
Mitochondrial DNA: Consensuses and Controversies



Abstract
{kind=link}
{kind=link}
{kind=link}
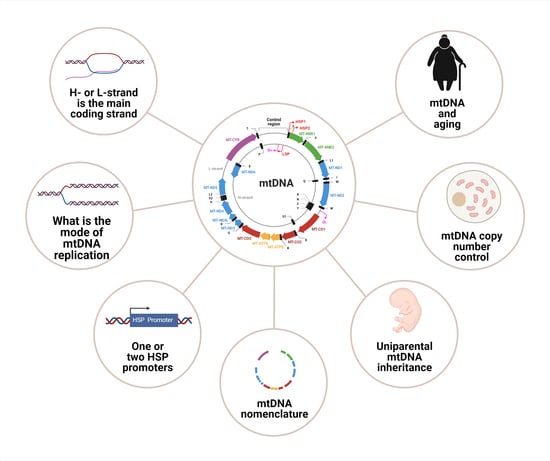
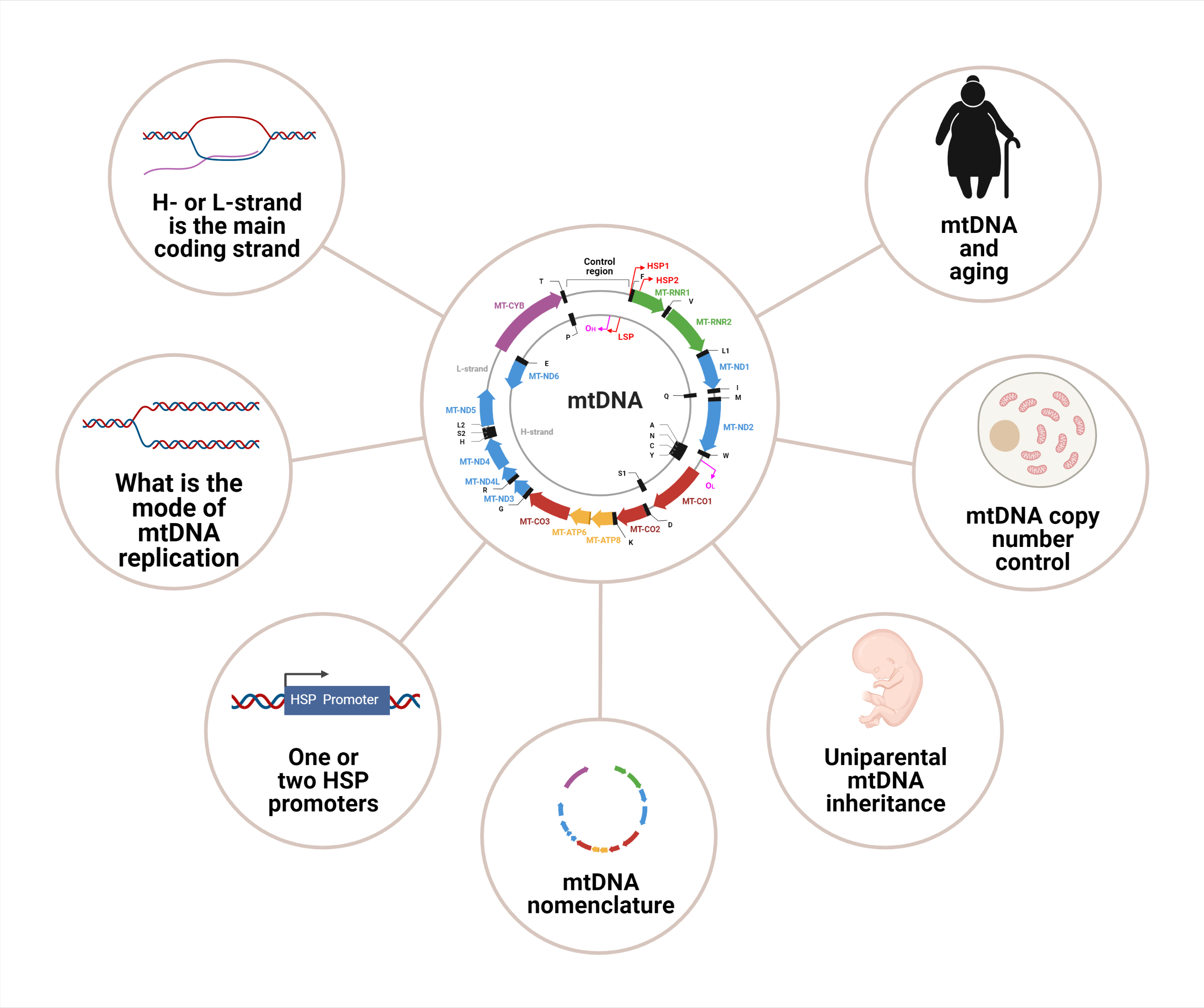
1. Introduction
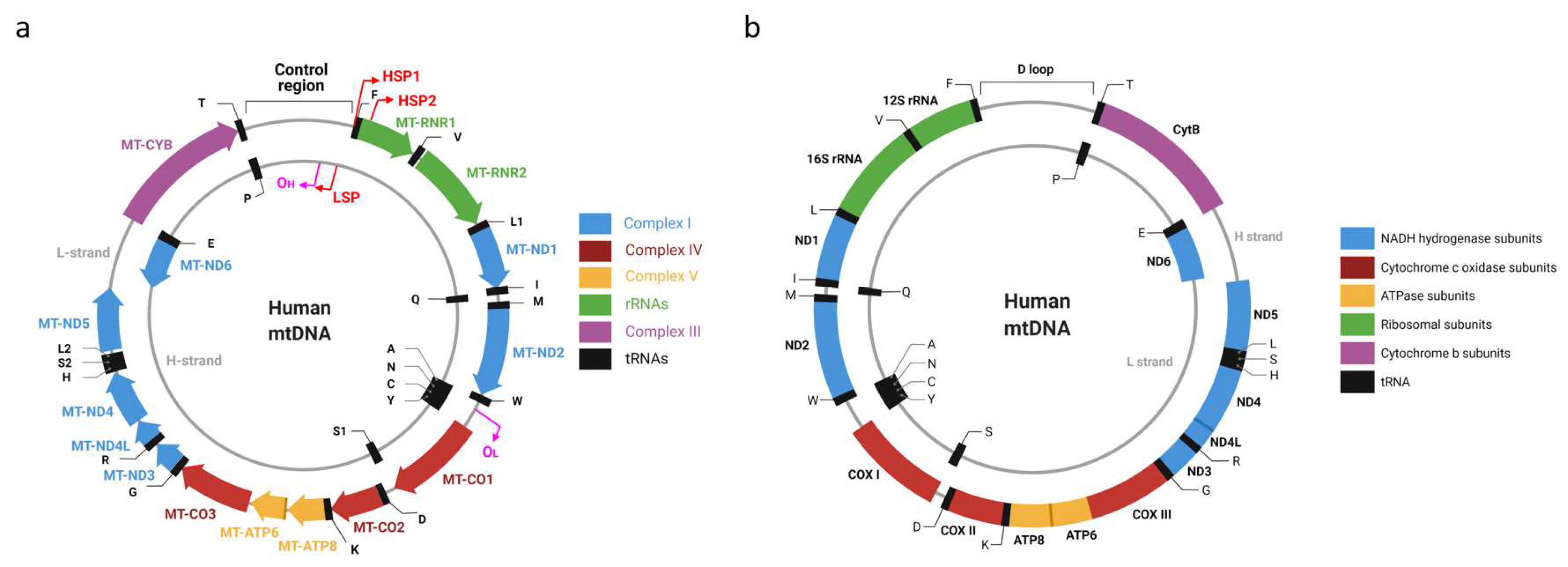
2. mtDNA Organization
3. mtDNA Nomenclature
4. Non-Canonical Mitochondrial Genes
5. mtDNA Replication
6. mtDNA Maintenance and Copy Number Control
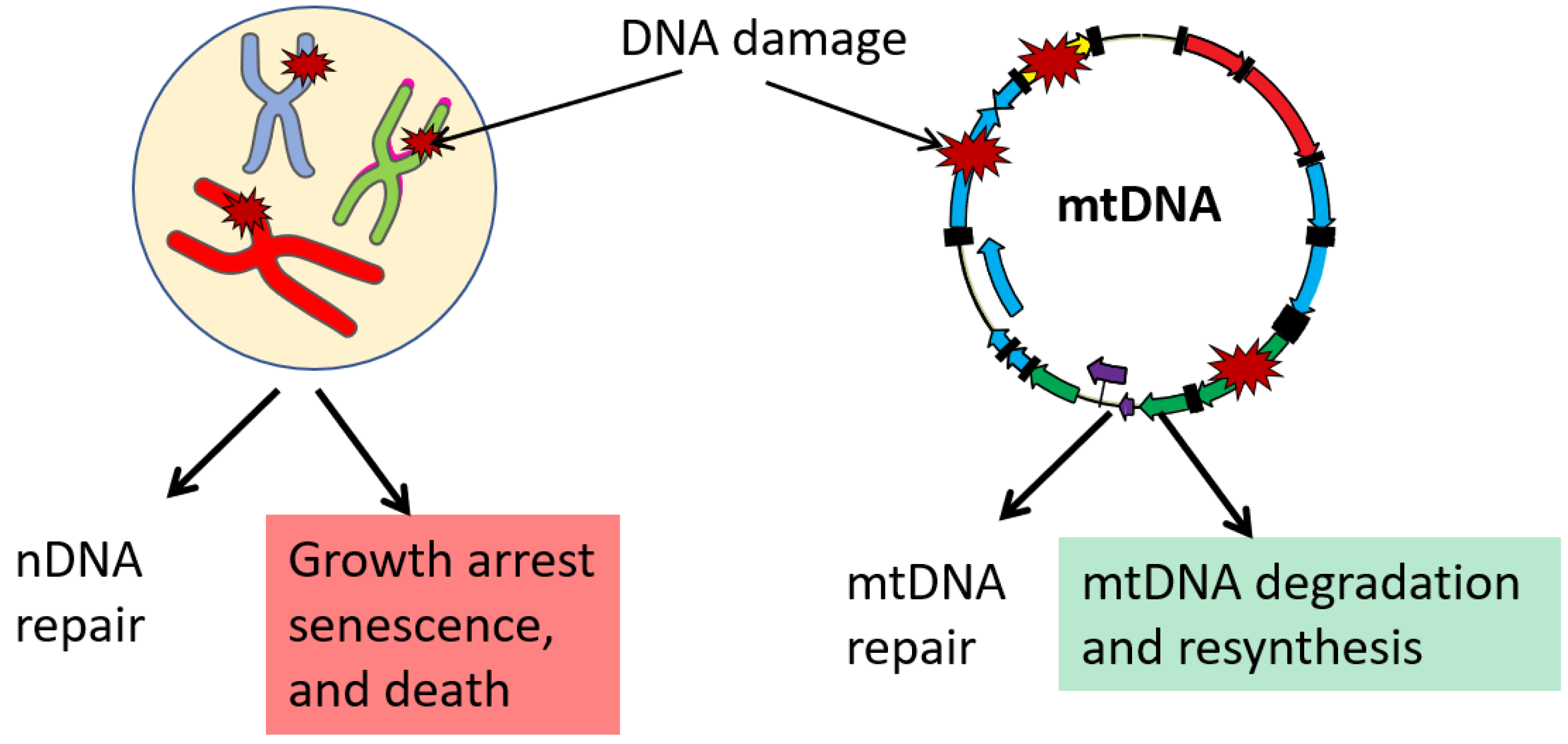
7. mtDNA Damage, Repair, and Degradation
8. mtDNA Transcription: One or Two Heavy Strand Promoters?
9. Uniparental mtDNA Inheritance
10. mtDNA and the Mitochondrial Theory of Aging
11. Extramitochondrial mtDNA
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Gabriel, M.S.; Chan, S.W.; Alhathal, N.; Chen, J.Z.; Zini, A. Influence of microsurgical varicocelectomy on human sperm mitochondrial DNA copy number: A pilot study. J. Assist. Reprod. Genet. 2012, 29, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Reynier, P.; May-Panloup, P.; Chretien, M.F.; Morgan, C.J.; Jean, M.; Savagner, F.; Barriere, P.; Malthiery, Y. Mitochondrial DNA content affects the fertilizability of human oocytes. Mol. Hum. Reprod. 2001, 7, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Nass, M.M.; Nass, S. Intramitochondrial Fibers with DNA Characteristics. I. Fixation and Electron Staining Reactions. J. Cell Biol. 1963, 19, 593–611. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.J.; Harding, A.E.; Morgan-Hughes, J.A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988, 331, 717–719. [Google Scholar] [CrossRef]
- Lestienne, P.; Ponsot, G. Kearns-Sayre syndrome with muscle mitochondrial DNA deletion. Lancet 1988, 1, 885. [Google Scholar] [CrossRef]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.; Elsas, L.J., 2nd; Nikoskelainen, E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef]
- Prakash, Y.S.; Pabelick, C.M.; Sieck, G.C. Mitochondrial Dysfunction in Airway Disease. Chest 2017, 152, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.; Wang, M.; Xiao, H.; Wei, X. Mitochondrial dysfunction and chronic lung disease. Cell Biol. Toxicol. 2019, 35, 493–502. [Google Scholar] [CrossRef]
- Bax, B.E. Mitochondrial neurogastrointestinal encephalomyopathy: Approaches to diagnosis and treatment. J. Transl. Genet. Genom. 2020, 4, 1. [Google Scholar] [CrossRef]
- Ray, K. Mitochondrial dysfunction in Crohn’s disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 260. [Google Scholar] [CrossRef]
- Meyers, D.E.; Basha, H.I.; Koenig, M.K. Mitochondrial cardiomyopathy: Pathophysiology, diagnosis, and management. Tex. Heart Inst. J. 2013, 40, 385–394. [Google Scholar] [PubMed]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Schijvens, A.M.; van de Kar, N.C.; Bootsma-Robroeks, C.M.; Cornelissen, E.A.; van den Heuvel, L.P.; Schreuder, M.F. Mitochondrial Disease and the Kidney With a Special Focus on CoQ10 Deficiency. Kidney Int. Rep. 2020, 5, 2146–2159. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, W.; Cao, Q.; Wang, Z.; Zhao, M.; Xu, L.; Zhuang, Q. Mitochondrial dysfunction in fibrotic diseases. Cell Death Discov. 2020, 6, 80. [Google Scholar] [CrossRef]
- Hussain, M.; Krishnamurthy, S.; Patel, J.; Kim, E.; Baptiste, B.A.; Croteau, D.L.; Bohr, V.A. Skin Abnormalities in Disorders with DNA Repair Defects, Premature Aging, and Mitochondrial Dysfunction. J. Investig. Dermatol. 2021, 141, 968–975. [Google Scholar] [CrossRef]
- Dobson, P.F.; Dennis, E.P.; Hipps, D.; Reeve, A.; Laude, A.; Bradshaw, C.; Stamp, C.; Smith, A.; Deehan, D.J.; Turnbull, D.M.; et al. Mitochondrial dysfunction impairs osteogenesis, increases osteoclast activity, and accelerates age related bone loss. Sci. Rep. 2020, 10, 11643. [Google Scholar] [CrossRef]
- Scotece, M.; Rego-Perez, I.; Lechuga-Vieco, A.V.; Cortes, A.C.; Jimenez-Gomez, M.C.; Filgueira-Fernandez, P.; Vaamonde-Garcia, C.; Enriquez, J.A.; Blanco, F.J. Mitochondrial DNA impact on joint damaged process in a conplastic mouse model after being surgically induced with osteoarthritis. Sci. Rep. 2021, 11, 9112. [Google Scholar] [CrossRef]
- Arena, I.G.; Pugliese, A.; Volta, S.; Toscano, A.; Musumeci, O. Molecular Genetics Overview of Primary Mitochondrial Myopathies. J. Clin. Med. 2022, 11, 632. [Google Scholar] [CrossRef]
- Kim, H.R.; Won, S.J.; Fabian, C.; Kang, M.G.; Szardenings, M.; Shin, M.G. Mitochondrial DNA aberrations and pathophysiological implications in hematopoietic diseases, chronic inflammatory diseases, and cancers. Ann. Lab. Med. 2015, 35, 1–14. [Google Scholar] [CrossRef]
- Sato, T.; Muroya, K.; Hanakawa, J.; Iwano, R.; Asakura, Y.; Tanaka, Y.; Murayama, K.; Ohtake, A.; Hasegawa, T.; Adachi, M. Clinical manifestations and enzymatic activities of mitochondrial respiratory chain complexes in Pearson marrow-pancreas syndrome with 3-methylglutaconic aciduria: A case report and literature review. Eur. J. Pediatr. 2015, 174, 1593–1602. [Google Scholar] [CrossRef]
- Lax, N.Z.; Gorman, G.S.; Turnbull, D.M. Review: Central nervous system involvement in mitochondrial disease. Neuropathol. Appl. Neurobiol. 2017, 43, 102–118. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Votruba, M.; Burte, F.; La Morgia, C.; Barboni, P.; Carelli, V. A neurodegenerative perspective on mitochondrial optic neuropathies. Acta Neuropathol. 2016, 132, 789–806. [Google Scholar] [CrossRef] [PubMed]
- Boguenet, M.; Bouet, P.E.; Spiers, A.; Reynier, P.; May-Panloup, P. Mitochondria: Their role in spermatozoa and in male infertility. Hum. Reprod. Update 2021, 27, 697–719. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Zhuo, G.; Zhang, C.; Leng, J. Point mutation in mitochondrial tRNA gene is associated with polycystic ovary syndrome and insulin resistance. Mol. Med. Rep. 2016, 13, 3169–3172. [Google Scholar] [CrossRef]
- Saeed, N.; Hamzah, I.H.; Al-Gharrawi, S.A.R. Polycystic ovary syndrome dependency on mtDNA mutation; copy Number and its association with insulin resistance. BMC Res. Notes 2019, 12, 455. [Google Scholar] [CrossRef]
- Shokolenko, I.N.; Alexeyev, M.F. Mitochondrial DNA: A disposable genome? Biochim. Biophys. Acta 2015, 1852, 1805–1809. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Bibb, M.J.; Van Etten, R.A.; Wright, C.T.; Walberg, M.W.; Clayton, D.A. Sequence and gene organization of mouse mitochondrial DNA. Cell 1981, 26, 167–180. [Google Scholar] [CrossRef]
- Wells, R.D.; Larson, J.E. Buoyant density studies on natural and synthetic deoxyribonucleic acids in neutral and alkaline solutions. J. Biol. Chem. 1972, 247, 3405–3409. [Google Scholar] [CrossRef]
- Liu, X.; Shan, G. Mitochondria Encoded Non-coding RNAs in Cell Physiology. Front. Cell Dev. Biol. 2021, 9, 713729. [Google Scholar] [CrossRef]
- Barroso Lima, N.C.; Prosdocimi, F. The heavy strand dilemma of vertebrate mitochondria on genome sequencing age: Number of encoded genes or G + T content? Mitochondrial DNA. Part A DNA Mapp. Seq. Anal. 2018, 29, 300–302. [Google Scholar] [CrossRef] [PubMed]
- Menger, K.E.; Rodriguez-Luis, A.; Chapman, J.; Nicholls, T.J. Controlling the topology of mammalian mitochondrial DNA. Open Biol. 2021, 11, 210168. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Han, J. Mitochondrial Nucleoid: Shield and Switch of the Mitochondrial Genome. Oxid. Med. Cell. Longev. 2017, 2017, 8060949. [Google Scholar] [CrossRef] [PubMed]
- Kukat, C.; Wurm, C.A.; Spahr, H.; Falkenberg, M.; Larsson, N.G.; Jakobs, S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc. Natl. Acad. Sci. USA 2011, 108, 13534–13539. [Google Scholar] [CrossRef]
- Brown, T.A.; Tkachuk, A.N.; Shtengel, G.; Kopek, B.G.; Bogenhagen, D.F.; Hess, H.F.; Clayton, D.A. Superresolution fluorescence imaging of mitochondrial nucleoids reveals their spatial range, limits, and membrane interaction. Mol. Cell. Biol. 2011, 31, 4994–5010. [Google Scholar] [CrossRef]
- Gilkerson, R.W.; Schon, E.A.; Hernandez, E.; Davidson, M.M. Mitochondrial nucleoids maintain genetic autonomy but allow for functional complementation. J. Cell Biol. 2008, 181, 1117–1128. [Google Scholar] [CrossRef]
- Bogenhagen, D.F. Mitochondrial DNA nucleoid structure. Biochim. Biophys. Acta 2012, 1819, 914–920. [Google Scholar] [CrossRef]
- Campbell, C.T.; Kolesar, J.E.; Kaufman, B.A. Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim. Biophys. Acta 2012, 1819, 921–929. [Google Scholar] [CrossRef]
- Farge, G.; Laurens, N.; Broekmans, O.D.; van den Wildenberg, S.M.; Dekker, L.C.; Gaspari, M.; Gustafsson, C.M.; Peterman, E.J.; Falkenberg, M.; Wuite, G.J. Protein sliding and DNA denaturation are essential for DNA organization by human mitochondrial transcription factor A. Nat. Commun. 2012, 3, 1013. [Google Scholar] [CrossRef]
- Lu, B.; Lee, J.; Nie, X.; Li, M.; Morozov, Y.I.; Venkatesh, S.; Bogenhagen, D.F.; Temiakov, D.; Suzuki, C.K. Phosphorylation of human TFAM in mitochondria impairs DNA binding and promotes degradation by the AAA+ Lon protease. Mol. Cell 2013, 49, 121–132. [Google Scholar] [CrossRef]
- Wang, Y.E.; Marinov, G.K.; Wold, B.J.; Chan, D.C. Genome-wide analysis reveals coating of the mitochondrial genome by TFAM. PLoS ONE 2013, 8, e74513. [Google Scholar] [CrossRef] [PubMed]
- Ngo, H.B.; Lovely, G.A.; Phillips, R.; Chan, D.C. Distinct structural features of TFAM drive mitochondrial DNA packaging versus transcriptional activation. Nat. Commun. 2014, 5, 3077. [Google Scholar] [CrossRef] [PubMed]
- Shoffner, J.M.; Wallace, D.C. Mitochondrial genetics: Principles and practice. Am. J. Hum. Genet. 1992, 51, 1179–1186. [Google Scholar] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondrial DNA sequence variation in human evolution and disease. Proc. Natl. Acad. Sci. USA 1994, 91, 8739–8746. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Niikura, T.; Tajima, H.; Yasukawa, T.; Sudo, H.; Ito, Y.; Kita, Y.; Kawasumi, M.; Kouyama, K.; Doyu, M.; et al. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer’s disease genes and Abeta. Proc. Natl. Acad. Sci. USA 2001, 98, 6336–6341. [Google Scholar] [CrossRef]
- Ikonen, M.; Liu, B.; Hashimoto, Y.; Ma, L.; Lee, K.W.; Niikura, T.; Nishimoto, I.; Cohen, P. Interaction between the Alzheimer’s survival peptide humanin and insulin-like growth factor-binding protein 3 regulates cell survival and apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 13042–13047. [Google Scholar] [CrossRef]
- Niikura, T.; Tajima, H.; Kita, Y. Neuronal cell death in Alzheimer’s disease and a neuroprotective factor, humanin. Curr. Neuropharmacol. 2006, 4, 139–147. [Google Scholar] [CrossRef]
- Miller, B.; Kim, S.J.; Kumagai, H.; Yen, K.; Cohen, P. Mitochondria-derived peptides in aging and healthspan. J. Clin. Investig. 2022, 132, e158449. [Google Scholar] [CrossRef]
- Lee, C.; Zeng, J.; Drew, B.G.; Sallam, T.; Martin-Montalvo, A.; Wan, J.; Kim, S.J.; Mehta, H.; Hevener, A.L.; de Cabo, R.; et al. The Mitochondrial-Derived Peptide MOTS-c Promotes Metabolic Homeostasis and Reduces Obesity and Insulin Resistance. Cell Metab. 2015, 21, 443–454. [Google Scholar] [CrossRef]
- Reynolds, J.C.; Lai, R.W.; Woodhead, J.S.T.; Joly, J.H.; Mitchell, C.J.; Cameron-Smith, D.; Lu, R.; Cohen, P.; Graham, N.A.; Benayoun, B.A.; et al. MOTS-c is an exercise-induced mitochondrial-encoded regulator of age-dependent physical decline and muscle homeostasis. Nat. Commun. 2021, 12, 470. [Google Scholar] [CrossRef]
- Mercer, T.R.; Neph, S.; Dinger, M.E.; Crawford, J.; Smith, M.A.; Shearwood, A.M.; Haugen, E.; Bracken, C.P.; Rackham, O.; Stamatoyannopoulos, J.A.; et al. The human mitochondrial transcriptome. Cell 2011, 146, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Rackham, O.; Shearwood, A.M.; Mercer, T.R.; Davies, S.M.; Mattick, J.S.; Filipovska, A. Long noncoding RNAs are generated from the mitochondrial genome and regulated by nuclear-encoded proteins. RNA 2011, 17, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Gusic, M.; Prokisch, H. ncRNAs: New Players in Mitochondrial Health and Disease? Front. Genet. 2020, 11, 95. [Google Scholar] [CrossRef] [PubMed]
- Kumarswamy, R.; Bauters, C.; Volkmann, I.; Maury, F.; Fetisch, J.; Holzmann, A.; Lemesle, G.; de Groote, P.; Pinet, F.; Thum, T. Circulating long noncoding RNA, LIPCAR, predicts survival in patients with heart failure. Circ. Res. 2014, 114, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Kasiviswanathan, R.; Collins, T.R.; Copeland, W.C. The interface of transcription and DNA replication in the mitochondria. Biochim. Biophys. Acta 2012, 1819, 970–978. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McKinney, E.A.; Oliveira, M.T. Replicating animal mitochondrial DNA. Genet. Mol. Biol. 2013, 36, 308–315. [Google Scholar] [CrossRef]
- Roy, A.; Kandettu, A.; Ray, S.; Chakrabarty, S. Mitochondrial DNA replication and repair defects: Clinical phenotypes and therapeutic interventions. Biochim. Biophys Acta Bioenerg. 2022, 1863, 148554. [Google Scholar] [CrossRef]
- Clayton, D.A. Replication of animal mitochondrial DNA. Cell 1982, 28, 693–705. [Google Scholar] [CrossRef]
- Clayton, D.A. Transcription of the mammalian mitochondrial genome. Annu. Rev. Biochem. 1984, 53, 573–594. [Google Scholar] [CrossRef]
- Clayton, D.A. Transcription and replication of mitochondrial DNA. Hum. Reprod. 2000, 15 (Suppl. S2), 11–17. [Google Scholar] [CrossRef] [PubMed]
- Agaronyan, K.; Morozov, Y.I.; Anikin, M.; Temiakov, D. Mitochondrial biology. Replication-transcription switch in human mitochondria. Science 2015, 347, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Contreras, M.; Sweetwyne, M.T.; Kohrn, B.F.; Tsantilas, K.A.; Hipp, M.J.; Schmidt, E.K.; Fredrickson, J.; Whitson, J.A.; Campbell, M.D.; Rabinovitch, P.S.; et al. A replication-linked mutational gradient drives somatic mutation accumulation and influences germline polymorphisms and genome composition in mitochondrial DNA. Nucleic Acids Res. 2021, 49, 11103–11118. [Google Scholar] [CrossRef] [PubMed]
- Shokolenko, I.N.; Fayzulin, R.Z.; Katyal, S.; McKinnon, P.J.; Wilson, G.L.; Alexeyev, M.F. Mitochondrial DNA ligase is dispensable for the viability of cultured cells but essential for mtDNA maintenance. J. Biol. Chem. 2013, 288, 26594–26605. [Google Scholar] [CrossRef]
- Yasukawa, T.; Reyes, A.; Cluett, T.J.; Yang, M.Y.; Bowmaker, M.; Jacobs, H.T.; Holt, I.J. Replication of vertebrate mitochondrial DNA entails transient ribonucleotide incorporation throughout the lagging strand. EMBO J. 2006, 25, 5358–5371. [Google Scholar] [CrossRef]
- Miralles Fuste, J.; Shi, Y.; Wanrooij, S.; Zhu, X.; Jemt, E.; Persson, O.; Sabouri, N.; Gustafsson, C.M.; Falkenberg, M. In vivo occupancy of mitochondrial single-stranded DNA binding protein supports the strand displacement mode of DNA replication. PLoS Genet. 2014, 10, e1004832. [Google Scholar] [CrossRef]
- DeLuca, S.Z.; O’Farrell, P.H. Barriers to male transmission of mitochondrial DNA in sperm development. Dev. Cell 2012, 22, 660–668. [Google Scholar] [CrossRef]
- Luo, S.M.; Ge, Z.J.; Wang, Z.W.; Jiang, Z.Z.; Wang, Z.B.; Ouyang, Y.C.; Hou, Y.; Schatten, H.; Sun, Q.Y. Unique insights into maternal mitochondrial inheritance in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 13038–13043. [Google Scholar] [CrossRef]
- Jin, Y.X.; Zheng, Z.; Yu, X.F.; Zhang, J.B.; Namgoong, S.; Cui, X.S.; Hyun, S.H.; Kim, N.H. Autophagy and ubiquitin-mediated proteolysis may not be involved in the degradation of spermatozoon mitochondria in mouse and porcine early embryos. Zygote 2016, 24, 31–41. [Google Scholar] [CrossRef]
- Al Rawi, S.; Louvet-Vallee, S.; Djeddi, A.; Sachse, M.; Culetto, E.; Hajjar, C.; Boyd, L.; Legouis, R.; Galy, V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science 2011, 334, 1144–1147. [Google Scholar] [CrossRef]
- Stringer, H.A.; Sohi, G.K.; Maguire, J.A.; Cote, H.C. Decreased skeletal muscle mitochondrial DNA in patients with statin-induced myopathy. J. Neurol. Sci. 2013, 325, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Murayama, K.; Tsuruoka, T.; Aizawa, M.; Nagasaka, H.; Horie, H.; Ohtake, A.; Saitou, K. Fatal case of mitochondrial DNA depletion with severe asphyxia in a newborn. Pediatr. Int. 2011, 53, 240–242. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, R.; Tedone, E.; Ludlow, A.; Huang, E.; Arosio, B.; Mari, D.; Shay, J.W. Quantitative mitochondrial DNA copy number determination using droplet digital PCR with single-cell resolution. Genome Res. 2019, 29, 1878–1888. [Google Scholar] [CrossRef] [PubMed]
- Kozhukhar, N.; Fant, A.; Alexeyev, M.F. Quantification of mtDNA content in cultured cells by direct droplet digital PCR. Mitochondrion 2021, 61, 102–113. [Google Scholar] [CrossRef]
- Nogueira, C.; Almeida, L.S.; Nesti, C.; Pezzini, I.; Videira, A.; Vilarinho, L.; Santorelli, F.M. Syndromes associated with mitochondrial DNA depletion. Ital. J. Pediatr. 2014, 40, 34. [Google Scholar] [CrossRef]
- Lee, I.C.; Lee, N.C.; Lu, J.J.; Su, P.H. Mitochondrial depletion causes neonatal-onset leigh syndrome, myopathy, and renal tubulopathy. J. Child Neurol. 2013, 28, 404–408. [Google Scholar] [CrossRef]
- Vu, T.H.; Tanji, K.; Valsamis, H.; DiMauro, S.; Bonilla, E. Mitochondrial DNA depletion in a patient with long survival. Neurology 1998, 51, 1190–1193. [Google Scholar] [CrossRef]
- Finsterer, J.; Kovacs, G.G.; Ahting, U. Adult mitochondrial DNA depletion syndrome with mild manifestations. Neurol. Int. 2013, 5, 28–30. [Google Scholar] [CrossRef]
- Maniura-Weber, K.; Goffart, S.; Garstka, H.L.; Montoya, J.; Wiesner, R.J. Transient overexpression of mitochondrial transcription factor A (TFAM) is sufficient to stimulate mitochondrial DNA transcription, but not sufficient to increase mtDNA copy number in cultured cells. Nucleic Acids Res. 2004, 32, 6015–6027. [Google Scholar] [CrossRef]
- Matsushima, Y.; Matsumura, K.; Ishii, S.; Inagaki, H.; Suzuki, T.; Matsuda, Y.; Beck, K.; Kitagawa, Y. Functional domains of chicken mitochondrial transcription factor A for the maintenance of mitochondrial DNA copy number in lymphoma cell line DT40. J. Biol. Chem. 2003, 278, 31149–31158. [Google Scholar] [CrossRef]
- Ekstrand, M.I.; Falkenberg, M.; Rantanen, A.; Park, C.B.; Gaspari, M.; Hultenby, K.; Rustin, P.; Gustafsson, C.M.; Larsson, N.G. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum. Mol. Genet. 2004, 13, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, Y.; Garesse, R.; Kaguni, L.S. Drosophila mitochondrial transcription factor B2 regulates mitochondrial DNA copy number and transcription in schneider cells. J. Biol. Chem. 2004, 279, 26900–26905. [Google Scholar] [CrossRef] [PubMed]
- Kanki, T.; Ohgaki, K.; Gaspari, M.; Gustafsson, C.M.; Fukuoh, A.; Sasaki, N.; Hamasaki, N.; Kang, D. Architectural role of mitochondrial transcription factor A in maintenance of human mitochondrial DNA. Mol. Cell. Biol. 2004, 24, 9823–9834. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Lezza, A.M. Regulation of mitochondrial biogenesis through TFAM-mitochondrial DNA interactions: Useful insights from aging and calorie restriction studies. Mitochondrion 2015, 25, 67–75. [Google Scholar] [CrossRef]
- Matsuda, T.; Kanki, T.; Tanimura, T.; Kang, D.; Matsuura, E.T. Effects of overexpression of mitochondrial transcription factor A on lifespan and oxidative stress response in Drosophila melanogaster. Biochem. Biophys. Res. Commun. 2013, 430, 717–721. [Google Scholar] [CrossRef]
- Seidel-Rogol, B.L.; Shadel, G.S. Modulation of mitochondrial transcription in response to mtDNA depletion and repletion in HeLa cells. Nucleic Acids Res. 2002, 30, 1929–1934. [Google Scholar] [CrossRef]
- Franko, A.; Mayer, S.; Thiel, G.; Mercy, L.; Arnould, T.; Hornig-Do, H.T.; Wiesner, R.J.; Goffart, S. CREB-1alpha is recruited to and mediates upregulation of the cytochrome c promoter during enhanced mitochondrial biogenesis accompanying skeletal muscle differentiation. Mol. Cell. Biol. 2008, 28, 2446–2459. [Google Scholar] [CrossRef]
- Kozhukhar, N.; Alexeyev, M.F. Limited predictive value of TFAM in mitochondrial biogenesis. Mitochondrion 2019, 49, 156–165. [Google Scholar] [CrossRef]
- Barth, E.; Stammler, G.; Speiser, B.; Schaper, J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J. Mol. Cell. Cardiol. 1992, 24, 669–681. [Google Scholar] [CrossRef]
- Brinckmann, A.; Weiss, C.; Wilbert, F.; von Moers, A.; Zwirner, A.; Stoltenburg-Didinger, G.; Wilichowski, E.; Schuelke, M. Regionalized pathology correlates with augmentation of mtDNA copy numbers in a patient with myoclonic epilepsy with ragged-red fibers (MERRF-syndrome). PLoS ONE 2010, 5, e13513. [Google Scholar] [CrossRef]
- Cotney, J.; Wang, Z.; Shadel, G.S. Relative abundance of the human mitochondrial transcription system and distinct roles for h-mtTFB1 and h-mtTFB2 in mitochondrial biogenesis and gene expression. Nucleic Acids Res. 2007, 35, 4042–4054. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.P.; Clayton, D.A. Purification and characterization of human mitochondrial transcription factor 1. Mol. Cell. Biol. 1988, 8, 3496–3509. [Google Scholar] [PubMed]
- Pellegrini, M.; Asin-Cayuela, J.; Erdjument-Bromage, H.; Tempst, P.; Larsson, N.G.; Gustafsson, C.M. MTERF2 is a nucleoid component in mammalian mitochondria. Biochim. Biophys. Acta 2009, 1787, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Alam, T.I.; Kanki, T.; Muta, T.; Ukaji, K.; Abe, Y.; Nakayama, H.; Takio, K.; Hamasaki, N.; Kang, D. Human mitochondrial DNA is packaged with TFAM. Nucleic Acids Res. 2003, 31, 1640–1645. [Google Scholar] [CrossRef]
- Kuhl, I.; Miranda, M.; Posse, V.; Milenkovic, D.; Mourier, A.; Siira, S.J.; Bonekamp, N.A.; Neumann, U.; Filipovska, A.; Polosa, P.L.; et al. POLRMT regulates the switch between replication primer formation and gene expression of mammalian mtDNA. Sci. Adv. 2016, 2, e1600963. [Google Scholar] [CrossRef]
- Sitarz, K.S.; Yu-Wai-Man, P.; Pyle, A.; Stewart, J.D.; Rautenstrauss, B.; Seeman, P.; Reilly, M.M.; Horvath, R.; Chinnery, P.F. MFN2 mutations cause compensatory mitochondrial DNA proliferation. Brain 2012, 135, e219. [Google Scholar] [CrossRef]
- Vielhaber, S.; Debska-Vielhaber, G.; Peeva, V.; Schoeler, S.; Kudin, A.P.; Minin, I.; Schreiber, S.; Dengler, R.; Kollewe, K.; Zuschratter, W.; et al. Mitofusin 2 mutations affect mitochondrial function by mitochondrial DNA depletion. Acta Neuropathol. 2012, 125, 245–256. [Google Scholar] [CrossRef]
- Navarro-Sastre, A.; Tort, F.; Garcia-Villoria, J.; Pons, M.R.; Nascimento, A.; Colomer, J.; Campistol, J.; Yoldi, M.E.; Lopez-Gallardo, E.; Montoya, J.; et al. Mitochondrial DNA depletion syndrome: New descriptions and the use of citrate synthase as a helpful tool to better characterise the patients. Mol. Genet. Metab. 2012, 107, 409–415. [Google Scholar] [CrossRef]
- Mengel-From, J.; Thinggaard, M.; Dalgard, C.; Kyvik, K.O.; Christensen, K.; Christiansen, L. Mitochondrial DNA copy number in peripheral blood cells declines with age and is associated with general health among elderly. Hum. Genet. 2014, 133, 1149–1159. [Google Scholar] [CrossRef]
- Van Leeuwen, N.; Beekman, M.; Deelen, J.; van den Akker, E.B.; de Craen, A.J.; Slagboom, P.E.; t Hart, L.M. Low mitochondrial DNA content associates with familial longevity: The Leiden Longevity Study. Age 2014, 36, 9629. [Google Scholar] [CrossRef]
- Lopez, S.; Buil, A.; Souto, J.C.; Casademont, J.; Blangero, J.; Martinez-Perez, A.; Fontcuberta, J.; Lathrop, M.; Almasy, L.; Soria, J.M. Sex-specific regulation of mitochondrial DNA levels: Genome-wide linkage analysis to identify quantitative trait Loci. PLoS ONE 2012, 7, e42711. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Johnson, J.; Gough, D.J.; Donoghue, J.; Cagnone, G.L.; Vaghjiani, V.; Brown, K.A.; Johns, T.G.; St John, J.C. Mitochondrial DNA copy number is regulated by DNA methylation and demethylation of POLGA in stem and cancer cells and their differentiated progeny. Cell Death Dis. 2015, 6, e1664. [Google Scholar] [CrossRef] [PubMed]
- Khozhukhar, N.; Spadafora, D.; Rodriguez, Y.; Alexeyev, M. Elimination of Mitochondrial DNA from Mammalian Cells. Curr. Protoc. Cell Biol. 2018, 78, 20.11.1–20.11.14. [Google Scholar] [CrossRef] [PubMed]
- Spadafora, D.; Kozhukhar, N.; Alexeyev, M.F. Presequence-Independent Mitochondrial Import of DNA Ligase Facilitates Establishment of Cell Lines with Reduced mtDNA Copy Number. PLoS ONE 2016, 11, e0152705. [Google Scholar] [CrossRef]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity—Critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar] [CrossRef]
- Mason, P.A.; Matheson, E.C.; Hall, A.G.; Lightowlers, R.N. Mismatch repair activity in mammalian mitochondria. Nucleic Acids Res. 2003, 31, 1052–1058. [Google Scholar] [CrossRef]
- Lakshmipathy, U.; Campbell, C. Double strand break rejoining by mammalian mitochondrial extracts. Nucleic Acids Res. 1999, 27, 1198–1204. [Google Scholar] [CrossRef]
- Krasich, R.; Copeland, W.C. DNA polymerases in the mitochondria: A critical review of the evidence. Front. Biosci. Landmark Ed. 2017, 22, 692–709. [Google Scholar] [CrossRef]
- Furda, A.M.; Marrangoni, A.M.; Lokshin, A.; Van Houten, B. Oxidants and not alkylating agents induce rapid mtDNA loss and mitochondrial dysfunction. DNA Repair 2012, 11, 684–692. [Google Scholar] [CrossRef]
- Spadafora, D.; Kozhukhar, N.; Chouljenko, V.N.; Kousoulas, K.G.; Alexeyev, M.F. Methods for Efficient Elimination of Mitochondrial DNA from Cultured Cells. PLoS ONE 2016, 11, e0154684. [Google Scholar] [CrossRef]
- Shokolenko, I.N.; Wilson, G.L.; Alexeyev, M.F. Persistent damage induces mitochondrial DNA degradation. DNA Repair 2013, 12, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Shokolenko, I.N.; Wilson, G.L.; Alexeyev, M.F. The “fast” and the “slow” modes of mitochondrial DNA degradation. Mitochondrial DNA 2014, 27, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Peeva, V.; Blei, D.; Trombly, G.; Corsi, S.; Szukszto, M.J.; Rebelo-Guiomar, P.; Gammage, P.A.; Kudin, A.P.; Becker, C.; Altmuller, J.; et al. Linear mitochondrial DNA is rapidly degraded by components of the replication machinery. Nat. Commun. 2018, 9, 1727. [Google Scholar] [CrossRef] [PubMed]
- Nissanka, N.; Bacman, S.R.; Plastini, M.J.; Moraes, C.T. The mitochondrial DNA polymerase gamma degrades linear DNA fragments precluding the formation of deletions. Nat. Commun. 2018, 9, 2491. [Google Scholar] [CrossRef]
- Yu, Z.; O’Farrell, P.H.; Yakubovich, N.; DeLuca, S.Z. The Mitochondrial DNA Polymerase Promotes Elimination of Paternal Mitochondrial Genomes. Curr. Biol. 2017, 27, 1033–1039. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Gelfand, R.; Attardi, G. Synthesis and turnover of mitochondrial ribonucleic acid in HeLa cells: The mature ribosomal and messenger ribonucleic acid species are metabolically unstable. Mol. Cell. Biol. 1981, 1, 497–511. [Google Scholar]
- Montoya, J.; Christianson, T.; Levens, D.; Rabinowitz, M.; Attardi, G. Identification of initiation sites for heavy-strand and light-strand transcription in human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1982, 79, 7195–7199. [Google Scholar] [CrossRef]
- Montoya, J.; Gaines, G.L.; Attardi, G. The pattern of transcription of the human mitochondrial rRNA genes reveals two overlapping transcription units. Cell 1983, 34, 151–159. [Google Scholar] [CrossRef]
- Martin, M.; Cho, J.; Cesare, A.J.; Griffith, J.D.; Attardi, G. Termination factor-mediated DNA loop between termination and initiation sites drives mitochondrial rRNA synthesis. Cell 2005, 123, 1227–1240. [Google Scholar] [CrossRef]
- Lodeiro, M.F.; Uchida, A.; Bestwick, M.; Moustafa, I.M.; Arnold, J.J.; Shadel, G.S.; Cameron, C.E. Transcription from the second heavy-strand promoter of human mtDNA is repressed by transcription factor A in vitro. Proc. Natl. Acad. Sci. USA 2012, 109, 6513–6518. [Google Scholar] [CrossRef] [PubMed]
- Zollo, O.; Tiranti, V.; Sondheimer, N. Transcriptional requirements of the distal heavy-strand promoter of mtDNA. Proc. Natl. Acad. Sci. USA 2012, 109, 6508–6512. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, C.M.; Falkenberg, M.; Larsson, N.G. Maintenance and Expression of Mammalian Mitochondrial DNA. Annu. Rev. Biochem. 2016, 85, 133–160. [Google Scholar] [CrossRef] [PubMed]
- Terzioglu, M.; Ruzzenente, B.; Harmel, J.; Mourier, A.; Jemt, E.; Lopez, M.D.; Kukat, C.; Stewart, J.B.; Wibom, R.; Meharg, C.; et al. MTERF1 binds mtDNA to prevent transcriptional interference at the light-strand promoter but is dispensable for rRNA gene transcription regulation. Cell Metab. 2013, 17, 618–626. [Google Scholar] [CrossRef]
- Chang, D.D.; Clayton, D.A. Precise assignment of the light-strand promoter of mouse mitochondrial DNA: A functional promoter consists of multiple upstream domains. Mol. Cell. Biol. 1986, 6, 3253–3261. [Google Scholar]
- Rojansky, R.; Cha, M.Y.; Chan, D.C. Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on PARKIN and MUL1. eLife 2016, 5, e17896. [Google Scholar] [CrossRef]
- De Melo, K.P.; Camargo, M. Mechanisms for sperm mitochondrial removal in embryos. Biochim. Biophys Acta Mol. Cell Res. 2021, 1868, 118916. [Google Scholar] [CrossRef]
- Luo, S.; Valencia, C.A.; Zhang, J.; Lee, N.C.; Slone, J.; Gui, B.; Wang, X.; Li, Z.; Dell, S.; Brown, J.; et al. Biparental Inheritance of Mitochondrial DNA in Humans. Proc. Natl. Acad. Sci. USA 2018, 115, 13039–13044. [Google Scholar] [CrossRef]
- Schwartz, M.; Vissing, J. Paternal inheritance of mitochondrial DNA. N. Engl. J. Med. 2002, 347, 576–580. [Google Scholar] [CrossRef]
- Schwartz, M.; Vissing, J. New patterns of inheritance in mitochondrial disease. Biochem. Biophys. Res. Commun. 2003, 310, 247–251. [Google Scholar] [CrossRef]
- Annis, S.; Fleischmann, Z.; Khrapko, M.; Franco, M.; Wasko, K.; Woods, D.; Kunz, W.S.; Ellis, P.; Khrapko, K. Quasi-Mendelian paternal inheritance of mitochondrial DNA: A notorious artifact, or anticipated behavior? Proc. Natl. Acad. Sci. USA 2019, 116, 14797–14798. [Google Scholar] [CrossRef] [PubMed]
- Lutz-Bonengel, S.; Parson, W. No further evidence for paternal leakage of mitochondrial DNA in humans yet. Proc. Natl. Acad. Sci. USA 2019, 116, 1821–1822. [Google Scholar] [CrossRef] [PubMed]
- Salas, A.; Schonherr, S.; Bandelt, H.J.; Gomez-Carballa, A.; Weissensteiner, H. Extraordinary claims require extraordinary evidence in asserted mtDNA biparental inheritance. Forensic Sci. Int. Genet. 2020, 47, 102274. [Google Scholar] [CrossRef] [PubMed]
- Pyle, A.; Hudson, G.; Wilson, I.J.; Coxhead, J.; Smertenko, T.; Herbert, M.; Santibanez-Koref, M.; Chinnery, P.F. Extreme-Depth Re-sequencing of Mitochondrial DNA Finds No Evidence of Paternal Transmission in Humans. PLoS Genet. 2015, 11, e1005040. [Google Scholar] [CrossRef]
- Wei, W.; Pagnamenta, A.T.; Gleadall, N.; Sanchis-Juan, A.; Stephens, J.; Broxholme, J.; Tuna, S.; Odhams, C.A.; Genomics England Research Consortium; NIHR BioResource; et al. Nuclear-mitochondrial DNA segments resemble paternally inherited mitochondrial DNA in humans. Nat. Commun. 2020, 11, 1740. [Google Scholar] [CrossRef]
- Luo, S.; Valencia, C.A.; Zhang, J.; Lee, N.C.; Slone, J.; Gui, B.; Wang, X.; Li, Z.; Dell, S.; Brown, J.; et al. Reply to Lutz-Bonengel et al.: Biparental mtDNA transmission is unlikely to be the result of nuclear mitochondrial DNA segments. Proc. Natl. Acad. Sci. USA 2019, 116, 1823–1824. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Gyllensten, U.; Wharton, D.; Josefsson, A.; Wilson, A.C. Paternal inheritance of mitochondrial DNA in mice see comments]. Nature 1991, 352, 255–257. [Google Scholar] [CrossRef]
- Zhao, X.; Li, N.; Guo, W.; Hu, X.; Liu, Z.; Gong, G.; Wang, A.; Feng, J.; Wu, C. Further evidence for paternal inheritance of mitochondrial DNA in the sheep (Ovis aries). Hered. Edinb. 2004, 93, 399–403. [Google Scholar] [CrossRef]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef]
- Boveris, A.; Oshino, N.; Chance, B. The cellular production of hydrogen peroxide. Biochem. J. 1972, 128, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Hansford, R.G.; Hogue, B.A.; Mildaziene, V. Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. J. Bioenerg. Biomembr. 1997, 29, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Staniek, K.; Nohl, H. H(2)O(2) detection from intact mitochondria as a measure for one-electron reduction of dioxygen requires a non-invasive assay system. Biochim. Biophys. Acta 1999, 1413, 70–80. [Google Scholar] [CrossRef]
- Staniek, K.; Nohl, H. Are mitochondria a permanent source of reactive oxygen species? Biochim. Biophys. Acta 2000, 1460, 268–275. [Google Scholar] [CrossRef]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef]
- Miquel, J.; Binnard, R.; Fleming, J.E. Role of metabolic rate and DNA-repair in Drosophila aging: Implications for the mitochondrial mutation theory of aging. Exp. Gerontol. 1983, 18, 167–171. [Google Scholar] [CrossRef]
- Miquel, J.; Economos, A.C.; Fleming, J.; Johnson, J.E., Jr. Mitochondrial role in cell aging. Exp. Gerontol. 1980, 15, 575–591. [Google Scholar] [CrossRef]
- Fleming, J.E.; Miquel, J.; Cottrell, S.F.; Yengoyan, L.S.; Economos, A.C. Is cell aging caused by respiration-dependent injury to the mitochondrial genome? Gerontology 1982, 28, 44–53. [Google Scholar] [CrossRef]
- Itsara, L.S.; Kennedy, S.R.; Fox, E.J.; Yu, S.; Hewitt, J.J.; Sanchez-Contreras, M.; Cardozo-Pelaez, F.; Pallanck, L.J. Oxidative stress is not a major contributor to somatic mitochondrial DNA mutations. PLoS Genet. 2014, 10, e1003974. [Google Scholar] [CrossRef]
- Kennedy, S.R.; Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 2013, 9, e1003794. [Google Scholar] [CrossRef]
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martincorena, I.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife 2014, 3, e02935. [Google Scholar] [CrossRef] [PubMed]
- Cullis, P.M.; Jones, G.D.; Symons, M.C.; Lea, J.S. Electron transfer from protein to DNA in irradiated chromatin. Nature 1987, 330, 773–774. [Google Scholar] [CrossRef] [PubMed]
- Liang, R.; Senturker, S.; Shi, X.; Bal, W.; Dizdaroglu, M.; Kasprzak, K.S. Effects of Ni(II) and Cu(II) on DNA interaction with the N-terminal sequence of human protamine P2: Enhancement of binding and mediation of oxidative DNA strand scission and base damage. Carcinogenesis 1999, 20, 893–898. [Google Scholar] [CrossRef]
- Liang, Q.; Dedon, P.C. Cu(II)/H2O2-induced DNA damage is enhanced by packaging of DNA as a nucleosome. Chem. Res. Toxicol. 2001, 14, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Enright, H.U.; Miller, W.J.; Hebbel, R.P. Nucleosomal histone protein protects DNA from iron-mediated damage. Nucleic Acids Res. 1992, 20, 3341–3346. [Google Scholar] [CrossRef][Green Version]
- Ljungman, M.; Hanawalt, P.C. Efficient protection against oxidative DNA damage in chromatin. Mol. Carcinog. 1992, 5, 264–269. [Google Scholar] [CrossRef]
- Enright, H.; Miller, W.J.; Hays, R.; Floyd, R.A.; Hebbel, R.P. Preferential targeting of oxidative base damage to internucleosomal DNA. Carcinogenesis 1996, 17, 1175–1177. [Google Scholar] [CrossRef]
- Thorslund, T.; Sunesen, M.; Bohr, V.A.; Stevnsner, T. Repair of 8-oxoG is slower in endogenous nuclear genes than in mitochondrial DNA and is without strand bias. DNA Repair 2002, 1, 261–273. [Google Scholar] [CrossRef]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef]
- Shokolenko, I.N.; Wilson, G.L.; Alexeyev, M.F. Aging: A mitochondrial DNA perspective, critical analysis and an update. World J. Exp. Med. 2014, 4, 46–57. [Google Scholar] [CrossRef]
- Trifunovic, A.; Hansson, A.; Wredenberg, A.; Rovio, A.T.; Dufour, E.; Khvorostov, I.; Spelbrink, J.N.; Wibom, R.; Jacobs, H.T.; Larsson, N.G. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl. Acad. Sci. USA 2005, 102, 17993–17998. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Mott, J.L.; Zhang, D.; Stevens, M.; Chang, S.; Denniger, G.; Zassenhaus, H.P. Oxidative stress is not an obligate mediator of disease provoked by mitochondrial DNA mutations. Mutat. Res. 2001, 474, 35–45. [Google Scholar] [CrossRef]
- Zhong, S.; Ng, M.C.; Lo, Y.M.; Chan, J.C.; Johnson, P.J. Presence of mitochondrial tRNA(Leu(UUR)) A to G 3243 mutation in DNA extracted from serum and plasma of patients with type 2 diabetes mellitus. J. Clin. Pathol. 2000, 53, 466–469. [Google Scholar] [CrossRef]
- Arnalich, F.; Codoceo, R.; Lopez-Collazo, E.; Montiel, C. Circulating cell-free mitochondrial DNA: A better early prognostic marker in patients with out-of-hospital cardiac arrest. Resuscitation 2012, 83, e162–e163. [Google Scholar] [CrossRef]
- Kung, C.T.; Hsiao, S.Y.; Tsai, T.C.; Su, C.M.; Chang, W.N.; Huang, C.R.; Wang, H.C.; Lin, W.C.; Chang, H.W.; Lin, Y.J.; et al. Plasma nuclear and mitochondrial DNA levels as predictors of outcome in severe sepsis patients in the emergency room. J. Transl. Med. 2012, 10, 130. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Shock, L.S.; Thakkar, P.V.; Peterson, E.J.; Moran, R.G.; Taylor, S.M. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 3630–3635. [Google Scholar] [CrossRef]
- Bellizzi, D.; D’Aquila, P.; Scafone, T.; Giordano, M.; Riso, V.; Riccio, A.; Passarino, G. The control region of mitochondrial DNA shows an unusual CpG and non-CpG methylation pattern. DNA Res. 2013, 20, 537–547. [Google Scholar] [CrossRef]
- Dzitoyeva, S.; Chen, H.; Manev, H. Effect of aging on 5-hydroxymethylcytosine in brain mitochondria. Neurobiol. Aging 2012, 33, 2881–2891. [Google Scholar] [CrossRef]
- Pollack, Y.; Kasir, J.; Shemer, R.; Metzger, S.; Szyf, M. Methylation pattern of mouse mitochondrial DNA. Nucleic Acids Res. 1984, 12, 4811–4824. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.K.; Mangalhara, K.C.; Prakasam, G.; Bamezai, R.N.K. DNA Methyltransferase1 (DNMT1) Isoform3 methylates mitochondrial genome and modulates its biology. Sci. Rep. 2017, 7, 1525. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.; Zhang, L.; Gao, J.; Wen, S.; Zhou, H.; Feng, S. Methylation of mitochondrial DNA displacement loop region regulates mitochondrial copy number in colorectal cancer. Mol. Med. Rep. 2017, 16, 5347–5353. [Google Scholar] [CrossRef] [PubMed]
- Wolters, J.E.J.; van Breda, S.G.J.; Caiment, F.; Claessen, S.M.; de Kok, T.; Kleinjans, J.C.S. Nuclear and mitochondrial DNA methylation patterns induced by valproic acid in human hepatocytes. Chem. Res. Toxicol. 2017, 30, 1847–1854. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Du, Q.; Chen, L.; Fu, G.; Li, S.; Fu, L.; Zhang, X.; Ma, C.; Bin, C. CpG methylation patterns of human mitochondrial DNA. Sci. Rep. 2016, 6, 23421. [Google Scholar] [CrossRef]
- Owa, C.; Poulin, M.; Yan, L.; Shioda, T. Technical adequacy of bisulfite sequencing and pyrosequencing for detection of mitochondrial DNA methylation: Sources and avoidance of false-positive detection. PLoS ONE 2018, 13, e0192722. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- Rongvaux, A.; Jackson, R.; Harman, C.C.; Li, T.; West, A.P.; de Zoete, M.R.; Wu, Y.; Yordy, B.; Lakhani, S.A.; Kuan, C.Y.; et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 2014, 159, 1563–1577. [Google Scholar] [CrossRef]
- White, M.J.; McArthur, K.; Metcalf, D.; Lane, R.M.; Cambier, J.C.; Herold, M.J.; van Delft, M.F.; Bedoui, S.; Lessene, G.; Ritchie, M.E.; et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 2014, 159, 1549–1562. [Google Scholar] [CrossRef]
- Ader, N.R.; Hoffmann, P.C.; Ganeva, I.; Borgeaud, A.C.; Wang, C.; Youle, R.J.; Kukulski, W. Molecular and topological reorganizations in mitochondrial architecture interplay during Bax-mediated steps of apoptosis. eLife 2019, 8, e40712. [Google Scholar] [CrossRef] [PubMed]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; San Chin, H.; et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018, 359, eaao6047. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 2018, 37, e99238. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shokolenko, I.; Alexeyev, M. Mitochondrial DNA: Consensuses and Controversies. DNA 2022, 2, 131-148. https://doi.org/10.3390/dna2020010
Shokolenko I, Alexeyev M. Mitochondrial DNA: Consensuses and Controversies. DNA. 2022; 2(2):131-148. https://doi.org/10.3390/dna2020010
Chicago/Turabian StyleShokolenko, Inna, and Mikhail Alexeyev. 2022. "Mitochondrial DNA: Consensuses and Controversies" DNA 2, no. 2: 131-148. https://doi.org/10.3390/dna2020010
APA StyleShokolenko, I., & Alexeyev, M. (2022). Mitochondrial DNA: Consensuses and Controversies. DNA, 2(2), 131-148. https://doi.org/10.3390/dna2020010