
Biological Treatments in Inflammatory Bowel Disease: A Complex Mix of Mechanisms and Actions



Abstract
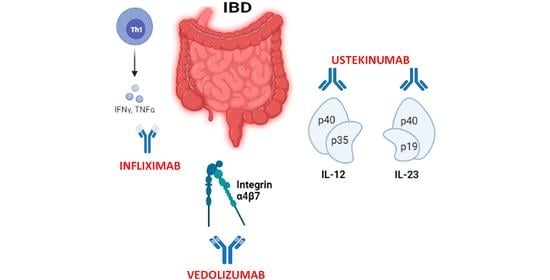
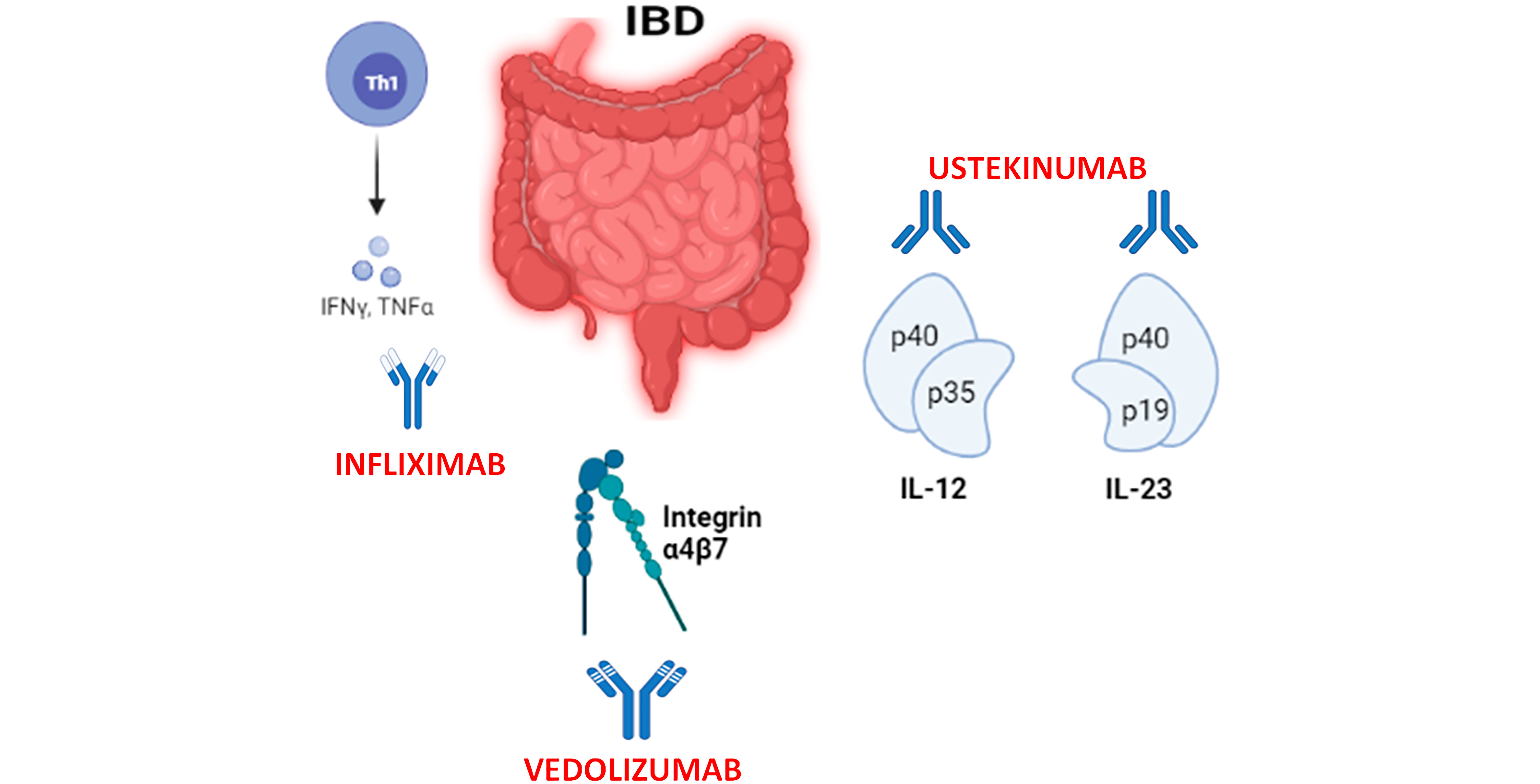
1. Introduction
2. Biological Therapies in IBD
2.1. Anti-TNF-α Therapy
2.1.1. TNF-α
2.1.2. Anti-TNF-α Antibodies in Current IBD Therapy
2.2. Anti-Integrin Therapy
2.3. Anti-Cytokine Therapy
2.4. New Biologics
3. A Deep Insight into the Mechanisms of Action of Biologics in IBD
3.1. Anti-TNF-α Antibodies
3.2. Anti-Integrins Antibodies
3.3. Anti-Cytokines Antibodies
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yeshi, K.; Ruscher, R.; Hunter, L.; Daly, N.L.; Loukas, A.; Wangchuk, P. Revisiting Inflammatory Bowel Disease: Pathology, Treatments, Challenges and Emerging Therapeutics Including Drug Leads from Natural Products. J. Clin. Med. 2020, 9, 1273. [Google Scholar] [CrossRef]
- Vavricka, S.R.; Schoepfer, A.; Scharl, M.; Lakatos, P.L.; Navarini, A.; Rogler, G. Extraintestinal manifestations of inflammatory bowel disease. Inflamm. Bowel Dis. 2015, 21, 1982–1992. [Google Scholar] [CrossRef]
- Garber, A.; Regueiro, M. Extraintestinal Manifestations of Inflammatory Bowel Disease: Epidemiology, Etiopathogenesis, and Management. Curr. Gastroenterol. Rep. 2019, 21, 31. [Google Scholar] [CrossRef] [PubMed]
- Tejeda Taveras, N.; Rivera Martínez, A.; Kumar, R.; Jamil, A.; Kumar, B. Pulmonary Manifestations of Inflammatory Bowel Disease. Cureus 2021, 13, e14216. [Google Scholar] [CrossRef]
- Molodecky, N.A.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; Kaplan, G.G. Challenges associated with identifying the environmental determinants of the inflammatory bowel diseases. Inflamm. Bowel Dis. 2011, 17, 1792–1799. [Google Scholar] [CrossRef]
- Uranga, J.A.; López-Miranda, V.; Lombó, F.; Abalo, R. Food, nutrients and nutraceuticals affecting the course of inflammatory bowel disease. Pharmacol. Rep. 2016, 68, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.; Rhodes, J.M.; Lindsay, J.O.; Abreu, M.T.; Kamm, M.A.; Gibson, P.R.; Gasche, C.; Silverberg, M.S.; Mahadevan, U.; Boneh, R.S.; et al. Dietary Guidance From the International Organization for the Study of Inflammatory Bowel Diseases. Clin. Gastroenterol. Hepatol. 2020, 18, 1381–1392. [Google Scholar] [CrossRef] [PubMed]
- Melinder, C.; Hiyoshi, A.; Fall, K.; Halfvarson, J.; Montgomery, S. Stress resilience and the risk of inflammatory bowel disease: A cohort study of men living in Sweden. BMJ Open 2017, 7, e014315. [Google Scholar] [CrossRef]
- Labanski, A.; Langhorst, J.; Engler, H.; Elsenbruch, S. Stress and the brain-gut axis in functional and chronic-inflammatory gastrointestinal diseases: A transdisciplinary challenge. Psychoneuroendocrinology 2020, 111, 104501. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.C.; Tang, W.; Ching, J.Y.; Wong, M.; Chow, C.M.; Hui, A.J.; Wong, T.C.; Leung, V.K.; Tsang, S.W.; Yu, H.H.; et al. Incidence and phenotype of inflammatory bowel disease based on results from the Asia-Pacific Crohn’s and colitis epidemiology study. Gastroenterology 2013, 145, 158–165.e2. [Google Scholar] [CrossRef]
- Loftus, E.V. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology 2004, 126, 1504–1517. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, K.; Kamada, N. Diet–Microbiota Interactions in Inflammatory Bowel Disease. Nutrients 2021, 13, 1533. [Google Scholar] [CrossRef]
- Aldars-garcía, L.; Marin, A.C.; Chaparro, M.; Gisbert, J.P. The interplay between immune system and microbiota in inflammatory bowel disease: A narrative review. Int. J. Mol. Sci. 2021, 22, 3076. [Google Scholar] [CrossRef] [PubMed]
- Gasaly, N.; de Vos, P.; Hermoso, M.A. Impact of Bacterial Metabolites on Gut Barrier Function and Host Immunity: A Focus on Bacterial Metabolism and Its Relevance for Intestinal Inflammation. Front. Immunol. 2021, 12, 658354. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, D.; Chaparro, M.; Gisbert, J.P. Human Intestinal Dendritic Cells in Inflammatory Bowel Diseases. Mol. Nutr. Food Res. 2018, 62, 1700931. [Google Scholar] [CrossRef] [PubMed]
- Tindemans, I.; Joosse, M.E.; Samsom, J.N. Dissecting the Heterogeneity in T-Cell Mediated Inflammation in IBD. Cells 2020, 9, 110. [Google Scholar] [CrossRef]
- Greuter, T.; Vavricka, S.; König, A.O.; Beaugerie, L.; Scharl, M. Malignancies in Inflammatory Bowel Disease. Digestion 2020, 101, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Axelrad, J.E.; Lichtiger, S.; Yajnik, V. Inflammatory bowel disease and cancer: The role of inflammation, immunosuppression, and cancer treatment. World J. Gastroenterol. 2016, 22, 4794–4801. [Google Scholar] [CrossRef]
- Magro, F.; Gionchetti, P.; Eliakim, R.; Ardizzone, S.; Armuzzi, A.; Barreiro-de Acosta, M.; Burisch, J.; Gecse, K.B.; Hart, A.L.; Hindryckx, P.; et al. Third European evidence-based consensus on diagnosis and management of ulcerative colitis. Part 1: Definitions, diagnosis, extra-intestinal manifestations, pregnancy, cancer surveillance, surgery, and ileo-anal pouch disorders. J. Crohn’s Colitis 2017, 11, 649–670. [Google Scholar] [CrossRef]
- Scharl, S.; Barthel, C.; Rossel, J.B.; Biedermann, L.; Misselwitz, B.; Schoepfer, A.M.; Straumann, A.; Vavricka, S.R.; Rogler, G.; Scharl, M.; et al. Malignancies in Inflammatory Bowel Disease: Frequency, Incidence and Risk Factors—Results from the Swiss IBD Cohort Study. Am. J. Gastroenterol. 2019, 114, 116–126. [Google Scholar] [CrossRef]
- Hazel, K.; O’Connor, A. Emerging treatments for inflammatory bowel disease. Ther. Adv. Chronic Dis. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gisbert, J.P.; Marín, A.C.; Chaparro, M. The risk of relapse after Anti-TNF discontinuation in inflammatory bowel disease: Systematic review and meta-analysis. Am. J. Gastroenterol. 2016, 111, 632–647. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Grunert, P.C.; Stallmach, A. An Update for Pharmacologists on New Treatment Options for Inflammatory Bowel Disease: The Clinicians’ Perspective. Front. Pharmacol. 2021, 12, 655054. [Google Scholar] [CrossRef]
- Macaluso, F.S.; Fries, W.; Privitera, A.C.; Siringo, S.; Inserra, G.; Magnano, A.; Di Mitri, R.; Mocciaro, F.; Belluardo, N.; Scarpulla, G.; et al. A propensity score-matched comparison of infliximab and adalimumab in TNF-α inhibitors naïve and non-naïve patients with Crohn’s disease: Real-life data from the Sicilian Network for Inflammatory Bowel Disease (SN-IBD). J. Crohn’s Colitis 2019, 13, 209–217. [Google Scholar] [CrossRef]
- Misselwitz, B.; Juillerat, P.; Sulz, M.C.; Siegmund, B.; Brand, S. Emerging Treatment Options in Inflammatory Bowel Disease: Janus Kinases, Stem Cells, and More. Digestion 2020, 101, 69–82. [Google Scholar] [CrossRef]
- Al-Bawardy, B.; Shivashankar, R.; Proctor, D.D. Novel and Emerging Therapies for Inflammatory Bowel Disease. Front. Pharmacol. 2021, 12, 651415. [Google Scholar] [CrossRef]
- Privitera, G.; Pugliese, D.; Lopetuso, L.R.; Scaldaferri, F.; Neri, M.; Guidi, L.; Gasbarrini, A.; Armuzzi, A. Novel trends with biologics in inflammatory bowel disease: Sequential and combined approaches. Ther. Adv. Gastroenterol. 2021, 14, 1–19. [Google Scholar] [CrossRef]
- Nakase, H.; Uchino, M.; Shinzaki, S.; Matsuura, M.; Matsuoka, K.; Kobayashi, T. Evidence-based clinical practice guidelines for inflammatory bowel disease. J. Gastroenterol. 2021, 56, 489–526. [Google Scholar] [CrossRef]
- Lamb, C.A.; Kennedy, N.A.; Raine, T.; Hendy, P.A.; Smith, P.J.; Limdi, J.K.; Hayee, B.; Lomer, M.C.E.; Parkes, G.C.; Selinger, C.; et al. British Society of Gastroenterology consensus guidelines on the management of inflammatory bowel disease in adults. Gut 2019, 68, s1–s106. [Google Scholar] [CrossRef] [PubMed]
- Burger, D.; Travis, S. Conventional medical management of inflammatory bowel disease. Gastroenterology 2011, 140, 1827–1837.e2. [Google Scholar] [CrossRef] [PubMed]
- Na, S.Y.; Moon, W. Perspectives on current and novel treatments for inflammatory bowel disease. Gut Liver 2019, 13, 604–616. [Google Scholar] [CrossRef]
- Hemperly, A.; Vande Casteele, N. Clinical Pharmacokinetics and Pharmacodynamics of Infliximab in the Treatment of Inflammatory Bowel Disease. Clin. Pharmacokinet. 2018, 57, 929–942. [Google Scholar] [CrossRef]
- Ordás, I.; Mould, D.R.; Feagan, B.G.; Sandborn, W.J. Anti-TNF monoclonal antibodies in inflammatory bowel disease: Pharmacokinetics-based dosing paradigms. Clin. Pharmacol. Ther. 2012, 91, 635–646. [Google Scholar] [CrossRef]
- Gisbert, J.P.; Chaparro, M. Predictors of Primary Response to Biologic Treatment [Anti-TNF, Vedolizumab, and Ustekinumab] in Patients with Inflammatory Bowel Disease: From Basic Science to Clinical Practice. J. Crohn’s Colitis 2020, 14, 694–709. [Google Scholar] [CrossRef] [PubMed]
- Sabino, J.; Verstockt, B.; Vermeire, S.; Ferrante, M. New biologics and small molecules in inflammatory bowel disease: An update. Ther. Adv. Gastroenterol. 2019, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Jang, D.I.; Lee, A.H.; Shin, H.Y.; Song, H.R.; Park, J.H.; Kang, T.B.; Lee, S.R.; Yang, S.H. The role of tumor necrosis factor alpha (Tnf-α) in autoimmune disease and current tnf-α inhibitors in therapeutics. Int. J. Mol. Sci. 2021, 22, 2719. [Google Scholar] [CrossRef]
- Friedrich, M.; Pohin, M.; Powrie, F. Cytokine Networks in the Pathophysiology of Inflammatory Bowel Disease. Immunity 2019, 50, 992–1006. [Google Scholar] [CrossRef]
- Neurath, M.F. Cytokines and inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Tracey, D.; Klareskog, L.; Sasso, E.H.; Salfeld, J.G.; Tak, P.P. Tumor necrosis factor antagonist mechanisms of action: A comprehensive review. Pharmacol. Ther. 2008, 117, 244–279. [Google Scholar] [CrossRef]
- Vos, A.C.W.; Wildenberg, M.E.; Duijvestein, M.; Verhaar, A.P.; Van Den Brink, G.R.; Hommes, D.W. AntiTumor necrosis factor-α antibodies induce regulatory macrophages in an Fc region-dependent manner. Gastroenterology 2011, 140, 221–230.e3. [Google Scholar] [CrossRef]
- Billmeier, U.; Dieterich, W.; Neurath, M.F.; Atreya, R. Molecular mechanism of action of anti-tumor necrosis factor antibodies in inflammatory bowel diseases. World J. Gastroenterol. 2016, 22, 9300–9313. [Google Scholar] [CrossRef] [PubMed]
- Atreya, R.; Zimmer, M.; Bartsch, B.; Waldner, M.J.; Atreya, I.; Neumann, H.; Hildner, K.; Hoffman, A.; Kiesslich, R.; Rink, A.D.; et al. Antibodies against tumor necrosis factor (TNF) induce T-cell apoptosis in patients with inflammatory bowel diseases via TNF receptor 2 and intestinal CD14+ macrophages. Gastroenterology 2011, 141, 2026–2038. [Google Scholar] [CrossRef]
- Digby-Bell, J.L.; Atreya, R.; Monteleone, G.; Powell, N. Interrogating host immunity to predict treatment response in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 9–20. [Google Scholar] [CrossRef]
- Slevin, S.M.; Egan, L.J. New Insights into the Mechanisms of Action of Anti-Tumor Necrosis Factor-alpha Monoclonal Antibodies in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 2909–2920. [Google Scholar] [CrossRef] [PubMed]
- Kaymakcalan, Z.; Sakorafas, P.; Bose, S.; Scesney, S.; Xiong, L.; Hanzatian, D.K.; Salfeld, J.; Sasso, E.H. Comparisons of affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin. Immunol. 2009, 131, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Mitoma, H.; Horiuchi, T.; Tsukamoto, H.; Ueda, N. Molecular mechanisms of action of anti-TNF-α agents—Comparison among therapeutic TNF-α antagonists. Cytokine 2018, 101, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Lee, S.H.; Lee, H.T.; Lee, J.U.; Son, J.Y.; Shin, W.; Heo, Y.S. Structural biology of the TNFα antagonists used in the treatment of rheumatoid arthritis. Int. J. Mol. Sci. 2018, 19, 768. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Hanauer, S.B.; Katz, S.; Safdi, M.; Wolf, D.G.; Baerg, R.D.; Tremaine, W.J.; Johnson, T.; Diehl, N.N.; Zinsmeister, A.R. Etanercept for active Crohn’s disease: A randomized, double-blind, placebo-controlled trial. Gastroenterology 2001, 121, 1088–1094. [Google Scholar] [CrossRef]
- Van Den Brande, J.M.H.; Koehler, T.C.; Zelinkova, Z.; Bennink, R.J.; Te Velde, A.A.; Ten Cate, F.J.W.; Van Deventer, S.J.H.; Peppelenbosch, M.P.; Hommes, D.W. Prediction of antitumour necrosis factor clinical efficacy by real-time visualisation of apoptosis in patients with Crohn’s disease. Gut 2007, 56, 509–517. [Google Scholar] [CrossRef]
- Leppkes, M.; Neurath, M.F. Cytokines in inflammatory bowel diseases—Update 2020. Pharmacol. Res. 2020, 158, 104835. [Google Scholar] [CrossRef]
- Rehman, M.; Cancarevic, I.; Iskander, B.; Lalani, S.; Malik, B.H. Biologics Targeting in the Treatment of Inflammatory Bowel Disease: A Conundrum. Cureus 2020, 12, e10621. [Google Scholar] [CrossRef]
- De Vries, H.S.; Van Oijen, M.G.H.; Driessen, R.J.B.; De Jong, E.M.G.J.; Creemers, M.C.W.; Kievit, W.; De Jong, D.J. Appropriate infliximab infusion dosage and monitoring: Results of a panel meeting of rheumatologists, dermatologists and gastroenterologists. Br. J. Clin. Pharmacol. 2011, 71, 7–19. [Google Scholar] [CrossRef]
- Present, D.H.; Rutgeerts, P.; Targan, S.; Hanauer, S.B.; Mayer, L.; Van Hogezand, R.A. Infliximab for the Treatment of Fistulas in Patients with Crohn’s Disease. N. Engl. J. Med. 1999, 340, 1398–1405. [Google Scholar] [CrossRef]
- Wong, U.; Cross, R.K. Primary and secondary nonresponse to infliximab: Mechanisms and countermeasures. Expert Opin. Drug Metab. Toxicol. 2017, 13, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Martineau, C.; Flourié, B.; Wils, P.; Vaysse, T.; Altwegg, R.; Buisson, A.; Amiot, A.; Pineton de Chambrun, G.; Abitbol, V.; Fumery, M.; et al. Efficacy and safety of golimumab in Crohn’s disease: A French national retrospective study. Aliment. Pharmacol. Ther. 2017, 46, 1077–1084. [Google Scholar] [CrossRef]
- Gibson, P.R.; Feagan, B.G.; Sandborn, W.J.; Marano, C.; Strauss, R.; Johanns, J.; Padgett, L.; Collins, J.; Tarabar, D.; Hebzda, Z.; et al. Maintenance of Efficacy and Continuing Safety of Golimumab for Active Ulcerative Colitis: PURSUIT-SC Maintenance Study Extension through 1 Year. Clin. Transl. Gastroenterol. 2016, 7, e168. [Google Scholar] [CrossRef]
- Knight, D.M.; Trinh, H.A.N.; Le, J.; Siegel, S.; Shealy, D.; Mcdonough, M.; Scallon, B.; Moore, M.A.; Vilcek, J.A.N.; Daddona, P.; et al. Construction and Initial Characterization of a Mouse-Human Chimeric Anti-TNF Antibody. Mol. Immunol. 1993, 30, 1443–1453. [Google Scholar] [CrossRef]
- Moon, W.; Pestana, L.; Becker, B.; Loftus, E.V.; Hanson, K.A.; Bruining, D.H.; Tremaine, W.J.; Kane, S.V. Efficacy and safety of certolizumab pegol for Crohn’s disease in clinical practice. Aliment. Pharmacol. Ther. 2015, 42, 428–440. [Google Scholar] [CrossRef]
- Wang, W.; Wang, E.Q.; Balthasar, J.P. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin. Pharmacol. Ther. 2008, 84, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Peyrin-Biroulet, L.; Deltenre, P.; de Suray, N.; Branche, J.; Sandborn, W.J.; Colombel, J.F. Efficacy and Safety of Tumor Necrosis Factor Antagonists in Crohn’s Disease: Meta-Analysis of Placebo-Controlled Trials. Clin. Gastroenterol. Hepatol. 2008, 6, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Modigliani, R. Picture of Attacks of Crohn’ Evolution on Prednisolone. Gastroenterology 1990, 98, 811–818. [Google Scholar] [CrossRef]
- Long, M.; Martin, C.; Pipkin, C.; Herfarth, H.; Sandler, R.; Kappelman, M. Risk of Melanoma and Nonmelanoma Skin Cancer Among Patients with Inflammatory Bowel Disease. Gastroenerology 2012, 143, 390–399. [Google Scholar] [CrossRef]
- Annese, V.; Beaugerie, L.; Egan, L.; Biancone, L.; Bolling, C.; Dierickx, D. European Evidence-Based Consensus: Inflammatory Bowel Disease and Malignancies. JCC 2015, 9, 945–965. [Google Scholar] [CrossRef]
- Butter, M.; Weiler, S.; Biedermann, L.; Scharl, M.; Rogler, G.; Bischoff-Ferrari, H.A.; Misselwitz, B. Clinical manifestations, pathophysiology, treatment and outcome of inflammatory bowel diseases in older people. Maturitas 2018, 110, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Park, S.C.; Jeen, Y.T. Anti-integrin therapy for inflammatory bowel disease. World J. Gastroenterol. 2018, 24, 1868–1880. [Google Scholar] [CrossRef]
- Binion, D.G.; West, G.A.; Ina, K.; Ziats, N.P.; Emancipator, S.N.; Fiocchi, C. Enhanced leukocyte binding by intestinal microvascular endothelial cells in inflammatory bowel disease. Gastroenterology 1997, 112, 1895–1907. [Google Scholar] [CrossRef]
- Zundler, S.; Becker, E.; Weidinger, C.; Siegmund, B. Anti-adhesion therapies in inflammatory bowel disease-molecular and clinical aspects. Front. Immunol. 2017, 8, 891. [Google Scholar] [CrossRef]
- Bamias, G.; Clark, D.; Rivera-Nieves, J. Leukocyte Traffic Blockade in Inflammatory Bowel Disease. Curr. Drug Targets 2013, 14, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Jeldres, T.; Tyler, C.J.; Boyer, J.D.; Karuppuchamy, T.; Bamias, G.; Dulai, P.S.; Boland, B.S.; Sandborn, W.J.; Patel, D.R.; Rivera-Nieves, J. Cell Trafficking Interference in Inflammatory Bowel Disease: Therapeutic Interventions Based on Basic Pathogenesis Concepts. Inflamm. Bowel Dis. 2019, 25, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Panés, J. Development of drugs to target interactions between leukocytes and endothelial cells and treatment algorithms for inflammatory bowel diseases. Gastroenterology 2014, 147, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Bellaguarda, E.; Keyashian, K.; Pekow, J.; Rubin, D.; Cohen, R.; Sakuraba, A. Prevalence of Antibodies Against JC Virus in Patients with Refractory Crohn’s Disease and Effects of Natalizumab Therapy. Physiol. Behav. 2015, 13, 1919–1925. [Google Scholar] [CrossRef]
- Sands, B.E.; Feagan, B.G.; Rutgeerts, P.; Colombel, J.F.; Sandborn, W.J.; Sy, R.; D’Haens, G.; Ben-Horin, S.; Xu, J.; Rosario, M.; et al. Effects of vedolizumab induction therapy for patients with Crohn’s disease in whom tumor necrosis factor antagonist treatment failed. Gastroenterology 2014, 147, 618–627.e3. [Google Scholar] [CrossRef]
- Feagan, B.G.; Rubin, D.T.; Danese, S.; Vermeire, S.; Abhyankar, B.; Sankoh, S.; James, A.; Smyth, M. Efficacy of Vedolizumab Induction and Maintenance Therapy in Patients With Ulcerative Colitis, Regardless of Prior Exposure to Tumor Necrosis Factor Antagonists. Clin. Gastroenterol. Hepatol. 2017, 15, 229–239.e5. [Google Scholar] [CrossRef]
- Vickers, A.D.; Ainsworth, C.; Mody, R.; Bergman, A.; Ling, C.S.; Medjedovic, J.; Smyth, M. Systematic review with network meta-analysis: Comparative efficacy of biologics in the treatment of moderately to severely active ulcerative colitis. PLoS ONE 2016, 11, e0165435. [Google Scholar] [CrossRef]
- Colombel, J.F.; Sands, B.E.; Rutgeerts, P.; Sandborn, W.; Danese, S.; D’Haens, G.; Panaccione, R.; Loftus, E.V.; Sankoh, S.; Fox, I.; et al. The safety of vedolizumab for ulcerative colitis and Crohn’s disease. Gut 2016, 66, 839–851. [Google Scholar] [CrossRef]
- Simon, E.G.; Ghosh, S.; Iacucci, M.; Moran, G.W. Ustekinumab for the treatment of Crohn’s disease: Can it find its niche? Ther. Adv. Gastroenterol. 2016, 9, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Benson, J.M.; Peritt, D.; Scallon, B.J.; Heavner, G.A.; Shealy, D.J.; Giles-Komar, J.M.; Mascelli, M.A. Discovery and mechanism of ustekinumab: A human monoclonal antibody targeting interleukin-12 and interleukin-23 for treatment of immune-mediated disorders. mAbs 2011, 3, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, A.; Rodríguez-Lago, I. How to Optimize Treatment With Ustekinumab in Inflammatory Bowel Disease: Lessons Learned From Clinical Trials and Real-World Data. Front. Med. 2021, 8, 640813. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.; Benson, J.; Blank, M.; Brodmerkel, C.; Baker, D.; Sharples, K.R.; Szapary, P. Ustekinumab: Lessons learned from targeting interleukin-1223p40 in immune-mediated diseases. Ann. N. Y. Acad. Sci. 2009, 1182, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Armuzzi, A.; Ardizzone, S.; Biancone, L.; Castiglione, F.; Danese, S.; Gionchetti, P.; Orlando, A.; Rizzello, F.; Scribano, M.L.; Vecchi, M.; et al. Ustekinumab in the management of Crohn’s disease: Expert opinion. Dig. Liver Dis. 2018, 50, 653–660. [Google Scholar] [CrossRef]
- Hanauer, S.B.; Sandborn, W.J.; Feagan, B.G.; Gasink, C.; Jacobstein, D.; Zou, B.; Johanns, J.; Adedokun, O.J.; Sands, B.E.; Rutgeerts, P.; et al. IM-UNITI: Three-year efficacy, safety, and immunogenicity of ustekinumab treatment of Crohn’s disease. J. Crohn’s Colitis 2020, 14, 23–32. [Google Scholar] [CrossRef]
- Sands, B.; Sandborn, W.; Panaccione, R.; O’Brien, C.; Zhang, H.; Johanns, J. Safety and Efficacy of Ustekinumab Induction Therapy in Patients with Moderate to Severe Ulcerative Colitis: Results from the Phase 3 Unifi Study. United Eur. Gastroenterol. J. 2018, 6, 1586–1597. [Google Scholar] [CrossRef]
- Vermeire, S.; O’Byrne, S.; Keir, M.; Williams, M.; Lu, T.T.; Mansfield, J.C.; Lamb, C.A.; Feagan, B.G.; Panes, J.; Salas, A.; et al. Etrolizumab as induction therapy for ulcerative colitis: A randomised, controlled, phase 2 trial. Lancet 2014, 384, 309–318. [Google Scholar] [CrossRef]
- Lichnog, C.; Klabunde, S.; Becker, E.; Fuh, F.; Tripal, P.; Atreya, R.; Klenske, E.; Erickson, R.; Chiu, H.; Reed, C.; et al. Cellular mechanisms of etrolizumab treatment in inflammatory bowel disease. Front. Pharmacol. 2019, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Selinger, C.; Sandborn, W.; Panes, J.; Jones, J.; Hassanali, A.; Jacob, R. Etrolizumab as Induction Therapy in Moderate to Severe Crohn’s Disease: Results From BERGAMOT Cohort 1. Gut 2018, 67, A53. [Google Scholar]
- Whibley, N.; Gaffen, S. Gut-busters—IL-17 Ain’t Afraid of No IL-23. Immunity 2015, 43, 620–622. [Google Scholar] [CrossRef]
- Kumar, P.; Monin, L.; Castillo, P.; Elsegeiny, W.; Horne, W.; Eddens, T. Intestinal interleukin-17 receptor signaling mediates reciprocal control of the gut microbiota and autoimmune inflammation. Immunity 2016, 44, 659–671. [Google Scholar] [CrossRef]
- Armstrong, A.W.; Read, C. Pathophysiology, Clinical Presentation, and Treatment of Psoriasis: A Review. JAMA J. Am. Med. Assoc. 2020, 323, 1945–1960. [Google Scholar] [CrossRef]
- Ten Hove, T.; Van Montfrans, C.; Peppelenbosch, M.P.; Van Deventer, S.J.H. Infliximab treatment induces apoptosis of lamina propria T lymphocytes in Crohn’s disease. Gut 2002, 50, 206–211. [Google Scholar] [CrossRef]
- Ueda, N.; Tsukamoto, H.; Mitoma, H.; Ayano, M.; Tanaka, A.; Ohta, S.I.; Inoue, Y.; Arinobu, Y.; Niiro, H.; Akashi, K.; et al. The cytotoxic effects of certolizumab pegol and golimumab mediated by transmembrane tumor necrosis factorα. Inflamm. Bowel Dis. 2013, 19, 1224–1231. [Google Scholar] [CrossRef]
- Fries, W.; Muja, C.; Crisafulli, C.; Cuzzocrea, S.; Mazzon, E. Dynamics of enterocyte tight junctions: Effect of experimental colitis and two different anti-TNF strategies. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Marini, M.; Bamias, G.; Rivera-Nieves, J.; Moskaluk, C.A.; Hoang, S.B.; Ross, W.G.; Pizarro, T.T.; Cominelli, F. TNF-α neutralization ameliorates the severity of murine Crohn’s-like ileitis by abrogation of intestinal epithelial cell apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 8366–8371. [Google Scholar] [CrossRef] [PubMed]
- Román, I.D.; Cano-Martínez, D.; Lobo, M.V.T.; Fernández-Moreno, M.D.; Hernández-Breijo, B.; Sacristán, S.; Sanmartín-Salinas, P.; Monserrat, J.; Gisbert, J.P.; Guijarro, L.G. Infliximab therapy reverses the increase of allograft inflammatory factor-1 in serum and colonic mucosa of rats with inflammatory bowel disease. Biomarkers 2017, 22, 133–144. [Google Scholar] [CrossRef]
- Rutella, S.; Fiorino, G.; Vetrano, S.; Correale, C.; Spinelli, A.; Pagano, N.; Arena, V.; Maggiano, N.; Repici, A.; Malesci, A.; et al. Infliximab therapy inhibits inflammation-induced angiogenesis in the mucosa of patients with crohn’s disease. Am. J. Gastroenterol. 2011, 106, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Vos, A.C.W.; Wildenberg, M.E.; Arijs, I.; Duijvestein, M.; Verhaar, A.P.; De Hertogh, G.; Vermeire, S.; Rutgeerts, P.; Van Den Brink, G.R.; Hommes, D.W. Regulatory macrophages induced by infliximab are involved in healing in vivo and in vitro. Inflamm. Bowel Dis. 2012, 18, 401–408. [Google Scholar] [CrossRef]
- Agnholt, J.; Kelsen, J.; Brandsborg, B.; Jakobsen, N.O.; Dahlerup, J.F. Increased production of granulocyte-macrophage colony-stimulating factor in Crohn’s disease—A possible target for infliximab treatment. Eur. J. Gastroenterol. Hepatol. 2004, 16, 649–655. [Google Scholar] [CrossRef]
- Eder, P.; Lykowska-Szuber, L.; Iwanik, K.; Krela-Kazmierczak, I.; Stawczyk-Eder, K.; Majewski, P.; Linke, K.; Kay, E.W.; Wozniak, A. The influence of anti-TNF therapy on CD31 and VEGF expression in colonic mucosa of crohn’s disease patients in relation to mucosal healing. Folia Histochem. Cytobiol. 2016, 54, 75–80. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, Z.; Arijs, I.; De Hertogh, G.; Vermeire, S.; Noman, M.; Bullens, D.; Coorevits, L.; Sagaert, X.; Schuit, F.; Rutgeerts, P.; et al. Reciprocal changes of Foxp3 expression in blood and intestinal mucosa in IBD patients responding to infliximab. Inflamm. Bowel Dis. 2010, 16, 1299–1310. [Google Scholar] [CrossRef]
- Fang, L.; Pang, Z.; Shu, W.; Wu, W.; Sun, M.; Cong, Y.; Liu, Z. Anti-TNF Therapy Induces CD4+ T-Cell Production of IL-22 and Promotes Epithelial Repairs in Patients with Crohn’s Disease. Inflamm. Bowel Dis. 2018, 24, 1733–1744. [Google Scholar] [CrossRef]
- Zhang, C.; Shu, W.; Zhou, G.; Lin, J.; Chu, F.; Wu, H.; Liu, Z. Anti-TNF-α therapy suppresses proinflammatory activities of mucosal neutrophils in inflammatory bowel disease. Mediat. Inflamm. 2018, 2018. [Google Scholar] [CrossRef]
- Slevin, S.M.; Dennedy, M.C.; Connaughton, E.P.; Ribeiro, A.; Ceredig, R.; Griffin, M.D.; Egan, L.J. Infliximab selectively modulates the circulating blood monocyte repertoire in Crohn’s Disease. Inflamm. Bowel Dis. 2016, 22, 2863–2878. [Google Scholar] [CrossRef]
- Orbán, C.; Szabó, D.; Bajnok, A.; Vásárhelyi, B.; Tulassay, T.; Arató, A.; Veres, G.; Toldi, G. Altered calcium influx of peripheral Th2 cells in pediatric Crohn’s disease: Infliximab may normalize activation patterns. Oncotarget 2016, 7, 44966–44974. [Google Scholar] [CrossRef]
- Orbán, C.; Szabó, D.; Bajnok, A.; Vásárhelyi, B.; Tulassay, T.; Arató, A.; Veres, G.; Toldi, G. Altered activation of peripheral CD8+ T cells in pediatric Crohn’s disease. Immunol. Lett. 2017, 185, 48–51. [Google Scholar] [CrossRef]
- Gareb, B.; Otten, A.T.; Frijlink, H.W.; Dijkstra, G.; Kosterink, J.G.W. Review: Local tumor necrosis factor-α inhibition in inflammatory bowel disease. Pharmaceutics 2020, 12, 539. [Google Scholar] [CrossRef]
- Bloemendaal, F.M.; Koelink, P.J.; Van Schie, K.A.; Rispens, T.; Peters, C.P.; Buskens, C.J.; Van Der Bilt, J.D.; Bemelman, W.A.; Korf, H.; Sabino, J.G.; et al. TNF-anti-TNF immune complexes inhibit IL-12/IL-23 secretion by inflammatory macrophages via an fc-dependent mechanism. J. Crohn’s Colitis 2018, 12, 1122–1130. [Google Scholar] [CrossRef]
- Gundersen, M.D.; Goll, R.; Hol, J.; Olsen, T.; Rismo, R.; Sørbye, S.W.; Sundnes, O.; Haraldsen, G.; Florholmen, J. Loss of interleukin 33 expression in colonic crypts-a potential marker for disease remission in ulcerative colitis. Sci. Rep. 2016, 6, 35403. [Google Scholar] [CrossRef]
- Timmermans, W.M.C.; Van Laar, J.A.M.; Van Der Houwen, T.B.; Kamphuis, L.S.J.; Bartol, S.J.W.; Lam, K.H.; Ouwendijk, R.J.; Sparrow, M.P.; Gibson, P.R.; Van Hagen, P.M.; et al. B-cell dysregulation in Crohn’s disease is partially restored with infliximab therapy. PLoS ONE 2016, 11, e0160103. [Google Scholar] [CrossRef]
- Zundler, S.; Becker, E.; Schulze, L.L.; Neurath, M.F. Immune cell trafficking and retention in inflammatory bowel disease: Mechanistic insights and therapeutic advances. Gut 2019, 68, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Soler, D.; Chapman, T.; Yang, L.L.; Wyant, T.; Egan, R.; Fedyk, E.R. The binding specificity and selective antagonism of vedolizumab, an anti-α4β7 integrin therapeutic antibody in development for inflammatory bowel diseases. J. Pharmacol. Exp. Ther. 2009, 330, 864–875. [Google Scholar] [CrossRef] [PubMed]
- Zundler, S.; Schillinger, D.; Fischer, A.; Atreya, R.; López-Posadas, R.; Watson, A.; Neufert, C.; Atreya, I.; Neurath, M.F. Blockade of αeβ7 integrin suppresses accumulation of CD8 + and Th9 lymphocytes from patients with IBD in the inflamed gut in vivo. Gut 2017, 66, 1936–1948. [Google Scholar] [CrossRef] [PubMed]
- Binder, M.T.; Becker, E.; Wiendl, M.; Schleier, L.; Fuchs, F.; Leppkes, M.; Atreya, R.; Neufert, C.; Atreya, I.; Neurath, M.F.; et al. Similar Inhibition of Dynamic Adhesion of Lymphocytes from IBD Patients to MAdCAM-1 by Vedolizumab and Etrolizumab-s. Inflamm. Bowel Dis. 2018, 24, 1237–1250. [Google Scholar] [CrossRef]
- Lord, J.D.; Long, S.A.; Shows, D.M.; Thorpe, J.; Schwedhelm, K.; Chen, J.; Kita, M.; Buckner, J.H. Circulating Integrin Alpha4/Beta7+ Lymphocytes Targeted by Vedolizumab Have a Pro-Inflammatory Phenotype. Clin. Inmunol. 2018, 193, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Uzzan, M.; Tokuyama, M.; Rosenstein, A.K.; Tomescu, C.; Ivo, N.; Ko, H.M.; Leyre, L.; Chokola, A.; Kaplan-Lewis, E.; Rodriguez, G.; et al. Anti-α4β7 therapy targets lymphoid aggregates in the gastrointestinal tract of HIV-1–infected individuals. Sci. Transl. Med. 2018, 10, eaau4711. [Google Scholar] [CrossRef] [PubMed]
- Zeissig, S.; Rosati, E.; Dowds, C.M.; Aden, K.; Bethge, J.; Schulte, B.; Pan, W.H.; Mishra, N.; Zuhayra, M.; Marx, M.; et al. Vedolizumab is associated with changes in innate rather than adaptive immunity in patients with inflammatory bowel disease. Gut 2019, 68, 25–39. [Google Scholar] [CrossRef]
- Arijs, I.; De Hertogh, G.; Lemmens, B.; Van Lommel, L.; De Bruyn, M.; Vanhove, W.; Cleynen, I.; MacHiels, K.; Ferrante, M.; Schuit, F.; et al. Effect of vedolizumab (anti-α4β7-integrin) therapy on histological healing and mucosal gene expression in patients with UC. Gut 2018, 67, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.A.; Mansfield, J.C.; Tew, G.W.; Gibbons, D.; Long, A.K.; Irving, P.; Diehl, L.; Eastham-Anderson, J.; Price, M.B.; Boyle, G.O.; et al. αEβ7 Integrin Identifies Subsets of Pro-Inflammatory Colonic CD4+ T Lymphocytes in Ulcerative Colitis. J. Crohn’s Colitis 2017, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Hassan-Zahraee, M.; Banerjee, A.; Cheng, J.B.; Zhang, W.; Ahmad, A.; Page, K.; von Schack, D.; Zhang, B.; Martin, S.W.; Nayak, S.; et al. Anti-MAdCAM antibody increases β7+ T cells and CCR9 gene expression in the peripheral blood of patients with Crohn’s disease. J. Crohn’s Colitis 2018, 12, 77–86. [Google Scholar] [CrossRef]
- Schippers, A.; Muschaweck, M.; Clahsen, T.; Tautorat, S.; Grieb, L.; Tenbrock, K.; Gaßler, N.; Wagner, N. β7-Integrin exacerbates experimental DSS-induced colitis in mice by directing inflammatory monocytes into the colon. Mucosal Immunol. 2016, 9, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Forkel, M.; van Tol, S.; Höö, C.; Michaëlsson, J.; Almer, S.; Mjösberg, J. Distinct aletrations in the composition of mucosal innate lymphoid cells in newly diagnosed and established Crohn´s disease and ulcerative colitis. J. Crohn’s Colitis 2019, 13, 67–78. [Google Scholar] [CrossRef]
- Pararasa, C.; Zhang, N.; Tull, T.J.; Chong, M.H.A.; Siu, J.H.Y.; Guesdon, W.; Chavele, K.M.; Sanderson, J.D.; Langmead, L.; Kok, K.; et al. Reduced CD27−IgD−B cells in blood and raised CD27−IgD−B cells in gut-associated lymphoid tissue in inflammatory bowel disease. Front. Immunol. 2019, 10, 361. [Google Scholar] [CrossRef]
- Matsuno, H.; Kayama, H.; Nishimura, J.; Sekido, Y.; Osawa, H.; Barman, S.; Ogino, T.; Takahashi, H.; Haraguchi, N.; Hata, T.; et al. CD103+ Dendritic Cell Function Is Altered in the Colons of Patients with Ulcerative Colitis. Inflamm. Bowel Dis. 2017, 23, 1524–1534. [Google Scholar] [CrossRef] [PubMed]
- Scharl, M.; Vavricka, S.; Rogler, G. Review: New Anti-Cytokines for IBD: What is in the Pipeline? Curr. Drug Targets 2013, 14, 1405–1420. [Google Scholar] [CrossRef] [PubMed]
- Gee, K.; Guzzo, C.; Mat, N.F.C.; Ma, W.; Kumar, A. The IL-12 family of cytokines in infection, inflammation and autoimmune disorders. Inflamm. Allergy Drug Targets 2009, 8, 40–52. [Google Scholar] [CrossRef]
- Parham, C.; Chirica, M.; Timans, J.; Vaisberg, E.; Travis, M.; Cheung, J.; Pflanz, S.; Zhang, R.; Singh, K.P.; Vega, F.; et al. A Receptor for the Heterodimeric Cytokine IL-23 Is Composed of IL-12Rβ1 and a Novel Cytokine Receptor Subunit, IL-23R. J. Immunol. 2002, 168, 5699–5708. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef]
- Langrish, C.L.; McKenzie, B.S.; Wilson, N.J.; De Waal Malefyt, R.; Kastelein, R.A.; Cua, D.J. IL-12 and IL-23: Master regulators of innate and adaptive immunity. Immunol. Rev. 2004, 202, 96–105. [Google Scholar] [CrossRef]
- Adedokun, O.J.; Xu, Z.; Gasink, C.; Jacobstein, D.; Szapary, P.; Johanns, J.; Gao, L.L.; Davis, H.M.; Hanauer, S.B.; Feagan, B.G.; et al. Pharmacokinetics and Exposure Response Relationships of Ustekinumab in Patients With Crohn’s Disease. Gastroenterology 2018, 154, 1660–1671. [Google Scholar] [CrossRef]
- Luo, J.; Wu, S.J.; Lacy, E.R.; Orlovsky, Y.; Baker, A.; Teplyakov, A.; Obmolova, G.; Heavner, G.A.; Richter, H.T.; Benson, J. Structural Basis for the Dual Recognition of IL-12 and IL-23 by Ustekinumab. J. Mol. Biol. 2010, 402, 797–812. [Google Scholar] [CrossRef]

{kind=link}
| Type of Antibody | Suffix | Anti-TNF-α | Anti-Integrin | Anti-Cytokine | |
|---|---|---|---|---|---|
 | Murine | -omab | |||
 | Human | -umab | Adalimumab (CD, UC) Golimumab (UC) | Ustekinumab (CD, UC) | |
 | Chimeric | -ximab | Infliximab (CD, UC) | ||
 | Humanized | -zumab | Certolizumab pegol (CD) | Natalizumab (CD) Vedolizumab (CD, UC) | |
| TNFR1 | TNFR2 | |||
|---|---|---|---|---|
| Alternative names | TNFRSF1A, CD120a, p55 | TNFRSF1B, CD120b, p75 | ||
| TNF-α form involved in activity | Soluble and transmembrane | Transmembrane | ||
| Intracellular transducer | TRADD | TRAF | ||
| Intracellular complexes | I | IIa and IIb (apoptosome) | IIc (necrosome) | I |
| Location of complex assemblage | Plasma membrane | Cytoplasm | Cytoplasm | Plasma membrane |
| Complex components | TNRF1, TRADD, RIPK1, TRAF (2 or 5), cIAP (1 or 2), LUBAC | TNRF1, TRADD, RIPK1, TRAF2, cIAP (1 or 2), pro-caspase-8, FADD (+ RIPK3 in complex IIb) | TNRF1, TRADD, RIPK (1 or 3) | TNFR2, TRAF2, TRAF2, cIAP1, cIAP2 |
| Final intracellular effector | NF-κB, MAPKs | Caspase-8 | MLKL | MAPKs, NF-κB, AKT |
| Biological effect | Inflammation, tissue degeneration, host defense, cell proliferation, cell survival | Apoptosis | Necroptosis, inflammation | Tissue regeneration, cell proliferation, cell survival, host defense, inflammation |
| Antibody | Experimental Samples # | Patients Group | Mechanism of Action | Reference |
|---|---|---|---|---|
| Infliximab | In vitro Jurkat T cell | - | Induction of apoptosis and increase in the Bax/Bcl-2 ration in the CD3/CD28 stimulated cells | [89] |
| In vitro Jurkat T cells | - | Induction of antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity | [90] | |
| Ileal epithelium | - | Prevention of the disappearance of occluding-1 and zonula occludens-1 and the increase of claudin-2 tight junction proteins induced by chemical-colitis and TNFα receptor-1 knockout model | [91] | |
| Intestinal epitelial cells * | - | Reduction of intestinal cell apoptosis with reduced expression of membrane bound FAS/CD95 | [92] | |
| Intestinal biopsies and serum * | - | Restoration of epithelial barrier integrity, mucus production and p38-decreased colon inflammation Reduced levels of allograft inflammatory factor-1 in serum and colon | [93] | |
| Intestinal biopsies | HC and CD | Reduction of proliferation marker Ki-67 in endothelial cells, mucosal levels of vascular endothelial growth factor-A, and migration capacity | [94] | |
| Intestinal biopsies | UC and CD | Induction of regulatory macrophages (CD206+/CD68+) in mucosal healing patients | [95] | |
| Intestinal biopsies | HC and CD | Inhibition of granulocyte-macrophage colony-stimulating factor intestinal content, as well as mucosal histology index and peripheral blood leucocyte count | [96] | |
| Intestinal biopsies | CD | Decrease in the immunohistochemical expression of CD31 and vascular endothelial growth factor, correlated to the endoscopic healing | [97] | |
| Intestinal biopsies | HC and CD | Induction of apoptosis of activated lamina propria T lymphocytes, in responding patients | [49] | |
| Intestinal biopsies | CD | Induction of apoptosis in activated T lymphocytes from lamina propria with increase in CD3 and TUNEL positive cells | [89] | |
| LPMC | HC, UC and CD | Induction of cell apoptosis in the co-culture of lamina propria TNFR2+ expressing CD4+ T cells with membrane-bound TNF+ CD14+ intestinal macrophages in the IBD patients | [42] | |
| Intestinal biopsies and PBMC | HC, UC and CD | Increase in CD4+ CD25+Foxp3+ T-regulatory cells and CD4+ CD25 Foxp3+ T-regulatory cells. Decrease of mucosal mRNA and protein expression of Foxp3 in responding patients, but not in non-responders | [98] | |
| Intestinal biopsies and PBMC | HC and CD | Upregulation of IL-22 gene expression in the gut mucosa. Promotion of IL-22 expression by CD4+ T cells through binding to membrane-bound TNF, and Th22 cell differentiation | [99] | |
| Intestinal biopsies and PBMC | HC, UC and CD | Reduction in the percentage of CD66b+ neutrophils and expression of CD66b in peripheral blood and inflamed mucosa of patients in the response group. Reduction of neutrophil-derived myeloperoxidase, calprotectin, and production of pro-inflammatory mediators, as well as migration of neutrophils | [100] | |
| PBMC | HC and CD | Reduction in the number of circulating monocytes, especially in the classical and intermediate subsets | [101] | |
| PBMC | CD | Lack of influence on the expression of activation markers, homing receptors, memory cells, Fas or Bax/Bcl-2 expression on peripheral blood T lymphocytes | [89] | |
| PBMC | HC and CD | Restoration of the calcium influx and potassium channel function in Th2 lymphocytes comparable to Th1 cells | [102] | |
| PBMC | HC and CD | Restoration of the calcium response and potassium channel function in CD8+ lymphocytes comparable to healthy controls | [103] | |
| Adalimumab | In vitro Jurkat T cells | - | Induction of antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity | [90] |
| Intestinal biopsies | CD | Decrease in the immunohistochemical expression of CD31 and vascular endothelial growth factor, correlated to the endoscopic healing | [97] | |
| LPMC | HC, UC and CD | Induction of cell apoptosis in the co-culture of lamina propria TNFR2+-expressing CD4+ T cells with membrane-bound TNF+ CD14+ intestinal macrophages in the IBD patients | [42] | |
| Golimumab | In vitro Jurkat T cells | - | Binding to transmembrane TNF-α Induction of antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity | [90] |
| Certolizumab pegol | In vitro Jurkat T cells | - | Binding to transmembrane TNF-α Induction of nonapoptotic cell death in transmembrane TNF-α-expressing cells | [90] |
| LPMC | HC, UC and CD | Induction of cell apoptosis in the co-culture of lamina propria TNFR2+-expressing CD4+ T cells with membrane-bound TNF+ CD14+ intestinal macrophages in the IBD patients | [42] |
| Antibody | Experimental Samples | Patients Group | Mechanism of Action | Reference |
|---|---|---|---|---|
| Vedolizumab | Blood | HC | Binding to a subset of peripheral blood memory CD4+ T cells including gut-homing IL-17 T-helper lymphocytes Binding to eosinophils at high levels, and to naïve T-helper lymphocytes, naïve and memory cytotoxic T lymphocytes, B lymphocytes, natural killer cells, and basophils at lower levels The highest level of binding was in the population α4β7high of memory CD45RO+ CD4+ T cells, specifically in competition with Act-1 | [109] |
| Intestinal biopsies and PBMC | HC, UC, CD | Blockade of the adhesion of T effector cells from CD patients to MadCAM-1 Increase in the expression of intestinal α4β1 integrin in CD | [110] | |
| PBMC | HC, UC, CD | Blockade of the adhesion of peripheral blood leukocytes including CD4+ T cells, CD8+ T cells, CD19+ B cells, and granulocytes to addressin molecules Adhesion was partially, but not completely, related to integrin expression Etrolizumab resulted in similar inhibition of adhesion to MAdCAM-1 than vedolizumab | [111] | |
| PBMC | HC, CD | Higher response of α4β7-expressing lymphocytes to pro-inflammatory cytokines IL-6, IL-7 and IL-21, but lower response to regulatory T cells Enrichment of cells bearing the circulating T follicular helper cell marker CXCR5, in relation with the selective effect of vedolizumab to replace pro-inflammatory effector cells with regulatory T cells and Th2 cells | [112] | |
| Intestinal biopsies and PBMC | HC, IBD (with concomitant HIV-1 infection) | Reduction of intestinal B subsets, naïve and activated CD4+ T cells in the terminal ileum, and lymphoid aggregates within the gastrointestinal tract | [113] | |
| Intestinal biopsies, LPMC and PBMC | HC, UC, CD | Minor effect on lamina propria T cell abundance and mucosal T cell receptor repertoire assessed by immunophenotyping, immunohistochemistry, T cell receptor profiling and RNA sequencing Notable alterations in innate immunity and macrophage populations correlated with the clinical efficacy | [114] | |
| Intestinal biopsies | HC, UC | Partial restoration of the colonic expression of mucosal immune-related genes in UC patients responding to vedolizumab at week 52 Significant reduction in the inflammatory cell infiltrate leading to mucosal healing, although persistent histological and gene expression abnormalities remain after therapy in responding patients | [115] | |
| Etrolizumab | Intestinal biopsies and PBMC | HC, UC, CD | Blockade of the adhesion of T effector cells from CD patients to VCAM-1 | [110] |
| Intestinal biopsies and PBMC | HC, UC | Enrichment of pro-inflammatory cytokines, Th17 and Th17/Th1 subsets, and lower expression of regulatory T cell-associated genes in colonic CD4+ T cells expressing higher levels of αEβ7 integrin | [116] | |
| Ontamalimab | Blood | CD | Decrease in soluble MAdCAM in serum Increase in β7+ central memory T cells, β7+ effector memory T cells, β7+ naïve T cells, in association with up-regulation of CCR9 gene expression | [117] |
| Experimental Techniques | Mechanism of Action | Reference |
|---|---|---|
| Cell lines: hIL-2 dependent T cell line kit225 (DNAX, Palo Alto, CA); mIL-3 dependent Ba/F3 cells; NKL cells Signal transduction: SDS-PAGE and immunoprecipitation. Western-blot | IL-12 and IL-23 have similar signal transduction mechanisms such as Jak2, Tyk2, Stat1, Stat3, Stat4, Stat5 IL-12 participates in phosphorylation of Stat4 and Stat 6, NK cell lytic functions IL-23 participates in phosphorylation of Stat3 and lymphocyte activation IL-12 and IL-23 produce pro-inflammatory cytokines | [77,124] |
| Human Ig transgenic mouse technology | Identification of monoclonal hybridoma clone that produces huma IgG that binds and neutralized IL-12: ustekinumab Description of IL-23 was later and due to the discovery of ustekinumab | [77] |
| Hu-Ig mice technology | Binds to p40 of IL-12 and IL-23 preventing their interaction with IL-12Rβ | [77,127] |
| Crystal structure studies | D1 of p40 binds epitope for ustekinumab | [77,128] |
| Isothermal titration colorimetry analysis | Ustekinumab binds to IL-12 and IL-23 equally | [77,128] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno, L.O.; Fernández-Tomé, S.; Abalo, R. Biological Treatments in Inflammatory Bowel Disease: A Complex Mix of Mechanisms and Actions. Biologics 2021, 1, 189-210. https://doi.org/10.3390/biologics1020012
Moreno LO, Fernández-Tomé S, Abalo R. Biological Treatments in Inflammatory Bowel Disease: A Complex Mix of Mechanisms and Actions. Biologics. 2021; 1(2):189-210. https://doi.org/10.3390/biologics1020012
Chicago/Turabian StyleMoreno, Lorena Ortega, Samuel Fernández-Tomé, and Raquel Abalo. 2021. "Biological Treatments in Inflammatory Bowel Disease: A Complex Mix of Mechanisms and Actions" Biologics 1, no. 2: 189-210. https://doi.org/10.3390/biologics1020012
APA StyleMoreno, L. O., Fernández-Tomé, S., & Abalo, R. (2021). Biological Treatments in Inflammatory Bowel Disease: A Complex Mix of Mechanisms and Actions. Biologics, 1(2), 189-210. https://doi.org/10.3390/biologics1020012