.; Fukushima, S.; Wanibuchi, H. Progression of hepatic adenoma to carcinoma in Ogg1 mutant mice induced by phenobarbital. Oxid. Med. Cell. Longev. 2017, 2017, 8541064. [Google Scholar] [CrossRef]
- Vartanian, V.; Lowell, B.; Minko, I.G.; Wood, T.G.; Ceci, J.D.; George, S.; Ballinger, S.W.; Corless, C.L.; McCullough, A.K.; Lloyd, R.S. The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase. Proc. Natl. Acad. Sci. USA 2006, 103, 1864–1869. [Google Scholar] [CrossRef]
- Chan, M.K.; Ocampo-Hafalla, M.T.; Vartanian, V.; Jaruga, P.; Kirkali, G.; Koenig, K.L.; Brown, S.; Lloyd, R.S.; Dizdaroglu, M.; Teebor, G.W. Targeted deletion of the genes encoding NTH1 and NEIL1 DNA N-glycosylases reveals the existence of novel carcinogenic oxidative damage to DNA. DNA Repair 2009, 8, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Rolseth, V.; Luna, L.; Olsen, A.K.; Suganthan, R.; Scheffler, K.; Neurauter, C.G.; Esbensen, Y.; Kusnierczyk, A.; Hildrestrand, G.A.; Graupner, A.; et al. No cancer predisposition or increased spontaneous mutation frequencies in NEIL DNA glycosylases-deficient mice. Sci. Rep. 2017, 7, 4384. [Google Scholar] [CrossRef]
- Takahashi, Y.; Nakatsuru, Y.; Zhang, S.; Shimizu, Y.; Kume, H.; Tanaka, K.; Ide, F.; Ishikawa, T. Enhanced spontaneous and aflatoxin-induced liver tumorigenesis in xeroderma pigmentosum group A gene-deficient mice. Carcinogenesis 2002, 23, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Calkins, M.J.; Vartanian, V.; Owen, N.; Kirkali, G.; Jaruga, P.; Dizdaroglu, M.; McCullough, A.K.; Lloyd, R.S. Enhanced sensitivity of Neil1−/− mice to chronic UVB exposure. DNA Repair 2016, 48, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Sampath, H.; Batra, A.K.; Vartanian, V.; Carmical, J.R.; Prusak, D.; King, I.B.; Lowell, B.; Earley, L.F.; Wood, T.G.; Marks, D.L.; et al. Variable penetrance of metabolic phenotypes and development of high-fat diet-induced adiposity in NEIL1-deficient mice. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E724–E734. [Google Scholar] [CrossRef] [PubMed]
- Sampath, H.; Vartanian, V.; Rollins, M.R.; Sakumi, K.; Nakabeppu, Y.; Lloyd, R.S. 8-Oxoguanine DNA glycosylase (OGG1) deficiency increases susceptibility to obesity and metabolic dysfunction. PLoS ONE 2012, 7, e51697. [Google Scholar] [CrossRef]
- Vartanian, V.; Tumova, J.; Dobrzyn, P.; Dobrzyn, A.; Nakabeppu, Y.; Lloyd, R.S.; Sampath, H. 8-oxoguanine DNA glycosylase (OGG1) deficiency elicits coordinated changes in lipid and mitochondrial metabolism in muscle. PLoS ONE 2017, 12, e0181687. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J.; Warmbrunn, M.V.; Nieuwdorp, M.; Clement, K. Metabolism and metabolic disorders and the microbiome: The intestinal microbiota associated with obesity, lipid metabolism, and metabolic health-pathophysiology and therapeutic strategies. Gastroenterology 2021, 160, 573–599. [Google Scholar] [CrossRef]
- Simon, H.; Vartanian, V.; Wong, M.H.; Nakabeppu, Y.; Sharma, P.; Lloyd, R.S.; Sampath, H. OGG1 deficiency alters the intestinal microbiome and increases intestinal inflammation in a mouse model. PLoS ONE 2020, 15, e0227501. [Google Scholar] [CrossRef]
- Komakula, S.S.B.; Tumova, J.; Kumaraswamy, D.; Burchat, N.; Vartanian, V.; Ye, H.; Dobrzyn, A.; Lloyd, R.S.; Sampath, H. The DNA repair protein OGG1 protects against obesity by altering mitochondrial energetics in white adipose tissue. Sci. Rep. 2018, 8, 14886. [Google Scholar] [CrossRef]
- Burchat, N.; Sharma, P.; Ye, H.; Komakula, S.S.B.; Dobrzyn, A.; Vartanian, V.; Lloyd, R.S.; Sampath, H. Maternal transmission of human OGG1 protects mice against genetically- and diet-Induced obesity through increased tissue mitochondrial content. Front. Cell Dev. Biol. 2021, 9, 718962. [Google Scholar] [CrossRef] [PubMed]
- Komakula, S.S.B.; Blaze, B.; Ye, H.; Dobrzyn, A.; Sampath, H. A Novel Role for the DNA Repair Enzyme 8-Oxoguanine DNA Glycosylase in Adipogenesis. Int. J. Mol. Sci. 2021, 22, 1152. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Galick, H.; Wallace, S.S. Attempted base excision repair of ionizing radiation damage in human lymphoblastoid cells produces lethal and mutagenic double strand breaks. DNA Repair 2004, 3, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Chaudhry, M.A.; Wallace, S.S. Base excision repair by hNTH1 and hOGG1: A two edged sword in the processing of DNA damage in g-irradiated human cells. DNA Repair 2006, 5, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yuan, K.; Yan, C.; Fox, J., 3rd; Gaid, M.; Breitwieser, W.; Bansal, A.K.; Zeng, H.; Gao, H.; Wu, M. 8-Oxoguanine-DNA glycosylase 1 deficiency modifies allergic airway inflammation by regulating STAT6 and IL-4 in cells and in mice. Free Radic. Biol. Med. 2012, 52, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Ba, X.; Aguilera-Aguirre, L.; Rashid, Q.T.; Bacsi, A.; Radak, Z.; Sur, S.; Hosoki, K.; Hegde, M.L.; Boldogh, I. The role of 8-oxoguanine DNA glycosylase-1 in inflammation. Int. J. Mol. Sci. 2014, 15, 16975–16997. [Google Scholar] [CrossRef] [PubMed]
- Mabley, J.G.; Pacher, P.; Deb, A.; Wallace, R.; Elder, R.H.; Szabo, C. Potential role for 8-oxoguanine DNA glycosylase in regulating inflammation. FASEB J. 2005, 19, 290–292. [Google Scholar] [CrossRef]
- Touati, E.; Michel, V.; Thiberge, J.M.; Ave, P.; Huerre, M.; Bourgade, F.; Klungland, A.; Labigne, A. Deficiency in OGG1 protects against inflammation and mutagenic effects associated with H. pylori infection in mouse. Helicobacter 2006, 11, 494–505. [Google Scholar] [CrossRef]
- Yang, J.; Mitra, A.; Dojer, N.; Fu, S.; Rowicka, M.; Brasier, A.R. A probabilistic approach to learn chromatin architecture and accurate inference of the NF-kB/RelA regulatory network using ChIP-Seq. Nucleic Acids Res. 2013, 41, 7240–7259. [Google Scholar] [CrossRef][Green Version]
- Pan, L.; Zhu, B.; Hao, W.; Zeng, X.; Vlahopoulos, S.A.; Hazra, T.K.; Hegde, M.L.; Radak, Z.; Bacsi, A.; Brasier, A.R.; et al. Oxidized guanine base lesions function in 8-oxoguanine DNA glycosylase-1-mediated epigenetic regulation of nuclear factor kB-driven gene expression. J. Biol. Chem. 2016, 291, 25553–25566. [Google Scholar] [CrossRef]
- Park, J.W.; Han, Y.I.; Kim, S.W.; Kim, T.M.; Yeom, S.C.; Kang, J.; Park, J. 8-OxoG in GC-rich Sp1 binding sites enhances gene transcription in adipose tissue of juvenile mice. Sci. Rep. 2019, 9, 15618. [Google Scholar] [CrossRef]
- Perillo, B.; Ombra, M.N.; Bertoni, A.; Cuozzo, C.; Sacchetti, S.; Sasso, A.; Chiariotti, L.; Malorni, A.; Abbondanza, C.; Avvedimento, E.V. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 2008, 319, 202–206. [Google Scholar] [CrossRef]
- Fleming, A.M.; Burrows, C.J. G-quadruplex folds of the human telomere sequence alter the site reactivity and reaction pathway of guanine oxidation compared to duplex DNA. Chem. Res. Toxicol. 2013, 26, 593–607. [Google Scholar] [CrossRef]
- Fleming, A.M.; Zhu, J.; Ding, Y.; Visser, J.A.; Zhu, J.; Burrows, C.J. Human DNA repair genes possess potential G-quadruplex sequences in their promoters and 5′-untranslated regions. Biochemistry 2018, 57, 991–1002. [Google Scholar] [CrossRef]
- Rogers, R.A.; Fleming, A.M.; Burrows, C.J. Rapid screen of potential i-motif forming sequences in DNA repair gene promoters. ACS Omega 2018, 3, 9630–9635. [Google Scholar] [CrossRef]
- Fleming, A.M.; Zhu, J.; Ding, Y.; Esders, S.; Burrows, C.J. Oxidative modification of guanine in a potential Z-DNA-forming sequence of a gene promoter impacts gene expression. Chem. Res. Toxicol. 2019, 32, 899–909. [Google Scholar] [CrossRef]
- Donley, N.; Jaruga, P.; Coskun, E.; Dizdaroglu, M.; McCullough, A.K.; Lloyd, R.S. Small molecule inhibitors of 8-oxoguanine DNA glycosylase-1 (OGG1). ACS Chem. Biol. 2015, 10, 2334–2343. [Google Scholar] [CrossRef]
- Tahara, Y.K.; Auld, D.; Ji, D.; Beharry, A.A.; Kietrys, A.M.; Wilson, D.L.; Jimenez, M.; King, D.; Nguyen, Z.; Kool, E.T. Potent and selective inhibitors of 8-oxoguanine DNA glycosylase. J. Am. Chem. Soc. 2018, 140, 2105–2114. [Google Scholar] [CrossRef]
- Visnes, T.; Cazares-Korner, A.; Hao, W.; Wallner, O.; Masuyer, G.; Loseva, O.; Mortusewicz, O.; Wiita, E.; Sarno, A.; Manoilov, A.; et al. Small-molecule inhibitor of OGG1 suppresses proinflammatory gene expression and inflammation. Science 2018, 362, 834–839. [Google Scholar] [CrossRef]
- Tahara, Y.K.; Kietrys, A.M.; Hebenbrock, M.; Lee, Y.; Wilson, D.L.; Kool, E.T. Dual inhibitors of 8-oxoguanine surveillance by OGG1 and NUDT1. ACS Chem. Biol. 2019, 14, 2606–2615. [Google Scholar] [CrossRef]
- Ling, H.; Song, C.; Fang, Y.; Yin, Y.; Wu, Z.; Wang, Y.; Xu, Z.; Gao, S.; Li, A.; Liu, G. TH5487, a small molecule inhibitor of OGG1, attenuates pulmonary fibrosis by NEDD4L-mediated OGG1 degradation. Chem. Biol. Interact. 2022, 362, 109999. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wu, S.; Wu, G.; Yang, J. Inhibition of 8-oxoguanine DNA glycosylase (OGG1) expression suppresses polycystic ovarian syndrome via the NF-kB signaling pathway. Reprod. Biol. 2022, 22, 100679. [Google Scholar] [CrossRef] [PubMed]
- Baquero, J.M.; Benitez-Buelga, C.; Rajagopal, V.; Zhenjun, Z.; Torres-Ruiz, R.; Muller, S.; Hanna, B.M.F.; Loseva, O.; Wallner, O.; Michel, M.; et al. Small molecule inhibitor of OGG1 blocks oxidative DNA damage repair at telomeres and potentiates methotrexate anticancer effects. Sci. Rep. 2021, 11, 3490. [Google Scholar] [CrossRef] [PubMed]
- Baquero, J.M.; Marchena-Perea, E.; Mirabet, R.; Torres-Ruiz, R.; Blanco-Aparicio, C.; Rodriguez-Perales, S.; Helleday, T.; Benitez-Buelga, C.; Benitez, J.; Osorio, A. OGG1 inhibition triggers synthetic lethality and enhances the effect of PARP inhibitor olaparib in BRCA1-deficient TNBC cells. Front. Oncol. 2022, 12, 888810. [Google Scholar] [CrossRef]
- Karsten, S. Targeting the DNA repair enzymes MTH1 and OGG1 as a novel approach to treat inflammatory diseases. Basic Clin. Pharmacol. Toxicol. 2022, 131, 95–103. [Google Scholar] [CrossRef]
- Gatto, E.M.; Rojas, N.G.; Persi, G.; Etcheverry, J.L.; Cesarini, M.E.; Perandones, C. Huntington disease: Advances in the understanding of its mechanisms. Clin. Park. Relat. Disord. 2020, 3, 100056. [Google Scholar] [CrossRef]
- Monckton, D.G. The contribution of somatic expansion of the CAG repeat to symptomatic development in Huntington’s disease: A historical perspective. J. Huntingtons. Dis. 2021, 10, 7–33. [Google Scholar] [CrossRef]
- Kovtun, I.V.; Liu, Y.; Bjoras, M.; Klungland, A.; Wilson, S.H.; McMurray, C.T. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature 2007, 447, 447–452. [Google Scholar] [CrossRef]
- Goula, A.V.; Berquist, B.R.; Wilson, D.M., 3rd; Wheeler, V.C.; Trottier, Y.; Merienne, K. Stoichiometry of base excision repair proteins correlates with increased somatic CAG instability in striatum over cerebellum in Huntington’s disease transgenic mice. PLoS Genet. 2009, 5, e1000749. [Google Scholar] [CrossRef]
- Kovtun, I.V.; Johnson, K.O.; McMurray, C.T. Cockayne syndrome B protein antagonizes OGG1 in modulating CAG repeat length in vivo. Aging 2011, 3, 509–514. [Google Scholar] [CrossRef]
- Iyer, R.R.; Pluciennik, A. DNA mismatch repair and its role in Huntington’s disease. J. Huntingtons. Dis. 2021, 10, 75–94. [Google Scholar] [CrossRef] [PubMed]
- Budworth, H.; Harris, F.R.; Williams, P.; Lee, D.Y.; Holt, A.; Pahnke, J.; Szczesny, B.; Acevedo-Torres, K.; Ayala-Pena, S.; McMurray, C.T. Suppression of somatic expansion delays the onset of pathophysiology in a mouse model of Huntington’s disease. PLoS Genet. 2015, 11, e1005267. [Google Scholar] [CrossRef] [PubMed]
- Mollersen, L.; Rowe, A.D.; Illuzzi, J.L.; Hildrestrand, G.A.; Gerhold, K.J.; Tveteras, L.; Bjolgerud, A.; Wilson, D.M., 3rd; Bjoras, M.; Klungland, A. Neil1 is a genetic modifier of somatic and germline CAG trinucleotide repeat instability in R6/1 mice. Hum. Mol. Genet. 2012, 21, 4939–4947. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Yoon, J.S.; Feldman, N.H.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. Endonuclease VIII-like 1 (NEIL1) promotes short-term spatial memory retention and protects from ischemic stroke-induced brain dysfunction and death in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 14948–14953. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Misiak, M.; Scheibye-Knudsen, M.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. Loss of NEIL1 causes defects in olfactory function in mice. Neurobiol. Aging 2015, 36, 1007–1012. [Google Scholar] [CrossRef]
- Yang, B.; Figueroa, D.M.; Hou, Y.; Babbar, M.; Baringer, S.L.; Croteau, D.L.; Bohr, V.A. NEIL1 stimulates neurogenesis and suppresses neuroinflammation after stress. Free Radic. Biol. Med. 2019, 141, 47–58. [Google Scholar] [CrossRef]
- Hildrestrand, G.A.; Rolseth, V.; Kunath, N.; Suganthan, R.; Jensen, V.; Bugaj, A.M.; Fernandez-Berrocal, M.S.; Sikko, S.B.; Vetlesen, S.; Kusnierczyk, A.; et al. NEIL1 and NEIL2 DNA glycosylases modulate anxiety and learning in a cooperative manner in mice. Commun. Biol. 2021, 4, 1354. [Google Scholar] [CrossRef]
- Hung, R.J.; Hall, J.; Brennan, P.; Boffetta, P. Genetic polymorphisms in the base excision repair pathway and cancer risk: A HuGE review. Am. J. Epidemiol. 2005, 162, 925–942. [Google Scholar] [CrossRef]
- Hill, J.W.; Evans, M.K. Dimerization and opposite base-dependent catalytic impairment of polymorphic S326C OGG1 glycosylase. Nucleic Acids Res. 2006, 34, 1620–1632. [Google Scholar] [CrossRef]
- Bravard, A.; Vacher, M.; Moritz, E.; Vaslin, L.; Hall, J.; Epe, B.; Radicella, J.P. Oxidation status of human OGG1-S326C polymorphic variant determines cellular DNA repair capacity. Cancer Res. 2009, 69, 3642–3649. [Google Scholar] [CrossRef]
- Baptiste, B.A.; Katchur, S.R.; Fivenson, E.M.; Croteau, D.L.; Rumsey, W.L.; Bohr, V.A. Enhanced mitochondrial DNA repair of the common disease-associated variant, Ser326Cys, of hOGG1 through small molecule intervention. Free Radic. Biol. Med. 2018, 124, 149–162. [Google Scholar] [CrossRef]
- Kabzinski, J.; Walczak, A.; Dziki, A.; Mik, M.; Majsterek, I. Impact of the Ser326Cys polymorphism of the OGG1 gene on the level of oxidative DNA damage in patients with colorectal cancer. Pol. Przegl. Chir. 2018, 90, 13–15. [Google Scholar] [CrossRef]
- Alanazi, M.; Pathan, A.A.K.; Shaik, J.P.; Alhadheq, A.; Khan, Z.; Khan, W.; Al Naeem, A.; Parine, N.R. The hOGG1 Ser326Cys gene polymorphism and breast cancer risk in Saudi population. Pathol. Oncol. Res. 2017, 23, 525–535. [Google Scholar] [CrossRef]
- Costa, E.F.; Santos, E.S.; Liutti, V.T.; Leal, F.; Santos, V.C.; Rinck-Junior, J.A.; Mariano, F.V.; Coutinho-Camillo, C.M.; Altemani, A.; Lima, C.S.; et al. Association between polymorphisms in genes related to DNA base-excision repair with risk and prognosis of oropharyngeal squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2016, 142, 1917–1926. [Google Scholar] [CrossRef]
- Gotoh, N.; Saitoh, T.; Takahashi, N.; Kasamatsu, T.; Minato, Y.; Lobna, A.; Oda, T.; Hoshino, T.; Sakura, T.; Shimizu, H.; et al. Association between OGG1 S326C CC genotype and elevated relapse risk in acute myeloid leukemia. Int. J. Hematol. 2018, 108, 246–253. [Google Scholar] [CrossRef]
- Smolarz, B.; Michalska, M.M.; Samulak, D.; Wojcik, L.; Romanowicz, H. Studies of correlations between single nucleotide polymorphisms of DNA repair genes and endometrial cancer in Polish women. Anticancer Res. 2018, 38, 5223–5229. [Google Scholar] [CrossRef]
- Xu, J.; Zheng, S.L.; Turner, A.; Isaacs, S.D.; Wiley, K.E.; Hawkins, G.A.; Chang, B.L.; Bleecker, E.R.; Walsh, P.C.; Meyers, D.A.; et al. Associations between hOGG1 sequence variants and prostate cancer susceptibility. Cancer Res. 2002, 62, 2253–2257. [Google Scholar]
- Chen, L.; Elahi, A.; Pow-Sang, J.; Lazarus, P.; Park, J. Association between polymorphism of human oxoguanine glycosylase 1 and risk of prostate cancer. J. Urol. 2003, 170, 2471–2474. [Google Scholar] [CrossRef]
- Le Marchand, L.; Donlon, T.; Lum-Jones, A.; Seifried, A.; Wilkens, L.R. Association of the hOGG1 Ser326Cys polymorphism with lung cancer risk. Cancer Epidemiol. Biomark. Prev. 2002, 11, 409–412. [Google Scholar]
- Geer, L.Y.; Marchler-Bauer, A.; Geer, R.C.; Han, L.; He, J.; He, S.; Liu, C.; Shi, W.; Bryant, S.H. The NCBI BioSystems database. Nucleic Acids Res. 2010, 38, D492–D496. [Google Scholar] [CrossRef]
- Hyun, J.W.; Choi, J.Y.; Zeng, H.H.; Lee, Y.S.; Kim, H.S.; Yoon, S.H.; Chung, M.H. Leukemic cell line, KG-1 has a functional loss of hOGG1 enzyme due to a point mutation and 8-hydroxydeoxyguanosine can kill KG-1. Oncogene 2000, 19, 4476–4479. [Google Scholar] [CrossRef]
- Hyun, J.W.; Cheon, G.J.; Kim, H.S.; Lee, Y.S.; Choi, E.Y.; Yoon, B.H.; Kim, J.S.; Chung, M.H. Radiation sensitivity depends on OGG1 activity status in human leukemia cell lines. Free Radic. Biol. Med. 2002, 32, 212–220. [Google Scholar] [CrossRef]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef]
- Owen, N.; Minko, I.G.; Moellmer, S.A.; Cammann, S.K.; Lloyd, R.S.; McCullough, A.K. Enhanced cytarabine-induced killing in OGG1-deficient acute myeloid leukemia cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2016833118. [Google Scholar] [CrossRef]
- Galick, H.A.; Marsden, C.G.; Kathe, S.; Dragon, J.A.; Volk, L.; Nemec, A.A.; Wallace, S.S.; Prakash, A.; Doublie, S.; Sweasy, J.B. The NEIL1 G83D germline DNA glycosylase variant induces genomic instability and cellular transformation. Oncotarget 2017, 8, 85883–85895. [Google Scholar] [CrossRef]
- Minko, I.G.; Vartanian, V.L.; Tozaki, N.N.; Linde, O.K.; Jaruga, P.; Coskun, S.H.; Coskun, E.; Qu, C.; He, H.; Xu, C.; et al. Characterization of rare NEIL1 variants found in East Asian populations. DNA Repair 2019, 79, 32–39. [Google Scholar] [CrossRef]
- Thameem, F.; Puppala, S.; Lehman, D.M.; Stern, M.P.; Blangero, J.; Abboud, H.E.; Duggirala, R.; Habib, S.L. The Ser(326)Cys polymorphism of 8-oxoguanine glycosylase 1 (OGG1) is associated with type 2 diabetes in Mexican Americans. Hum. Hered. 2010, 70, 97–101. [Google Scholar] [CrossRef]
- Anderson, S.M.; Naidoo, R.N.; Ramkaran, P.; Asharam, K.; Muttoo, S.; Chuturgoon, A.A. OGG1 Ser326Cys polymorphism, HIV, obesity and air pollution exposure influences adverse birth outcome susceptibility, within South African Women. Reprod. Toxicol. 2018, 79, 8–15. [Google Scholar] [CrossRef]
- Sliwinska, A.; Kwiatkowski, D.; Czarny, P.; Toma, M.; Wigner, P.; Drzewoski, J.; Fabianowska-Majewska, K.; Szemraj, J.; Maes, M.; Galecki, P.; et al. The levels of 7,8-dihydrodeoxyguanosine (8-oxoG) and 8-oxoguanine DNA glycosylase 1 (OGG1)—A potential diagnostic biomarkers of Alzheimer’s disease. J. Neurol. Sci. 2016, 368, 155–159. [Google Scholar] [CrossRef]
- Sanders, L.H.; Paul, K.C.; Howlett, E.H.; Lawal, H.; Boppana, S.; Bronstein, J.M.; Ritz, B.; Greenamyre, J.T. Editor’s highlight: Base excision repair variants and pesticide exposure increase Parkinson’s disease risk. Toxicol. Sci. 2017, 158, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Corella, D.; Ramirez-Sabio, J.B.; Coltell, O.; Ortega-Azorin, C.; Estruch, R.; Martinez-Gonzalez, M.A.; Salas-Salvado, J.; Sorli, J.V.; Castaner, O.; Aros, F.; et al. Effects of the Ser326Cys polymorphism in the DNA repair OGG1 gene on cancer, cardiovascular, and all-cause mortality in the PREDIMED study: Modulation by diet. J. Acad. Nutr. Diet. 2018, 118, 589–605. [Google Scholar] [CrossRef] [PubMed]
- Fromme, J.C.; Bruner, S.D.; Yang, W.; Karplus, M.; Verdine, G.L. Product-assisted catalysis in base-excision DNA repair. Nat. Struct. Biol. 2003, 10, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Katchur, S.R.; Jiang, Y.; Briand, J.; Schaber, M.; Kreatsoulas, C.; Schwartz, B.; Thrall, S.; Davis, A.M.; Duvall, S.; et al. Small molecule-mediated allosteric activation of the base excision repair enzyme 8-oxoguanine DNA glycosylase and its impact on mitochondrial function. Sci. Rep. 2022, 12, 14685. [Google Scholar] [CrossRef]
- Michel, M.; Benitez-Buelga, C.; Calvo, P.A.; Hanna, B.M.F.; Mortusewicz, O.; Masuyer, G.; Davies, J.; Wallner, O.; Sanjiv, K.; Albers, J.J.; et al. Small-molecule activation of OGG1 increases oxidative DNA damage repair by gaining a new function. Science 2022, 376, 1471–1476. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lloyd, R.S. Complex Roles of NEIL1 and OGG1: Insights Gained from Murine Knockouts and Human Polymorphic Variants. DNA 2022, 2, 279-301. https://doi.org/10.3390/dna2040020
Lloyd RS. Complex Roles of NEIL1 and OGG1: Insights Gained from Murine Knockouts and Human Polymorphic Variants. DNA. 2022; 2(4):279-301. https://doi.org/10.3390/dna2040020
Chicago/Turabian StyleLloyd, R. Stephen. 2022. "Complex Roles of NEIL1 and OGG1: Insights Gained from Murine Knockouts and Human Polymorphic Variants" DNA 2, no. 4: 279-301. https://doi.org/10.3390/dna2040020
APA StyleLloyd, R. S. (2022). Complex Roles of NEIL1 and OGG1: Insights Gained from Murine Knockouts and Human Polymorphic Variants. DNA, 2(4), 279-301. https://doi.org/10.3390/dna2040020