

Multi-Faceted Roles of ERCC1-XPF Nuclease in Processing Non-B DNA Structures



Abstract
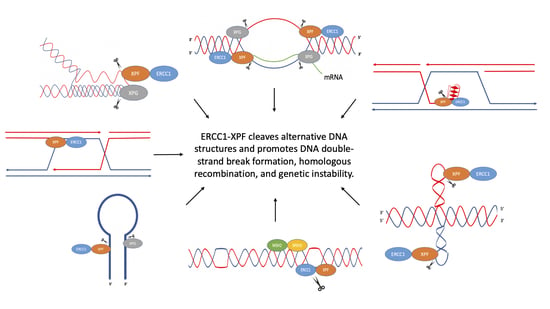
{kind=link}
{kind=link}
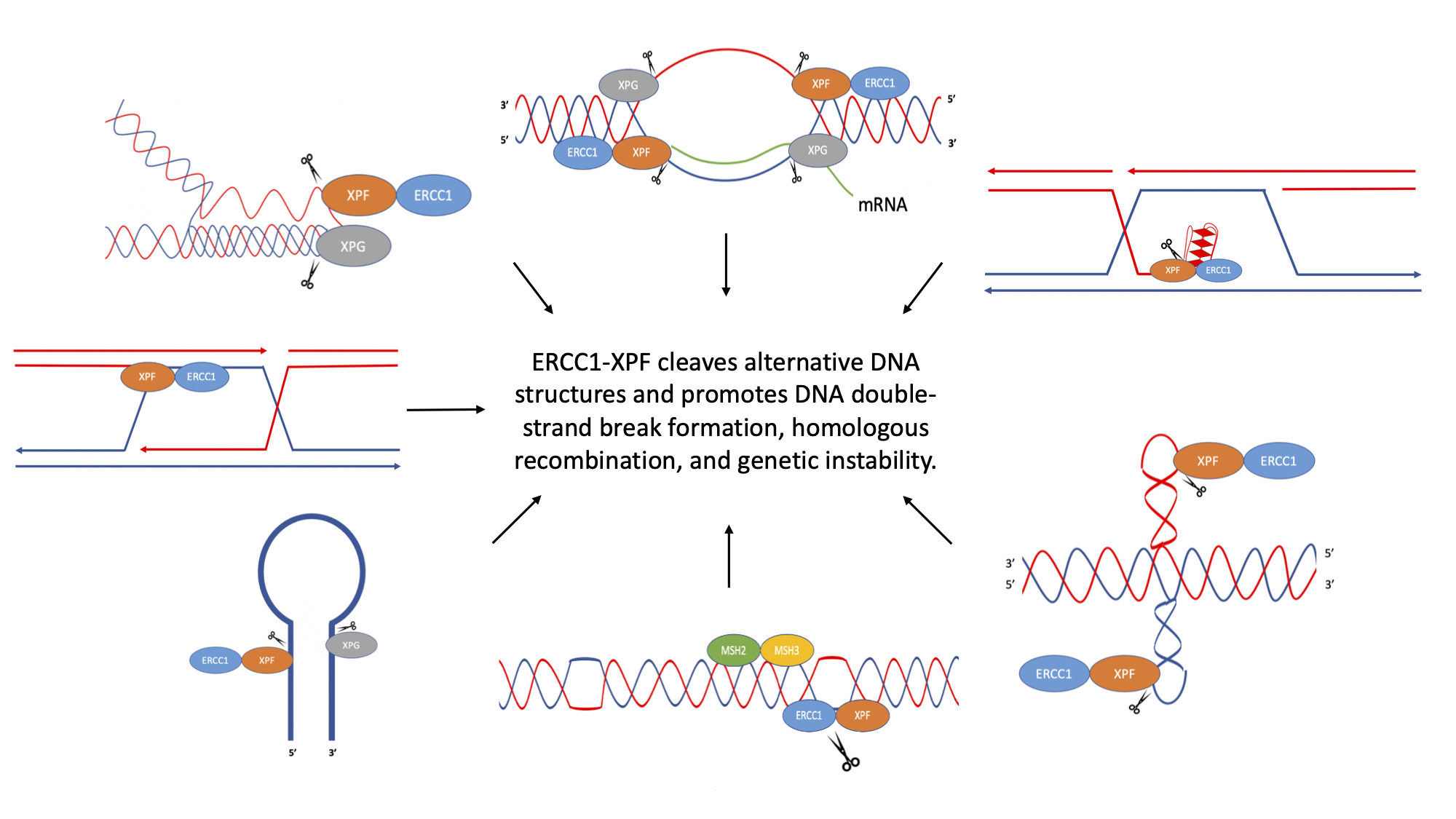
{kind=link}
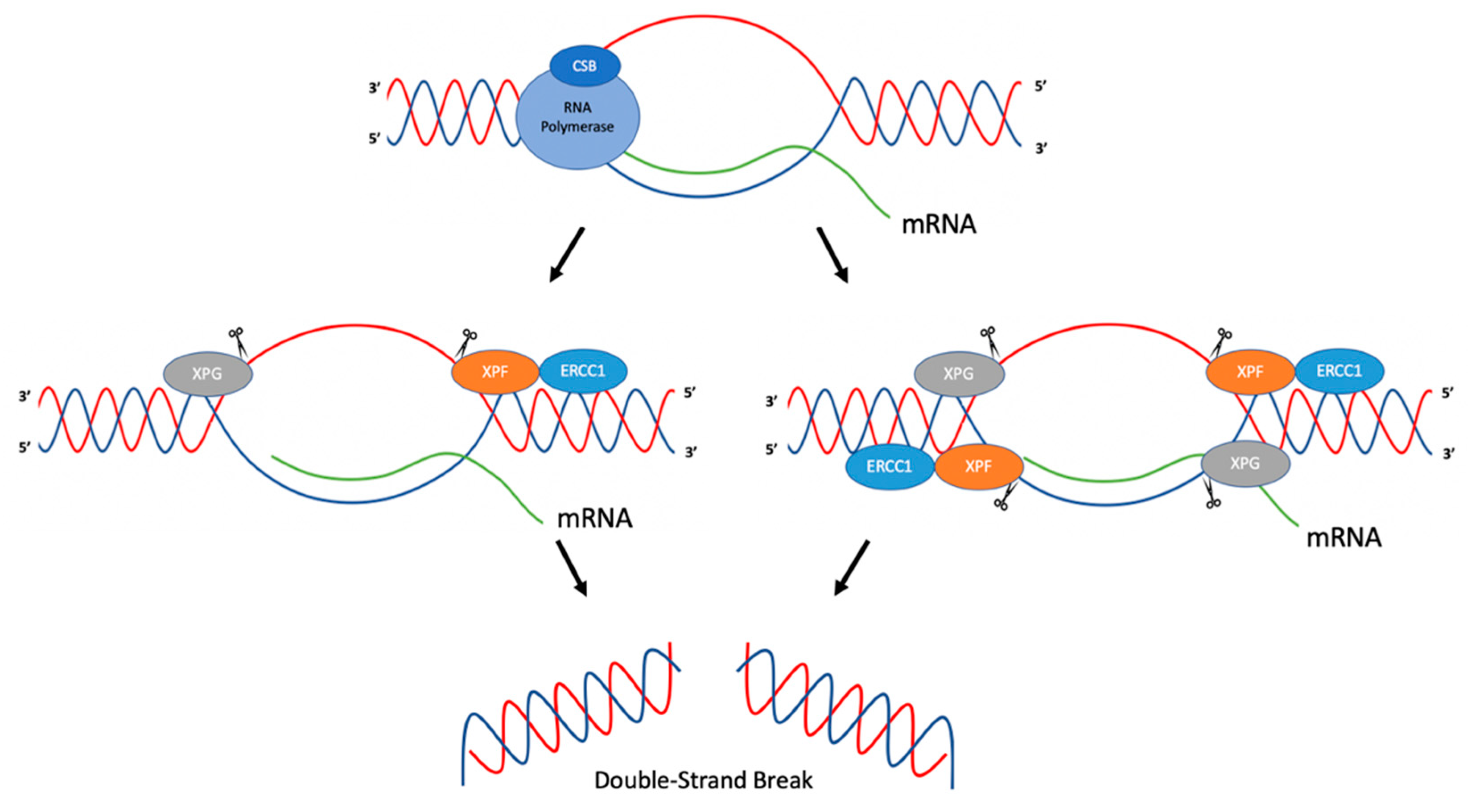
{kind=link}
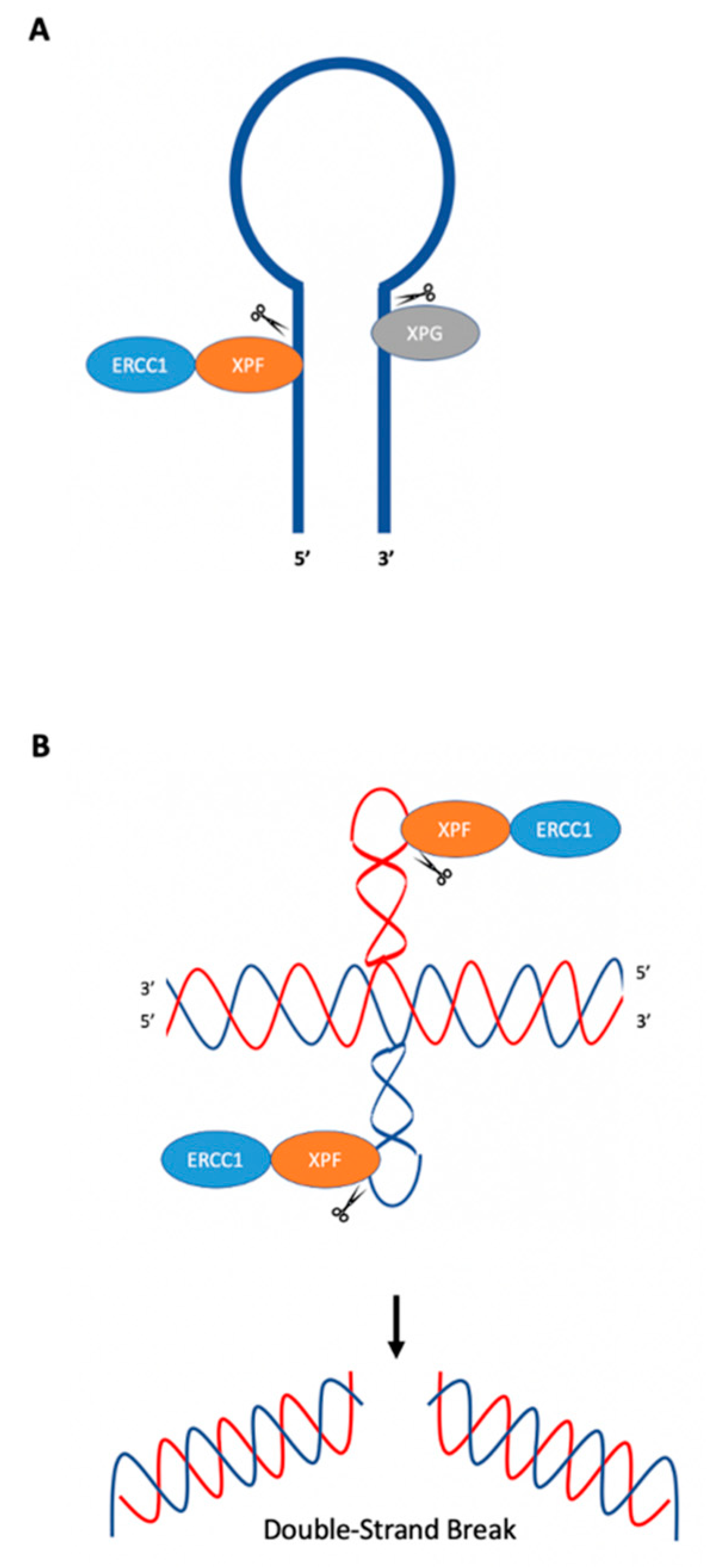{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Non-B DNA
3. R-Loops
4. D-Loops
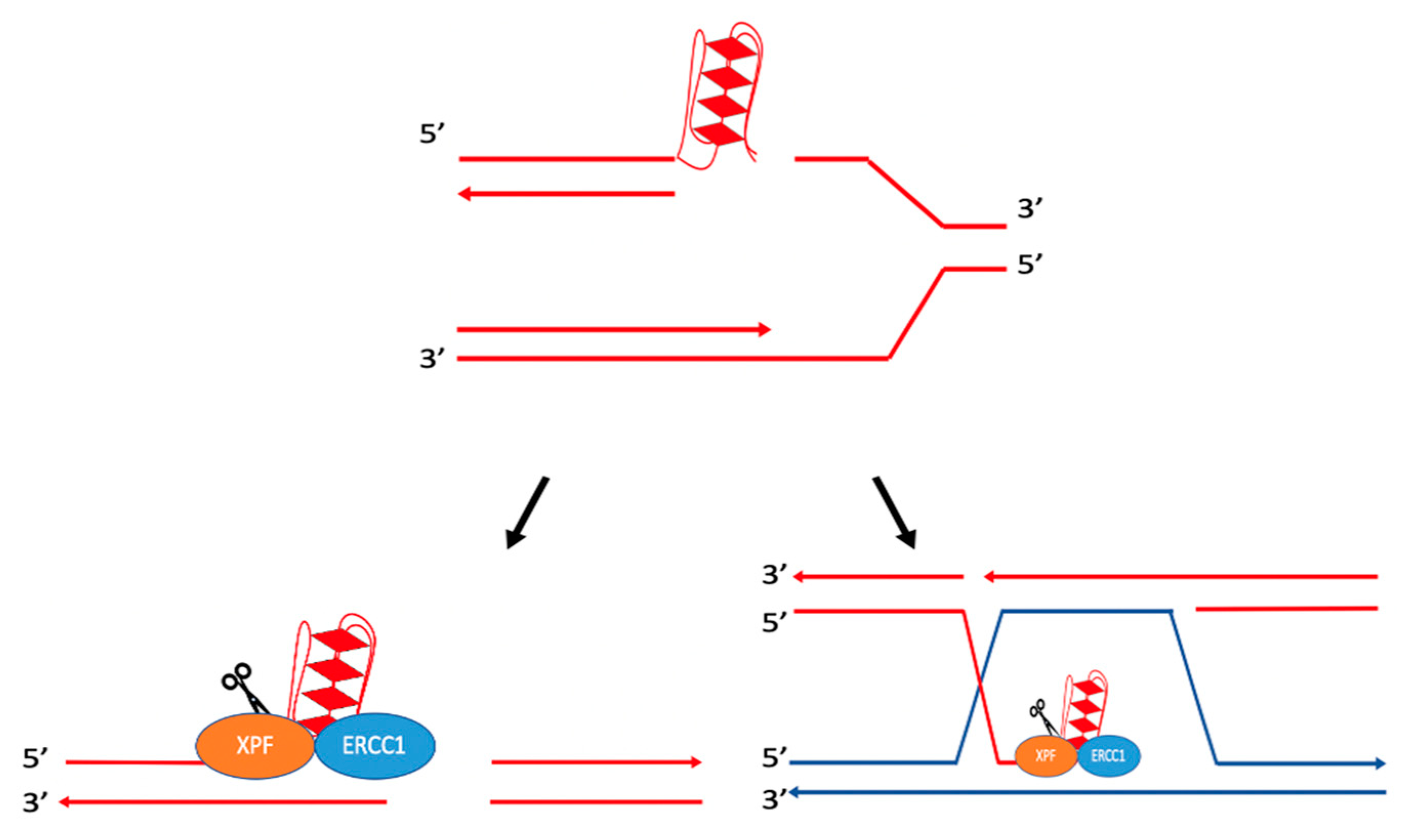
5. Hairpin/Stem-Loops and Cruciform DNA
6. Z-DNA
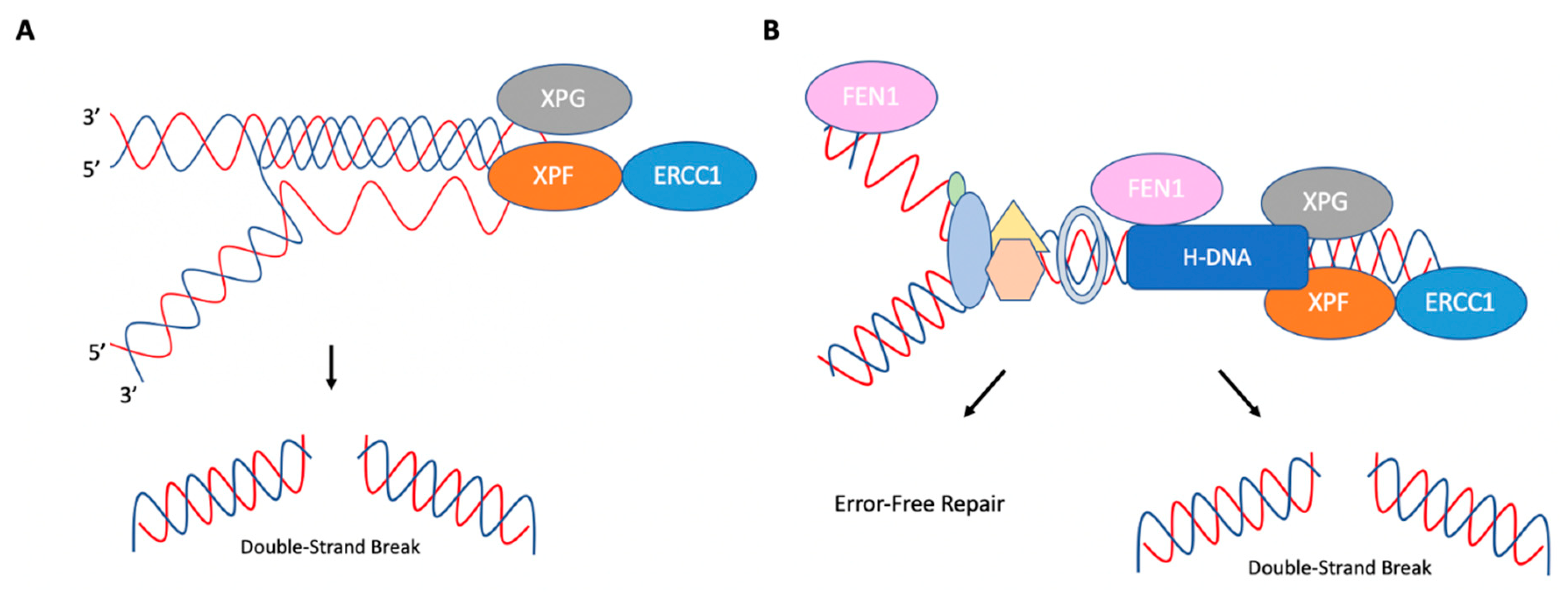
7. H-DNA

8. G-Quadruplex (G4 DNA)
9. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bartek, J. DNA damage response, genetic instability and cancer: From mechanistic insights to personalized treatment. Mol. Oncol. 2011, 5, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Bindra, R.S.; Glazer, P.M. Genetic instability and the tumor microenvironment: Towards the concept of microenvironment-induced mutagenesis. Mutat. Res. 2005, 569, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Beckman, R.A.; Loeb, L.A. Genetic instability in cancer: Theory and experiment. Semin. Cancer Biol. 2005, 15, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Breivik, J.; Gaudernack, G. Genomic instability, DNA methylation, and natural selection in colorectal carcinogenesis. Semin. Cancer Biol. 1999, 9, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Gregg, S.Q.; Robinson, A.R.; Niedernhofer, L.J. Physiological consequences of defects in ERCC1-XPF DNA repair endonuclease. DNA Repair 2011, 10, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, K.; Melton, D.W. Multiple roles of the ERCC1-XPF endonuclease in DNA repair and resistance to anticancer drugs. Anticancer Res. 2010, 30, 3223–3232. [Google Scholar] [PubMed]
- Manandhar, M.; Boulware, K.S.; Wood, R.D. The ERCC1 and ERCC4 (XPF) genes and gene products. Gene 2015, 569, 153–161. [Google Scholar] [CrossRef]
- Faridounnia, M.; Folkers, G.E.; Boelens, R. Function and Interactions of ERCC1-XPF in DNA Damage Response. Molecules 2018, 23, 3205. [Google Scholar] [CrossRef]
- Wang, G.; Vasquez, K.M. Non-B DNA structure-induced genetic instability. Mutat. Res. 2006, 598, 103–119. [Google Scholar] [CrossRef]
- Bacolla, A.; Wells, R.D. Non-B DNA conformations, genomic rearrangements, and human disease. J. Biol. Chem. 2004, 279, 47411–47414. [Google Scholar] [CrossRef]
- Zhao, J.; Bacolla, A.; Wang, G.; Vasquez, K.M. Non-B DNA structure-induced genetic instability and evolution. Cell Mol. Life Sci. 2010, 67, 43–62. [Google Scholar] [CrossRef]
- Wang, G.; Vasquez, K.M. Impact of alternative DNA structures on DNA damage, DNA repair, and genetic instability. DNA Repair 2014, 19, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.T.; Wang, G.; Thompson, A.C.; Wucherpfennig, J.I.; Reimchen, T.E.; MacColl, A.D.C.; Schluter, D.; Bell, M.A.; Vasquez, K.M.; Kingsley, D.M. DNA fragility in the parallel evolution of pelvic reduction in stickleback fish. Science 2019, 363, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Bacolla, A.; Tainer, J.A.; Vasquez, K.M.; Cooper, D.N. Translocation and deletion breakpoints in cancer genomes are associated with potential non-B DNA-forming sequences. Nucleic Acids Res. 2016, 44, 5673–5688. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, H.; Ohye, T.; Kogo, H.; Kato, T.; Bolor, H.; Taniguchi, M.; Shaikh, T.H.; Emanuel, B.S.; Kurahasi, H. Chromosomal instability mediated by non-B DNA: Cruciform conformation and not DNA sequence is responsible for recurrent translocation in humans. Genome Res. 2009, 19, 191–198. [Google Scholar] [CrossRef]
- Sollier, J.; Cimprich, K.A. Breaking bad: R-loops and genome integrity. Trends Cell Biol. 2015, 25, 514–522. [Google Scholar] [CrossRef]
- Hamperl, S.; Cimprich, K.A. The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair 2014, 19, 84–94. [Google Scholar] [CrossRef]
- Aguilera, A.; García-Muse, T. R loops: From transcription byproducts to threats to genome stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef]
- Li, X.; Manley, J.L. Cotranscriptional processes and their influence on genome stability. Genes Dev. 2006, 20, 1838–1847. [Google Scholar] [CrossRef]
- Skourti-Stathaki, K.; Proudfoot, N.J. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014, 28, 1384–1396. [Google Scholar] [CrossRef]
- Gan, W.; Guan, Z.; Liu, J.; Gui, T.; Shen, K.; Manley, J.L.; Li, X. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011, 25, 2041–2056. [Google Scholar] [CrossRef] [PubMed]
- Groh, M.; Lufino, M.M.P.; Wade-Martins, R.; Gromak, N. R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet. 2014, 10, e1004318. [Google Scholar] [CrossRef] [PubMed]
- Cerritelli, S.M.; Crouch, R.J. Ribonuclease H: The enzymes in eukaryotes. FEBS J. 2009, 276, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Hausen, P.; Stein, H. Ribonuclease H: An enzyme degrading the RNA moiety of DNA-RNA hybrids. Eur. J. Biochem. 1970, 14, 278–283. [Google Scholar] [CrossRef]
- Stein, H.; Hausen, P. Enzyme from calf thymus degrading the RNA moiety of DNA-RNA Hybrids: Effect on DNA-dependent RNA polymerase. Science 1969, 166, 393–395. [Google Scholar] [CrossRef]
- Wahba, L.; Amon, J.D.; Koshland, D.; Vuica-Ross, M. RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol. Cell 2011, 44, 978–988. [Google Scholar] [CrossRef]
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 2011, 42, 794–805. [Google Scholar] [CrossRef]
- Mischo, H.E.; Gómez-González, B.; Grzechnik, P.; Rondón, A.G.; Wei, W.; Steinmetz, L.; Aguilera, A.; Proudfoot, N.J. Yeast Sen1 helicase protects the genome from transcription-associated instability. Mol. Cell 2011, 41, 21–32. [Google Scholar] [CrossRef]
- Tuduri, S.; Crabbé, L.; Conti, C.; Tourrière, H.; Holtgreve-Grez, H.; Jauch, A.; Pantesco, V.; De Vos, J.; Thomas, A.; Theillet, C.; et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009, 11, 1315–1324. [Google Scholar] [CrossRef]
- Tian, M.; Alt, F.W. Transcription-induced cleavage of immunoglobulin switch regions by nucleotide excision repair nucleases in vitro. J. Biol. Chem. 2000, 275, 24163–24172. [Google Scholar] [CrossRef]
- Sollier, J.; Stork, C.T.; García-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.D.; Heyer, W.-D. Rad54 functions as a heteroduplex DNA pump modulated by its DNA substrates and Rad51 during D loop formation. Mol. Cell 2014, 53, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, E.K.; Heyer, W.-D. Processing of joint molecule intermediates by structure-selective endonucleases during homologous recombination in eukaryotes. Chromosoma 2011, 120, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Giaccherini, C.; Gaillard, P.-H. Control of structure-specific endonucleases during homologous recombination in eukaryotes. Curr. Opin. Genet. Dev. 2021, 71, 195–205. [Google Scholar] [CrossRef]
- Mazón, G.; Lam, A.F.; Ho, C.K.; Kupiec, M.; Symington, L.S. The Rad1-Rad10 nuclease promotes chromosome translocations between dispersed repeats. Nat. Struct. Mol. Biol. 2012, 19, 964–971. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.-D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef]
- Cejka, P.; Plank, J.L.; Bachrati, C.Z.; Hickson, I.D.; Kowalczykowski, S.C. Rmi1 stimulates decatenation of double Holliday junctions during dissolution by Sgs1-Top3. Nat. Struct. Mol. Biol. 2010, 17, 1377–1382. [Google Scholar] [CrossRef]
- Lyndaker, A.M.; Alani, E. A tale of tails: Insights into the coordination of 3′ end processing during homologous recombination. Bioessays 2009, 31, 315–321. [Google Scholar] [CrossRef]
- Nassif, N.; Penney, J.; Pal, S.; Engels, W.R.; Gloor, G.B. Efficient copying of nonhomologous sequences from ectopic sites via P-element-induced gap repair. Mol. Cell Biol. 1994, 14, 1613–1625. [Google Scholar]
- Wright, W.D.; Shah, S.S.; Heyer, W.-D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef]
- Fishman-Lobell, J.; Haber, J.E. Removal of nonhomologous DNA ends in double-strand break recombination: The role of the yeast ultraviolet repair gene RAD1. Science 1992, 258, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Pâques, F.; Haber, J.E. Two pathways for removal of nonhomologous DNA ends during double-strand break repair in Saccharomyces cerevisiae. Mol. Cell. Biol. 1997, 17, 6765–6771. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pâques, F.; Haber, J.E. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1999, 63, 349–404. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, N.; Pâques, F.; Colaiácovo, M.; Haber, J.E. Role of Saccharomyces cerevisiae Msh2 and Msh3 repair proteins in double-strand break-induced recombination. Proc. Natl. Acad. Sci. USA 1997, 94, 9214–9219. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.R. Meeting DNA palindromes head-to-head. Genes Dev. 2008, 22, 2612–2620. [Google Scholar] [CrossRef][Green Version]
- Bikard, D.; Loot, C.; Baharoglu, Z.; Mazel, D. Folded DNA in action: Hairpin formation and biological functions in prokaryotes. Microbiol. Mol. Biol. Rev. 2010, 74, 570–588. [Google Scholar] [CrossRef]
- Schroth, G.P.; Ho, P.S. Occurrence of potential cruciform and H-DNA forming sequences in genomic DNA. Nucleic Acids Res. 1995, 23, 1977–1983. [Google Scholar] [CrossRef]
- Kurahashi, H.; Inagaki, H.; Ohye, T.; Kogo, H.; Kato, T.; Emanuel, B.S. Palindrome-mediated chromosomal translocations in humans. DNA Repair 2006, 5, 1136–1145. [Google Scholar] [CrossRef]
- Kato, T.; Kurahashi, H.; Emanuel, B.S. Chromosomal translocations and palindromic AT-rich repeats. Curr. Opin. Genet. Dev. 2012, 22, 221–228. [Google Scholar] [CrossRef]
- Cromie, G.A.; Millar, C.B.; Schmidt, K.H.; Leach, D.R. Palindromes as substrates for multiple pathways of recombination in Escherichia coli. Genetics 2000, 154, 513–522. [Google Scholar] [CrossRef]
- De Laat, W.L.; Appeldoorn, E.; Jaspers, N.G.; Hoeijmakers, J.H. DNA structural elements required for ERCC1-XPF endonuclease activity. J. Biol. Chem. 1998, 273, 7835–7842. [Google Scholar] [CrossRef] [PubMed]
- Sijbers, A.M.; de Laat, W.L.; Ariza, R.R.; Biggerstaff, M.; Wei, Y.F.; Moggs, J.G.; Carter, K.C.; Shell, B.K.; Evans, E.; de Jong, M.C.; et al. Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell 1996, 86, 811–822. [Google Scholar] [CrossRef]
- Lu, S.; Wang, G.; Bacolla, A.; Zhao, J.; Spitser, S.; Vasquez, K.M. Short Inverted Repeats Are Hotspots for Genetic Instability: Relevance to Cancer Genomes. Cell Rep. 2015, 10, 1674–1680. [Google Scholar] [CrossRef]
- Habraken, Y.; Sung, P.; Prakash, L.; Prakash, S. Holliday junction cleavage by yeast Rad1 protein. Nature 1994, 371, 531–534. [Google Scholar] [CrossRef] [PubMed]
- West, S.C. Holliday junctions cleaved by Rad1? Nature 1995, 373, 27–28. [Google Scholar] [CrossRef] [PubMed]
- Bardwell, A.J.; Bardwell, L.; Tomkinson, A.E.; Friedberg, E.C. Specific cleavage of model recombination and repair intermediates by the yeast Rad1-Rad10 DNA endonuclease. Science 1994, 265, 2082–2085. [Google Scholar] [CrossRef]
- Rich, A.; Nordheim, A.; Wang, A.H.-J. The chemistry and biology of left-handed Z-DNA. Annu. Rev. Biochem. 1984, 53, 791–846. [Google Scholar] [CrossRef]
- Singleton, C.K.; Klysik, J.; Stirdivant, S.M.; Wells, R.D. Left-handed Z-DNA is induced by supercoiling in physiological ionic conditions. Nature 1982, 299, 312–316. [Google Scholar] [CrossRef]
- Singleton, C.K.; Klysik, J.; Wells, R.D. Conformational flexibility of junctions between contiguous B- and Z-DNAs in supercoiled plasmids. Proc. Natl. Acad. Sci. USA 1983, 80, 2447–2451. [Google Scholar] [CrossRef]
- Miesfeld, R.; Krystal, M.; Arnheim, N. A member of a new repeated sequence family which is conserved throughout eucaryotic evolution is found between the human delta and beta globin genes. Nucleic Acids Res. 1981, 9, 5931–5947. [Google Scholar] [CrossRef]
- Hamada, H.; Petrino, M.G.; Kakunaga, T. A novel repeated element with Z-DNA-forming potential is widely found in evolutionarily diverse eukaryotic genomes. Proc. Natl. Acad. Sci. USA 1982, 79, 6465–6469. [Google Scholar] [CrossRef] [PubMed]
- Hamada, H.; Kakunaga, T. Potential Z-DNA forming sequences are highly dispersed in the human genome. Nature 1982, 298, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Schroth, G.P.; Chou, P.-J.; Ho, P.S. Mapping Z-DNA in the human genome. Computer-aided mapping reveals a nonrandom distribution of potential Z-DNA-forming sequences in human genes. J. Biol. Chem. 1992, 267, 11846–11855. [Google Scholar] [CrossRef]
- Wölfl, S.; Wittig, B.; Rich, A. Identification of transcriptionally induced Z-DNA segments in the human c-myc gene. Biochim. Biophys. Acta 1995, 1264, 294–302. [Google Scholar] [CrossRef]
- Adachi, M.; Tsujimoto, Y. Potential Z-DNA elements surround the breakpoints of chromosome translocation within the 5′ flanking region of bcl-2 gene. Oncogene 1990, 5, 1653–1657. [Google Scholar]
- Seité, P.; Leroux, D.; Hillion, J.; Monteil, M.; Berger, R.; Mathieu-Mahul, D.; Larsen, C.-J. Molecular analysis of a variant 18;22 translocation in a case of lymphocytic lymphoma. Genes Chromosomes Cancer 1993, 6, 39–44. [Google Scholar] [CrossRef]
- Wang, G.; Christensen, L.A.; Vasquez, K.M. Z-DNA-forming sequences generate large-scale deletions in mammalian cells. Proc. Natl. Acad. Sci. USA 2006, 103, 2677–2682. [Google Scholar] [CrossRef]
- McKinney, J.A.; Wang, G.; Vasquez, K.M. Distinct mechanisms of mutagenic processing of alternative DNA structures by repair proteins. Mol. Cell. Oncol. 2020, 7, 1743807. [Google Scholar] [CrossRef]
- Kha, D.T.; Wang, G.; Natrajan, N.; Harrison, L.; Vasquez, K.M. Pathways for double-strand break repair in genetically unstable Z-DNA-forming sequences. J. Mol. Biol. 2010, 398, 471–480. [Google Scholar] [CrossRef]
- Wang, G.; Carbajal, S.; Vijg, J.; DiGiovanni, J.; Vasquez, K.M. DNA structure-induced genomic instability in vivo. J. Natl. Cancer Inst. 2008, 100, 1815–1817. [Google Scholar] [CrossRef]
- Lu, G.; Ferl, R.J. Homopurine/homopyrimidine sequences as potential regulatory elements in eukaryotic cells. Int. J. Biochem. 1993, 25, 1529–1537. [Google Scholar] [CrossRef]
- Jain, A.; Wang, G.; Vasquez, K.M. DNA triple helices: Biological consequences and therapeutic potential. Biochimie 2008, 90, 1117–1130. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.D.; Collier, D.A.; Hanvey, J.C.; Shimizu, M.; Wohlrab, F. The chemistry and biology of unusual DNA structures adopted by oligopurine·oligopyrimidine sequences. FASEB J. 1988, 2, 2939–2949. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, G.; Del Mundo, I.M.; McKinney, J.A.; Lu, X.; Bacolla, A.; Boulware, S.B.; Zhang, C.; Zhang, H.; Ren, P.; et al. Distinct Mechanisms of Nuclease-Directed DNA-Structure-Induced Genetic Instability in Cancer Genomes. Cell Rep. 2018, 22, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Kinniburgh, A.J. A cis-acting transcription element of the c-myc gene can assume an H-DNA conformation. Nucleic Acids Res. 1989, 17, 7771–7778. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.C.; Chastain, P.; Lee, J.S.; Hedge, B.G.; Houston, S.; Langen, R.; Hsieh, C.-L.; Haworth, I.S.; Lieber, M.R. Evidence for a triplex DNA conformation at the bcl-2 major breakpoint region of the t(14;18) translocation. J. Biol. Chem. 2005, 280, 22749–22760. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Vasquez, K.M. Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 13448–13453. [Google Scholar] [CrossRef] [PubMed]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA secondary structures: Stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef]
- Hazel, P.; Huppert, J.; Balasubramanian, S.; Neidle, S. Loop-length-dependent folding of G-quadruplexes. J. Am. Chem. Soc. 2004, 126, 16405–16415. [Google Scholar] [CrossRef]
- Hazel, P.; Parkinson, G.N.; Neidle, S. Predictive modelling of topology and loop variations in dimeric DNA quadruplex structures. Nucleic Acids Res. 2006, 34, 2117–2127. [Google Scholar] [CrossRef]
- Burge, S.; Parkinson, G.N.; Hazel, P.; Todd, A.K.; Neidle, S. Quadruplex DNA: Sequence, topology and structure. Nucleic Acids Res. 2006, 34, 5402–5415. [Google Scholar] [CrossRef] [PubMed]
- Kim, N. The Interplay between G-quadruplex and Transcription. Curr. Med. Chem. 2019, 26, 2898–2917. [Google Scholar] [CrossRef]
- Usdin, K.; Woodford, K.J. CGG repeats associated with DNA instability and chromosome fragility form structures that block DNA synthesis in vitro. Nucleic Acids Res. 1995, 23, 4202–4209. [Google Scholar] [CrossRef] [PubMed]
- Fry, M.; Loeb, L.A. The fragile X syndrome d(CGG)n nucleotide repeats form a stable tetrahelical structure. Proc. Natl. Acad. Sci. USA 1994, 91, 4950–4954. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Brosh, R.M., Jr. G-quadruplex nucleic acids and human disease. FEBS J. 2010, 277, 3470–3488. [Google Scholar] [CrossRef]
- Kettani, A.; Kumar, R.A.; Patel, D.J. Solution structure of a DNA quadruplex containing the fragile X syndrome triplet repeat. J. Mol. Biol. 1995, 254, 638–656. [Google Scholar] [CrossRef]
- Todd, A.K.; Johnston, M.; Neidle, S. Highly prevalent putative quadruplex sequence motifs in human DNA. Nucleic Acids Res. 2005, 33, 2901–2907. [Google Scholar] [CrossRef]
- Huppert, J.L.; Balasubramanian, S. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005, 33, 2908–2916. [Google Scholar] [CrossRef]
- Chambers, V.S.; Marsico, G.; Boutell, J.M.; Di Antonio, M.; Smith, G.P.; Balasubramanian, S. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. Biotechnol. 2015, 33, 877–881. [Google Scholar] [CrossRef]
- Fang, G.; Cech, T.R. The beta subunit of Oxytricha telomere-binding protein promotes G-quartet formation by telomeric DNA. Cell 1993, 74, 875–885. [Google Scholar] [CrossRef]
- Sundquist, W.I.; Klug, A. Telomeric DNA dimerizes by formation of guanine tetrads between hairpin loops. Nature 1989, 342, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Paeschke, K.; Simonsson, T.; Postberg, J.; Rhodes, D.; Lipps, H.J. Telomere end-binding proteins control the formation of G-quadruplex DNA structures in vivo. Nat. Struct. Mol. Biol. 2005, 12, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.; Gilbert, W. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature 1988, 334, 364–366. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.L.; Balasubramanian, S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007, 35, 406–413. [Google Scholar] [CrossRef]
- Du, Z.; Zhao, Y.; Li, N. Genome-wide analysis reveals regulatory role of G4 DNA in gene transcription. Genome Res. 2008, 18, 233–241. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef]
- Sun, D.; Hurley, L.H. The importance of negative superhelicity in inducing the formation of G-quadruplex and i-motif structures in the c-Myc promoter: Implications for drug targeting and control of gene expression. J. Med. Chem. 2009, 52, 2863–2874. [Google Scholar] [CrossRef]
- London, T.B.; Barber, L.J.; Mosedale, G.; Kelly, G.P.; Balasubramanian, S.; Hickson, I.D.; Boulton, S.J.; Hiom, K. FANCJ is a structure-specific DNA helicase associated with the maintenance of genomic G/C tracts. J. Biol. Chem. 2008, 283, 36132–36139. [Google Scholar] [CrossRef]
- Mohaghegh, P.; Karow, J.K.; Brosh, R.M., Jr.; Bohr, V.A.; Hickson, I.D. The Bloom’s and Werner’s syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 2001, 29, 2843–2849. [Google Scholar] [CrossRef]
- Huber, M.D.; Lee, D.C.; Maizels, N. G4 DNA unwinding by BLM and Sgs1p: Substrate specificity and substrate-specific inhibition. Nucleic Acids Res. 2002, 30, 3954–3961. [Google Scholar] [CrossRef]
- Ribeyre, C.; Lopes, J.; Boulé, J.-B.; Piazza, A.; Guédin, A.; Zakian, V.A.; Mergny, J.-L.; Nicolas, A. The yeast Pif1 helicase prevents genomic instability caused by G-quadruplex-forming CEB1 sequences in vivo. PLoS Genet. 2009, 5, e1000475. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Lu, H.; Wang, Z.; Hu, Q.; Wang, H.; Xiang, R.; Chiba, T.; Wu, X. ERCC1/XPF Is Important for Repair of DNA Double-Strand Breaks Containing Secondary Structures. iScience 2019, 16, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, S.; Oaks, J.; Ren, J.; Li, L.; Wu, X. The concerted roles of FANCM and Rad52 in the protection of common fragile sites. Nat. Commun. 2018, 9, 2791. [Google Scholar] [CrossRef] [PubMed]
- Moruno-Manchon, J.F.; Koellhoffer, E.C.; Gopakumar, J.; Hambarde, S.; Kim, N.; McCullough, L.D.; Tsvetkov, A.S. The G-quadruplex DNA stabilizing drug pyridostatin promotes DNA damage and downregulates transcription of Brca1 in neurons. Aging 2017, 9, 1957–1970. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.T.; Vasquez, K.M. Multi-Faceted Roles of ERCC1-XPF Nuclease in Processing Non-B DNA Structures. DNA 2022, 2, 231-247. https://doi.org/10.3390/dna2040017
Li TT, Vasquez KM. Multi-Faceted Roles of ERCC1-XPF Nuclease in Processing Non-B DNA Structures. DNA. 2022; 2(4):231-247. https://doi.org/10.3390/dna2040017
Chicago/Turabian StyleLi, Tonia T., and Karen M. Vasquez. 2022. "Multi-Faceted Roles of ERCC1-XPF Nuclease in Processing Non-B DNA Structures" DNA 2, no. 4: 231-247. https://doi.org/10.3390/dna2040017
APA StyleLi, T. T., & Vasquez, K. M. (2022). Multi-Faceted Roles of ERCC1-XPF Nuclease in Processing Non-B DNA Structures. DNA, 2(4), 231-247. https://doi.org/10.3390/dna2040017