Abstract
Overuse or misuse of broad-spectrum antibiotics increases the risk of the emergence of antibiotic-resistant bacteria, which increases the possibility of antimicrobial-resistant (AMR) bacterial infections, and subsequently raises healthcare costs. The excessive use of broad-spectrum antibiotics has also been linked to increased death rates, whilst the benefits that they offer against antibiotic-resistant bacterial pathogens are minimal. Patients infected with antibiotic-resistant bacterial pathogens frequently receive inadequate antimicrobial therapies due to a lack of effective options than those with non-resistant infections, resulting in poor health outcomes and longer recovery times, especially among patients who are critically ill. Broad-spectrum antibiotics also disturb the gut microbiome, which is increasingly recognized as a regulator of immune health. This study offers insights into the use of targeted antimicrobial therapies for bacterial infections, focusing on strategies that mitigate the risk of antibiotic resistance and unwanted side effects associated with the use of broad-spectrum antibiotics. We focus on identifying the genotype and phenotype of bacterial pathogens and then using either nanoparticle-based, vaccine-based, bacteriophage-based, monoclonal antibody-based, and CRISPR-based targeted therapies to directly kill those pathogens and reduce collateral damage. Furthermore, the mechanisms of action of these targeted therapies and their advantages and disadvantages are discussed.
1. Introduction
The development of antimicrobial therapies stands as one of the most crucial medical advancements of the 20th century, serving as a cornerstone of contemporary medicine. Indeed, antimicrobial therapies prevent millions of premature deaths caused by microbial infections. For example, before the development of modern antibiotic therapies, the mortality rate of pneumoniae due to Streptococcus pneumoniae infections was approximately 40% of the infected individuals [1]. Similarly, the mortality rate caused by Staphylococcus aureus infections was approximately 80% of infected individuals [2], and as high as 97% for patients suffering from endocarditis [3]. Before the discovery of microbial-derived antibiotic chemotherapies, infected wounds were commonly treated by amputation of the affected tissue. Indeed, 70% of the amputations that were performed during World War I resulted from wound infections [4]. Antibiotics have dramatically changed the outcomes for patients with infections, revolutionizing the treatment and cure of infectious diseases. This has enabled advancements in modern medicine, including organ transplants, complex surgeries, and chemotherapy. Unfortunately, the evolution of antibiotic resistance, both in communities as well as in healthcare settings, threatens to undermine the significant progress achieved through the increased availability of effective antimicrobial therapies [5]. The development of multidrug-resistant (MDR) strains of Gram-negative and Gram-positive pathogens has led to infections that are challenging to treat, or that may even be untreatable using the current range of clinical antibiotics [6]. In the late 1950s and early 1960s, resistance to multiple antibiotics was first detected in enteric bacteria, particularly in Shigella spp., Salmonella spp., and in Escherichia coli [7]. These resistant pathogen strains led to significant reductions in clinical efficacy and also resulted in economic losses, primarily in the developing world. However, in the developed world, infections of these bacteria have largely still been regarded as mild health problems, confined only to enteric pathogens [7]. This misconception changed in the 1970s when it was observed that Haemophilus influenza and Neisseria gonorrhoeae had developed resistance to ampicillin [7]. Additionally, H. influenza strains that were resistant to chloramphenicol and tetracycline were also detected, further restricting the therapeutic options [7]. The increasing use of antibiotics combined with poor hygienic settings has facilitated the transmission of antibiotic resistance and has resulted in limited antibiotic therapy options for some infections [8].
Broad-spectrum antibiotics possess inhibitory activity against multiple bacterial species, which has generally been considered to be advantageous, particularly when the bacterial species/strain responsible for the illness is not immediately apparent. However, whilst broad-spectrum activity offers substantial advantages, it also has several inherent disadvantages. One of the most challenging issues associated with using broad-spectrum antibiotics is the promotion of antibiotic resistance as bacteria develop mutations conferring resistance, coupled with selective pressure from the use of antibiotics, which provides a competitive advantage for the mutated strain. The increased use of antibiotics with broad specificities directs the selection of pathogens with antibiotic resistance. Resistance genes may be located on bacterial chromosomal DNA, although more frequently, they are found on transmissible extrachromosomal elements (plasmids) [9]. As a result, resistant bacterial strains, including methicillin-resistant S. aureus (MRSA), Klebsiella ST258, and E. coli ST131, spread rapidly globally. The swift development of microbial resistance is particularly evident for the β-lactam class of antibiotics. Nearly 1000 resistance-related β-lactamase enzymes, which render this class of antibiotics ineffective, have been identified, equating to a tenfold increase in the incidence of antibiotic-resistant pathogens since 1990 [10].
Another drawback of using indiscriminate broad-spectrum antibiotics is their harmful impact on the host microbiome. As these therapies are broad-spectrum, they affect multiple bacteria, including beneficial members of the microbiome, that would otherwise outcompete the pathogenic strains. These effects extend beyond the period of antibiotic treatment. Even temporary exposure (seven days) to broad-spectrum antibiotics may alter the makeup of the gut microbiota for extended periods of up to two years following completion of the treatment [11]. In more extreme instances of antibiotic exposure, the gastrointestinal microbiome may never revert to its original composition, providing ongoing health impacts [12]. Damage to the microbiome can influence many of its important functions, including the production of vitamins, the supply of nutrients, and immunological protection from pathogens [13], thereby increasing susceptibility to multiple infectious diseases [14]. Exposure to some broad-spectrum clinical antibiotics during the early ages of development may be particularly harmful to the microbiome as the early microbiota is less stable and diverse, making it susceptible to disruption [15]. Besides the gut microbiome, the complex oral microbiome is crucial for maintaining both systemic as well as oral health, and dysbiosis in the gut and/or oral microbiome(s) is associated with various medical issues that include cardiovascular, cerebrovascular, and respiratory diseases [16].
Developing targeted antimicrobial therapies that do not promote the development of antibiotic resistance in non-targeted bacteria, and that cause minimum collateral damage to the natural microflora, is a promising strategy for combating antimicrobial resistance. A switch to targeted antimicrobial therapies would be beneficial once the causative pathogen has been identified. At that point, antimicrobial therapies such as nanoparticles, vaccines, bacteriophages, monoclonal antibodies and CRISPR-based therapies can target these pathogens specifically, thereby decreasing the negative effects associated with broad-spectrum therapies. This review summarises five targeted antimicrobial technologies that are useful for treating antibiotic-resistant infections in humans. Thus, it is limited to bacterial pathogens and does not consider fungal and viral pathogens.
2. Overview of Acquisition and Mechanism of Antibiotic Resistance
Improper and overuse of antibiotics selects for antibiotic-resistant genes, which confer competitive advantages to the mutated strain. Bacteria transfer resistant genes to other bacteria through several mechanisms, including conjugation, transduction, and transformation (Figure 1). When a bacterium acquires antibiotic-resistance genes, it may render the antibiotic ineffective via several mechanisms, including enzymatic degradation of the antibiotic; alterations of the antibiotic structure or the target structure so that they can no longer bind, pumping antibiotics out of the cell through the action of an efflux pump; or by modifying the cell wall and/or cell membrane structure so that the entry of antibiotics into the cell is inhibited (Figure 1). Previous studies have outlined the mechanisms of antibiotic-resistance gene transfer and how it impacts the efficacy of antibiotics in greater detail, and the reader is referred to those studies for a greater understanding of resistance mechanisms and development [17,18].
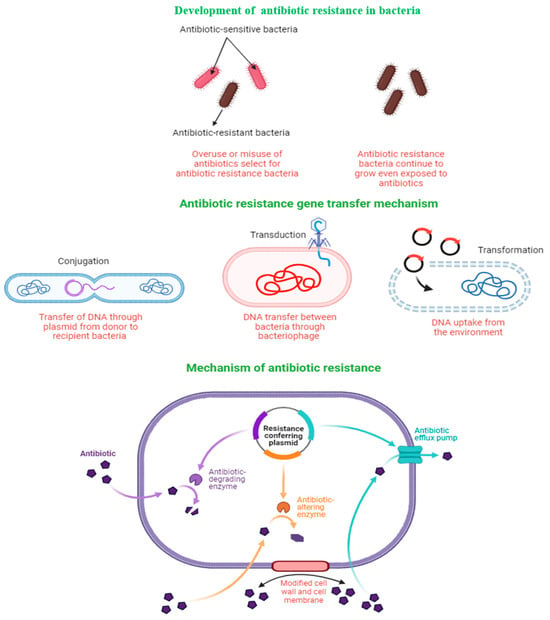
Figure 1.
Gene acquisition and mechanisms of antibiotic resistance in bacteria.
3. Genotypic and Phenotypic Methods to Identify Bacterial Pathogens
The clinical availability of broad-spectrum antibiotic therapies has allowed physicians to treat many microbial infections effectively and safely without the need to specifically identify the disease-causing pathogens. As a result, the need to develop diagnostic tests that can quickly and accurately identify pathogens to enable the use of effective targeted antimicrobial therapies was not widely pursued [19]. However, as noted above, such broad-spectrum approaches have inherent problems, including the development of further resistance and disruption of the microbiome. Targeted approaches afford substantial benefits, although they require fast and accurate pathogen identification [20]. Culture-based methods for identifying infecting pathogens are time-consuming, with initial culturing steps requiring 24–48 h (or substantially longer for some species), during which time broad-spectrum antibiotics are typically administered. After identifying the microbe, determining antibiotic sensitivity may require an additional 24–48 h [21]. Of concern, a 2015 survey revealed that for 31% of infectious diseases, physicians believed that patients receive incorrect antibiotic therapies whilst waiting for blood culture results [22].
Recent enhancements in nucleic acid-based amplification technologies (NAATs), including next-generation sequencing (NGS) and polymerase chain reaction (PCR), along with the development of rapid, high-resolution technologies such as matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS), have transformed the diagnostic approaches used to identify pathogens [23]. NAAT-based approaches to identify pathogens are now routinely used in clinical microbiological laboratories. These approaches are particularly promising for bloodstream infections as they significantly accelerate pathogen identification in comparison with culture-based methods, allowing for species identification within 3–6 h [23]. PCR-based diagnostic methods have now been successfully established for a broad range of bacterial pathogens [24]. PCR is highly regarded by infectious disease experts for its remarkable specificity, sensitivity, and speed of amplification [25]. It is particularly valuable for the detection of pathogens that cannot be cultured in vitro, or in cases where current culture methodologies lack sensitivity or require prolonged incubation durations [26]. For a deeper discussion of the advancements in these technologies, the reader is referred to a recent review of these methods [27].
Despite the substantial improvements in sensitivity, specificity, reproducibility and rapid throughput provided by NAAT, NGS and MALDI-MS technologies, several factors have limited their routine application. These methods require complex sample purification and preparation steps, as well as access to expensive equipment. Additionally, the use of fluorescent dyes in some methods can lead to the formation of primer dimers, which can provide false-positive detections [27]. Furthermore, the methods not only require expensive equipment, but costly reagents are also used, making them less attractive for less well-funded laboratories.
MS fingerprinting methods using MALDI-TOF have been developed to assist in the identification of many bacterial pathogens, and these have been widely adopted by clinical microbiological laboratories worldwide [28]. Detection of bacteria by MALDI-TOF MS may be achieved either by comparing the peptide mass fingerprint (PMF) of an unknown bacterium with the PMFs in published databases, or by matching the biomarker masses of the unknown bacterium with proteome databases [29]. In PMF matching, the mass spectral properties of the unknown bacteria are compared with the mass spectra (MS) of known bacterial isolates in MS databases [29]. Directly identifying pathogens by MALDI-TOF MS techniques may substantially shorten the identification process by a day or more, compared to using conventional identification methods [23]. This method is commonly used in pathogen identification as it is straightforward and is easily implemented in diagnostic laboratories due to the development of numerous commercial PMF libraries [29]. Conversely, identifying microbes through matching measured biomarker masses with predicted protein molecular masses calculated from genome sequences is less favourable as a diagnostic tool, since it requires the complete bacterial genome sequence to establish the required database.
4. Nanoparticle-Based Targeted Antimicrobial Therapies
Over the past decade, nanotechnology has garnered significant attention in antimicrobial therapies, demonstrating great potential to enhance the treatment of antibiotic-resistant bacteria [30,31,32]. Targeted nanoparticles (NPs) that can selectively interact with pathogens may significantly enhance therapeutic outcomes for some antimicrobial therapies and reduce the side effects associated with broad-spectrum antibiotics. Active targeting methods utilize targeting ligands to recognize and bind to the target structure or substrate [33]. Specific ligands that have affinity for cell surface receptors, or bacterial surface molecules that are overexpressed in infected tissues, can be attached to nanoparticle surfaces to direct their interaction with specific pathogens [34]. To specifically target the inhibition of pathogens, as an alternative to broad-spectrum antibiotics, peptide toxins, bacterial toxins, CRISPR, and bacteriophage-based antimicrobial therapies have been investigated in various studies [35,36,37,38,39]. For example, the coating of gold nanoparticles with IgG molecules selectively inhibits the growth of MRSA [40]. The specificity and sensitivity of the treatment were assessed using contrast-phase microscopy, flow cytometry, and fluorescence microscopy; the substantial and sensitive killing of MRSA was reported.
Bacteria naturally release vesicles from their membranes into the extracellular environment. Vesicles secreted by the Gram-negative bacteria are called outer-membrane vehicles [41], whilst vesicles released by Gram-positive bacterial membranes are called extracellular vesicles [42]. Notably, extracellular membrane vesicles contain numerous immunogenic antigens [43] and display varied pathogen-associated molecular patterns that modulate the host immune response [44]. The coating of gold nanoparticles with membranes from outer-membrane vehicles secreted by E. coli results in a nano-vaccine that can generate a specific and significantly stronger immune response [45]. Indeed, antibiotic-loaded nanoparticles (NPs-antibiotic) have already been developed using membranes from extracellular vesicles that are secreted by S. aureus [46]. The resulting NP-antibiotic-linked constructs mimic S. aureus and allow for the host’s immune system to target and eliminate S. aureus [46]. Nanoparticles have also been conjugated with targeting ligands (including fructose and/or mannose-specific lectins) to target carbohydrate receptors on Helicobacter pylori cell surfaces [47]. Such targeted nanoparticles provide site-specific release and gastro-retentive properties. Collectively, these properties enhance localised drug levels, resulting in greater bactericidal potency. Clarithromycin-loaded nanoparticles that are coated with plasma membranes derived from the gastric epithelial cells promote pathogen–host adhesion and specifically target H. pylori [48]. Notably, clarithromycin-loaded nanoparticles demonstrate superior activity against H. pylori compared with free clarithromycin or non-targeted nanoparticles.
The effectiveness of actively targeted nanoparticles relies on several factors, including the ligand properties, the nanoparticle design, as well as the functionalization process. Physical properties, including the shape, size, or charge of nanoparticles have substantial effects on their accumulation in target tissues [49]. However, in biological environments such as the bloodstream, the targeted effects of functionalized nanoparticles may be hindered by the nonspecific biomolecule interactions. Notably, nanoparticle surfaces adsorb biological macromolecules, forming a “biomolecular corona” which covers the NPs’ targeting ligands, lowering the surface energy of the NP [50]. The biomolecular corona may hinder the binding of nanoparticles to their targeted pathogen, thereby reducing the efficacy of the nanoparticles, potentially inducing bacterial resistance [51]. Proteins are essential components in biomolecular corona formation, although sugars and lipids also play important roles [52]. Nanoparticles covered by biomolecular corona exhibit altered physiochemical and functional characteristics which give them new biological identities [53]. The proteins surrounding nanoparticles can either mitigate or stimulate immune responses, or alternatively they may induce pathological and physiological changes [54]. Protein corona is considered to be a major factor that limits nanoparticle-targeted delivery, as it reduces the targeting capability in vivo [55]. Several strategies have been developed to mitigate the effects of biological fluids including blood nanoparticle’s functionality. For example, functionalizing copper sulphide NPs with the glycan-binding protein jacalin reduces protein corona interference and enhances the targeting ability of the NP via microbial cell surface glycan recognition. This results in substantially higher antimicrobial activity compared to pristine nanoparticles [56].
Nanoparticle-based targeted antimicrobial therapies use a range of delivery systems based on metals, liposomes or lipids, mesoporous silica materials or polymers [57,58,59,60]. Antibacterial-targeted nanoparticles may possess inherent antimicrobial activities, or they may carry therapeutic payloads such as antibiotics [61]. The cell envelope surrounding bacteria acts as a physical barrier, which prevents the accumulation and internalization of antibiotic molecules within bacteria [61]. Silica NPs containing gluconamide-targeting moieties have improved binding affinity to Gram-negative bacteria cell membranes [62]. Therefore, the use of nanoparticles for targeted antimicrobial therapies offers a promising solution to targeted therapies, although substantially more work is required in this field to further refine the nanoparticle therapies.
5. Bacteriophage-Based Targeted Antimicrobial Therapies
The potential of bacteriophages in antimicrobial therapy (commonly referred to as phage therapy), has been discussed, and in some regions of the world, utilized, since these bacterial viruses were discovered over a century ago [63]. Bacteriophages specifically target bacteria and cannot infect mammalian cells. Furthermore, bacteriophages are generally specific to one bacterium, or to relatively few bacterial species/strains. Lytic bacteriophages are self-replicating and self-limiting, and can be administered through different routes, making them an attractive option for targeted antibacterial therapies (Figure 2). Additionally, bacteriophages have different mechanisms to those of antibiotics, and therefore their effects are not reduced by bacterial antibiotic resistance mechanisms. Furthermore, as bacteriophage therapies are specific to a single bacterial clone, they generally do not adversely affect the normal bacterial flora, and therefore present reduced risks for secondary infections, which are frequently associated with the use of antibiotics [64]. Some bacteriophages bind to single bacterial receptors, although other phages may also require secondary receptors. These receptors are most frequently identical to antigens that determine the bacterial serotype of the pathogen [64]. A single phage unit will therefore generally only infect one bacterial strain, or a limited number of strains. The narrow specificity of bacteriophages typically necessitates identifying the bacteria causing the infection before initiating the appropriate phage treatment. The specificity of the phage means it will not typically impact beneficial bacteria such as those residing in the gastrointestinal tract. Phage therapy challenges current pharmacokinetic studies because it acts as a self-amplifying drug, replicating in vivo when the target bacteria are present, unlike antimicrobial therapies that require multiple doses [65].
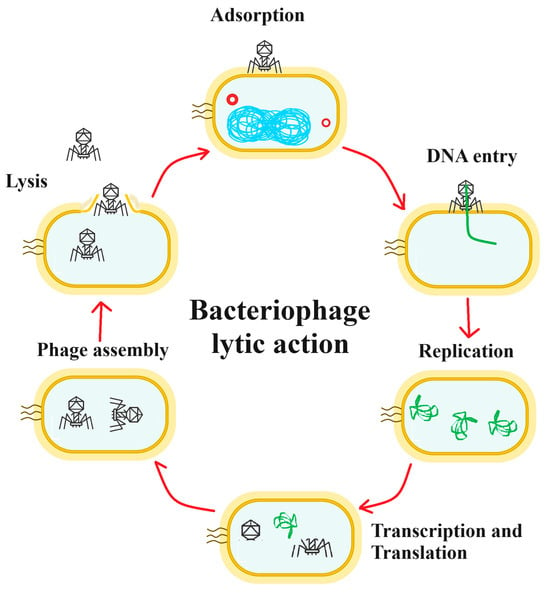
Figure 2.
A schematic for antibacterial phage therapy. The phage particle attaches to the bacterial cell surface in the adsorption stage and the DNA enters the cell (DNA entry phase), leaving the viral protein components outside the cell. The DNA is replicated (replication phase). The viral DNA is then transcribed and translated (transcription and translation phase), resulting in the production of the viral protein components. The viral DNA is incorporated into the viral shell proteins and the mature viruses are assembled (phage assembly phase). Finally, bacterial cell lysis occurs (lysis phase), releasing the new viral particles.
Before the discovery and common usage of clinical antibiotics, it was recognized that many microbial infections could be treated and/or prevented using bacteriophage therapies [66]. However, early clinical bacteriophage studies were relatively neglected in Western Europe and the United States. In contrast, phage therapy continued to be used in Eastern Europe and the former Soviet Union and is still currently used in some parts of Eastern Europe. Early studies reported that phages specific to diarrhea-causing E. coli can substantially reduce the levels of pathogenic E. coli in the gastrointestinal tract by many orders of magnitude in several in vivo animal models [67,68,69]. Bacteriophages have also been used to prevent and treat Acinetobacter and Pseudomonas aeruginosa infections [70], as well Klebsiella pneumoniae, Klebsiella rhinoscleromatis, and Klebsiella ozaenae infections [71]. However, bacteriophage-targeted antimicrobial therapies require the identification of the disease-causing bacteria before application of the presumptive treatment. Bacteriophages can be used in combination with other phages in ‘phage cocktails’ to increase their target ranges, whilst remaining substantially more selective and specific than typical narrow-spectrum antibiotic therapies [72].
The use of temperate bacteriophages therapeutically may pose challenges because of their ability to display superinfection immunity, which converts sensitive phage bacterial targets into insensitive ones and alters their potential to encode bacterial virulence factors (including the production of toxins) [73]. Bacteriophages may also exhibit low virulence and decreased bactericidal potency against their target bacteria due to several factors, including their limited ability to evade bacterial defences, inadequate adsorption properties or poor replication characteristics [74].
6. Vaccine-Based Antimicrobial Therapies
Modern medicine has greatly benefited from antimicrobial drugs and vaccines [75,76]. Whilst bacterial resistance to antibiotics undermines their efficacy, vaccine resistance has evolved substantially less readily than antibiotics [77]. Vaccines are used preventively, whilst antibiotics are typically used for treatment post-infection. This timing difference means that vaccines (unlike antibiotics) generally prevent pathogens from reaching large population sizes within hosts [77]. Additionally, antibiotics typically target pathogens via a single mechanism [18], whereas vaccines may affect their targets in multiple ways, by inducing T-cell responses and/or inducing host-specific antibodies [78]. Due to the differences in the number of target sites affected, more mutations are generally needed for pathogens to develop resistance to vaccines, compared to antibiotics. Several vaccines are licensed and available clinically, with others currently undergoing clinical trials (Table 1).
Streptococcus pneumoniae can harmlessly colonize the nasopharynx, although it may also cause a variety of illnesses under specific conditions [79]. The incidence of infections caused by S. pneumoniae dropped substantially following the introduction of the PCV7 vaccine, which is specific to that bacterium. Notably, that single vaccine conferred immunity against more than 90 known antigens of that bacterium [77]. Before the introduction of the vaccine, there were concerns that the benefits of PCV7 might be quickly diminished by the evolvement of new antigens [80]. This concern was heightened because S. pneumoniae is naturally competent, and antigen replacement was observed in clinical trials [81]. However, an improved conjugate vaccine (PCV13) was developed and introduced in 2010, which protected against the original antigens as well as six additional ones. In its first three years of use, this updated vaccine has prevented an estimated 30,000 additional infections [82]. Vaccinations can also reduce the use of antibiotics by inhibiting viral infections. For instance, influenza vaccines have reduced antibiotic prescriptions from approximately 13% to 64%, indirectly decreasing the spread of antibiotic-resistance genes [83]. It is therefore reasonable to suggest that a major contribution of vaccines against the spread of AMR comes from indirect mechanisms, such as reducing the use of antibiotics, consequently lowering the selection pressure on pathogens.
Despite the benefits of vaccines developed to target antibiotic-resistant bacterial pathogens and their impact on controlling the use of widespread antibiotics, vaccines are yet to be developed against some major antibiotic-resistant pathogens. Currently, no vaccine is available against Clostridioides difficile, a major cause of gastrointestinal illness. Vaccines targeting the pathogenic factors toxin A (TcdA) and toxin B (TcdB) are currently in development, but are not yet available clinically. Sanofi initiated phase III clinical trials of a vaccine against C. difficile that was targeted for use in individuals over the age 50 years who, due to lifestyle factors, had an increased risk of infection from this bacterium. The trial aimed to evaluate the vaccine’s efficacy in preventing primary symptomatic episodes, and therefore to reduce the incidence and effects of the infection. The trial was terminated before completion after an independent data monitoring committee reviewed interim data and determined that the likelihood of achieving the study’s objectives was low [84]. Similarly, a phase III clinical trial is currently being conducted to test a vaccine based on genetically modified full-length TcdA and TcdB toxoid molecules in adults over the age of 50 years who are at risk of C. difficile infections [85]. The efficacy of vaccines targeting only TcdA and TcdB is debatable. Whilst antibodies against these toxins may prevent disease, they do not inhibit the pathogen’s ability to colonize the intestine and therefore are of limited value. Consequently, vaccines that target specific C. difficile surface antigens required for colonization and spore formation of the bacterium are under investigation, although those studies are yet to pass the preclinical stages of drug development [86].
Neisseria gonorrhoeae surface proteins exhibit antigenic diversity and phase variation, making the development of a vaccine against this bacterium complicated. To date, four candidate vaccines have advanced to clinical trials, including a partially autolyzed vaccine, a whole-cell vaccine, a PorA-based vaccine, and a pilus-based vaccine, although none of these approaches have yet proven to be effective [87]. Despite significant efforts, no licensed vaccines are yet available for the prevention of P. aeruginosa or infections, and no candidates are currently in clinical trials [88]. Indeed, few candidates for P. aeruginosa vaccines are showing promise, and an effective vaccine is unlikely in the immediate future. The vaccine candidates tested in humans to date have targeted single virulence mechanisms, including the outer-membrane proteins Oprl and OprF, as well as flagella proteins [89]. None of these candidates have shown enough promise to progress to late-stage development. Indeed, even the most promising vaccine based on an OprF-Oprl fusion protein yielded disappointing clinical results, and studies have since been discontinued [90]. Similarly, despite extensive preclinical research on K. pneumoniae antigens, no vaccines against this bacterium advanced past early-stage trials [91]. Several plain capsule polysaccharide (CPS)-based vaccines have undergone preclinical studies and clinical testing, and have been used to produce hyperimmune human sera for therapeutic purposes [91]. Recently, bioconjugate vaccines developed using CPS from two K. pneumoniae serotypes have demonstrated immunogenicity and efficacy, successfully protecting mice from lethal infections [92]. Despite the ability of the tested vaccines to induce functional antibodies in preclinical trials, it is improbable that a vaccine developed in this way will be considered effective enough for clinical introduction due to the existence of 77 CPS serotypes, which have limited or no cross-reactivity. To provide immunity towards 70% of the K. pneumoniae strains, a vaccine candidate would need to include at least 24 major serotypes [93]. The currently marketed vaccine against Salmonella enterica subsp. enterica serovar Typhi is a live-attenuated Vi polysaccharide vaccine and Ty21a oral vaccine. These vaccines have intrinsic limitations, such as lacking immunological memory and affinity maturation, requiring multiple doses, and having a limited duration of antibody responses [94]. Despite a phase III study showing more than 90% efficacy for the Salmonella Typhi Vi conjugate vaccine when used in children aged 2–5 years, an absence of commercial incentives has hindered the market introduction of these vaccines [95]. There is currently no effective readily available vaccine targeting Shigella spp., although there is significant interest globally in its development [96]. Notably, natural immunity against bacteria of the Shigella genus is serotype-specific, with the O-polysaccharide being the primary vaccine target [97]. Protein-based vaccine candidates targeting Shigella spp. are also in development, including the DB Fusion vaccine, which is created by genetically fusing the type III secretion system proteins IpaD and IpaB [98].
The vaccine therapies listed in Table 1 and discussed above provide examples of some noteworthy bacterial vaccines. However, this list is not exhaustive, and vaccines have also been developed against other noteworthy bacterial pathogens including Haemophilus influenzae, Corynebacterium diphtheriae, and Clostridium tetani. For an in-depth discussion of vaccines prepared against those bacterial pathogens, the reader is referred to a recent review on bacterial vaccines [99].
7. Monoclonal Antibody-Based Antimicrobial Therapies
Monoclonal antibodies (mAbs) were initially discovered by Kilner and Milstein in the early 1970s [100]. Since that time, numerous advancements have been made to enhance the efficacy of mAbs as therapeutic agents. Table 1 summarizes the currently licensed mAB therapies. The isolation of mAbs from patients who had previously been infected and subsequently cleared an infection is useful for identifying human monoclonal antibodies (Hu-mAbs) capable of neutralizing the infection [101]. Once these Hu-mAbs are obtained, they can be sequenced and produced recombinantly, which allows for the manufacture of large quantities for downstream therapeutic use [102]. Advancements have also been made to encompass the creation of libraries of bacteriophages displaying engineered Hu-mAbs, significantly boosting the total number of Hu-mAbs available for screening trials, thereby enhancing the chances of identifying a unique and effective antibody [103]. In addition to the potential of Hu-mAbs as human therapeutic products (which reduce toxicity concerns), they also offer several other advantages that make their development a promising approach for antimicrobial treatment. Hu-mAbs provide specificity to target pathogenic bacteria, ensuring that commensal bacteria remain unharmed [104]. Hu-mAbs offer longevity as their clearance by the host immune system is generally slow, with IgG subtypes typically having a half-life of 21 days [101]. Hu-mAbs enable both sustained and rapid microbial killing through various mechanisms, including anti-virulence activity, direct killing, complement deposition, neutralization, and opsonization by phagocytes [102,104]. Eliminating bacteria through these multiple mechanisms reduces the emergence of resistant strains and reduces the risk of toxic shock. Hu-mAbs disrupt the activity of bacteria through different mechanisms including biofilm formation, bactericidal effects, or via attachment/adhesion, iron acquisition, opsonophagocytosis, and anti-toxin/anti-virulence [104,105,106,107,108,109].
Anthim® (obiltoxaximab) was the first injectable Hu-mAb for antimicrobial treatment, approved by the FDA in 2016 as a treatment for inhalation anthrax. Hu-mAb is used in combination with antibiotics, typically ciprofloxacin. Anthim® was approved for use under the Animal Rule for biothreat organisms when alternative therapies are inappropriate or unavailable [110]. Inhalation of Bacillus anthracis spores causes the most severe form of anthrax infection (inhalation infection), and the ability of the spores to endure harsh environments allows them to avoid the effects of many physical and chemical treatments [111]. Thus, the clinical introduction of Anthim is particularly promising and underscores the potential of Hu-mAb therapeutics. Similarly, in October 2016, the FDA approved Zinplava™ (bezlotoxumab) for the treatment of C. difficile infections in adults [112]. This product is not intended for preventing or treating primary C. difficile infection, but instead was approved to reduce the recurrence of infections, which is common for C. difficile infections [112].

Table 1.
Vaccines (licensed and in clinical trials, as well as licensed monoclonal therapies targeting specific bacterial pathogens).
Table 1.
Vaccines (licensed and in clinical trials, as well as licensed monoclonal therapies targeting specific bacterial pathogens).
| Bacterial Target Species | Vaccine/Antibody Name | Antigen Targeted/Specificity | Reference |
| Licensed | |||
| Mycobacterium tuberculosis | BCG vaccine | Low specificity, but still in use. Research is ongoing to develop a more specific vaccine. | [77] |
| Salmonella enterica serotype Typhi | >20 vaccines have been licensed | The WHO recommends typhoid conjugate vaccine (TCV) over an unconjugated Vi polysaccharide or live-attenuated bacteria vaccines. | [95] |
| Streptococcus pneumoniae | PCV7 | Targets 7 different antigens. Specific for S. pneumoniae, but as it targets many antigens, it provides some immunity, even with bacterial mutations. | [77,78,79,80,81,82,83] |
| PCV10 | A 10-valent vaccine against S. pneumoniae polysaccharides. Although there are >100 serotypes, this vaccine covers most disease-forming serotypes. | ||
| PCV13 | A 13-valent vaccine against S. pneumoniae polysaccharides. Although there are >100 serotypes, this vaccine covers most disease-forming serotypes. | ||
| Vaccines in late-stage clinical trials | |||
| Clostridiodes diffcile | Three vaccines are in phase III clinical trials | The vaccines target TcdA and TcdB toxins for use in individuals >50 years old. The vaccines’ efficacy is dubious. | [85,86] |
| Neisseria gonorrhoeae | Bexsero vaccine | Bexsero vaccine was developed against type B menningococcal infections, but also provides protection against N. gonnorhoea. There is substantial research to develop vaccines with greater specificity. | [87] |
| Salmonella paratyphi | Three vaccines are in clinical trials | Targets bacterial polysaccharides. A 12-TT conjugate vaccine is in clinical use in China. | [77] |
| Vaccines in early-stage clinical trials | |||
| Enterotoxigenic Escherichia coli | Several vaccine candidates | Several vaccines are in phase 2 clinical trials. There is significant variability in strains, but it is hoped that a vaccine targeting heat labile toxoid and colonisation factors would cover 80% of strains. | [77] |
| Klebsiella pneumoniae | KlebV4 | 4-valent vaccine is currently in phase 1/2 clinical trials. | [91,92] |
| Salmonella spp. (non-Typhi) | iCVD1000 | A trivalent vaccine targeting S. typhi and two other non-typhoidal serotypes. Currently in phase 1 trials. | [95] |
| Shigella spp. | WRSS2/WRSS3 | Live-attenuated bacteria vaccine targeting S. sonnei. | [96] |
| GlycoShig3 | A 4-valent glucoconjugate vaccine is in development; it is believed it would successfully target 80% of diseases causing Shigella spp. | [97,98] | |
| Monoclonal antibody-based therapies | |||
| Bacillus anthracis | Anthim® (obiltoxaximab) | Usually used in combination with ciprofloxacin. Is effective against the endospore form of the bacterium. | [111] |
| Clostridium diffcile | Zinplava™ (bezlotoxumab) | Used to reduce and treat recurrent C. difficle infections. Not recommended for primary infections. | [112] |
| Staphylococcus aureus | Tefibazumab | The single antibody targets 200–400 surface antigens. | [113] |
Only licensed vaccines or mAbs; vaccines/mAbs in clinical trials are included in the table. Where trials have been discontinued, the treatment is not included in the table.
It is perhaps surprising that more Hu-mAbs are not in development for microbial infections, given the success of the previous examples. However, antibody research and production are generally costly, and the failures of some recent Hu-mAb trial may have dampened the enthusiasm of drug companies for further developing Hu-mAbs for antimicrobial treatment [113]. There have been multiple unsuccessful attempts to develop Hu-mAbs treatments for S. aureus, including tefibazumab, causing developers to reconsider the mAbs approach against this bacterial species [114]. Monoclonal antibodies have shown potential in animal studies, but later failed in phase II trials [114]. Using a single antibody against one target may be insufficient since bacteria have over 200–400+ surface targets involved in virulence. Additionally, most approaches overlook the fact that bacteria exhibit different lifestyles, such as encapsulated/unencapsulated, vegetative, intracellular, and biofilm-associated lifestyles, each with varying surface and secreted protein profiles [114]. The development of Hu-mAb can be costly, as extensive initial research is necessary to highlight the most effective bacterial surface targets, and to filter through numerous antibody binding studies to identify the Hu-mAbs that best address bacterial infection through various mechanisms of action [113].
8. CRISPR-Based Targeted Antimicrobial Therapies
Clustered regularly interspaced short palindromic repeats/CRISPR-associated protein (CRISPR/Cas) provides a mechanism to specifically target bacterial antibiotic resistance genes in a directed sequence-specific manner [115]. CRISPR/Cas systems are adaptive bacterial defence mechanisms that are directed against infectious viruses and mobile genetic elements (MGEs). Whilst a detailed discussion of the use of CRISPR-Cas technologies is beyond the scope of this study, several recent reviews provide comprehensive summaries of the methodology and recent advancements in the field [116]. Briefly (as summarized in Figure 3), the use of CRISPR-Cas against pathogens involves three phases: (i) CRISPR adaption, which involves the incorporation of spacer sequences into the CRISPR-Cas cassette; (ii) expression/biogenesis of crRNA and the Cas protein; (iii) and crRNA-guided cleavage of target sequences. When MGEs invade bacteria, specific CRISPR RNAs (crRNA) with complementary sequences direct effector proteins to the targets for enzymatic cleavage, resulting in the sequence-specific elimination of the invading molecule [117]. CRISPR/Cas systems are divided into two classes, and further classified into six subtypes (I-VI). Although the type I and II CRISPR systems are most commonly used as CRISPR antimicrobials due to their well-understood mechanisms of action and adaptability to genetic modifications, type VI systems have recently been suggested as an alternative. The type VI CRISPR system may inhibit bacterial growth by targeting either the chromosome, or a plasmid [118]. A notable feature of CRISPR/Cas is its precise targeting ability, as the guide RNA can focus on virulence, antibiotic resistance, or unique genes that are essential genes to pathogen’s survival [119]. Previous studies have successfully utilized the type II CRISPR/Cas9 system obtained from Streptococcus pyogenes to target S. aureus and E. coli, resulting in cell death in both in vivo and in vitro models [120]. CRISPR/Cas3 and CRISPR/Cas9 genome editing constructs kill bacteria carrying AMR genes when they are delivered into bacteria by packaging them into specific bacteriophages [121]. When the phage infects the bacteria, the CRISPR/Cas13a and CRISPR/Cas type VI class 2 systems detect the phage genome transcript, leading to the nonspecific degradation of the bacterial transcripts, thereby blocking bacterial cell growth [122]. CRISPR/Cas 13a antimicrobials show promise in three main areas: (1) as tools to modify the bacterial flora by targeting and removing specific bacterial populations, without adversely affecting other non-targeted bacteria; (2) as a cost-effective system for detecting and identifying bacterial genes in molecular epidemiological studies, without the need for nucleic acid amplifications or optical devices; (3) as effective antibacterial agents with the ability to target any bacterial gene (including AMR genes), or to selectively eliminate toxin-producing bacteria [118].
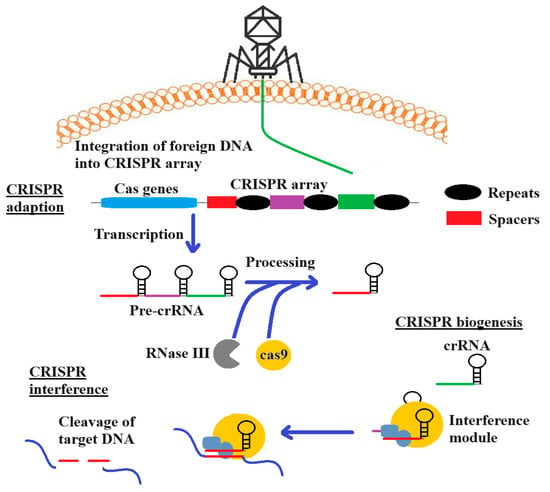
Figure 3.
The use of the CRISPR-Cas adaptive immunity system. The Cas complex selects spacers between repeats and incorporates them into the CRISPR locus (upstream). During crRNA biogenesis, transcription of the locus results in pre-crRNA, which forms crRNA. This aids in the recognition by RNase III, leading to RNA cutting during the interference phase.
Some studies have reported that although the CRISPR/Cas genome targeting construct lowered bacterial load, they also noted the presence of escape mutants, which evaded cell death [123]. This highlights the potential that CRISPR antimicrobials might face similar challenges when used to treat bacterial infection in vivo, and future studies are required to test this hypothesis. Unlike antibiotics, which can induce dysbiosis and promote the spread of resistant bacteria, CRISPR antimicrobials target and kill only a small fraction of the bacterial population. It is likely that this enables commensal bacteria to occupy the niche left by the targeted bacteria, and to further limit their growth [120]. Several countermeasures have been proposed to address pathogen “escape” from CRISPR antimicrobials. One suggestion is to repeat to prevent recombination and subsequent spacer deletion by reducing the CRISPR array to a single spacer [123]. Additionally, overexpressing Cas9 in Enterococcus faecalis has been shown to increase the lethality of self-targeting CRISPR [124].
9. Challenges and Outlook
Broad-spectrum antibiotic therapies have provided medical science with numerous effective and safe therapies to treat bacterial infections. They have provided the ability to treat pathogen infections, without a definitive diagnosis of the disease-causing bacterium, which allows for rapid and effective treatment [1,2,3,4]. However, such untargeted therapies also affect the natural microbiome and may cause dysbiosis, resulting in other illnesses [11,12,13]. Additionally, the use of broad-spectrum antibiotics has allowed the spread of antibiotic-resistance genes through the microbiome (including pathogens), thereby limiting the efficacy of those therapies [5,6,7,8,9,10]. More specific, directed therapies are required to address this problem to mitigate the spread of antibiotic resistance, retaining the efficacy of the available antibiotics for when they are required. Substantial recent research has focused on new targeted technologies, including nanoparticle-, bacteriophage-, vaccine-, monoclonal antibody- and CRISPR-Cas-based therapies, and multiple effective new therapies have been developed.
However, targeted antimicrobial therapies also face limitations due to the delay in identifying pathogens responsible for the disease [21,22], which risks the prescription of specific agents to save critically ill patients [20]. In many cases, this means that the patient may still require broad-spectrum antibiotic therapy whilst the disease-causing infective agent is identified, so that the correct/directed therapy can be applied [23]. Better diagnostic methods are urgently required to speed up the identification processes to allow medical professionals to bypass the use of broad-spectrum antibiotics whilst the causative pathogen is identified. This would not only provide more effective patient care but would also decrease the spread of further antibiotic resistance. Fortunately, several improvements have been proposed to address these challenges. Using nucleic acid [24,25,26,27] or mass spectrometry-based technologies [28] significantly speeds up pathogen diagnosis before specifically targeting those pathogens. With the development of these methods to provide rapid and accurate pathogen identification, targeted therapies are likely to attain greater clinical significance.
Notably, some of the targeted therapies discussed in this review may be better viewed as preventions, rather than treatments [77]. In particular, vaccines prime the recipient’s immune system, allowing it to produce antibodies directed at specific bacterial antigens [78]. This provides the recipient with the ability to quickly respond to infection from the pathogen, allowing them to counteract the pathogen before illness occurs (or to decrease the severity and/or duration of illness). This not only provides effective healthcare for the patient, but also decreases the need for broad-spectrum antibiotic therapies, thereby reducing the development of further resistance. However, vaccines are of little value once an infection has occurred and illness has begun. Furthermore, the development of specific vaccines that are effective and safe is time-consuming and expensive [83,84,85]. Despite this, several vaccines that specifically target bacterial pathogens are already licensed, and others are either undergoing clinical trials, or are in development [86,87,88,89,90,91,92,93,94,95]. Similarly, mAbs offer an additional immunity-related therapeutic pathway [102,104]. Unlike vaccines, mAbs are suitable as therapies rather than just as preventative treatments, although they also suffer from several of the same issues. Whilst mAbs are also expensive and time-consuming to produce, they offer a novel and specific way to target infections, and substantially more work is required to develop more of these therapies.
Nanoparticles have also garnered substantial recent attention for their exceptional performance. However, their synthesis requires stringent conditions unsuitable for large-scale industrial production. Additionally, challenges related to dispersion, storage, cytotoxicity, stability, and degradation significantly hinder their progress [125]. Regarding bacteriophage therapies, large-scale sequencing in the early diagnostic stages necessitates substantial initial investment and time. Furthermore, clearance by the innate immune system directly impacts phage pharmacokinetics. Uncertainties about safety, challenges in penetrating mammalian cells, and increasing resistance present significant challenges [125]. Encapsulating bacteriophages in non-immunogenic polyethylene glycol and liposomes may enhance their in vivo stability against proteolytic and acidic environments and help them evade elimination using the innate immune system, although substantially more work is required to confirm this and develop viable clinical bacteriophage options [126,127]. CRISPR/Cas technology is reliable, but the “escape” phenomenon may occasionally occur, which can impact its efficiency [128]. However, the adaptability of this technology provides it with substantial promise for the development of effective and highly specific therapies.
10. Conclusions
The increasing use of broad-spectrum antibiotics contributes to the evolvement of AMR and hence calls for novel ways to target pathogenic bacteria, especially targeted antimicrobial therapies. In this review, an overview of five promising targeted antimicrobial therapies—nanoparticle-, bacteriophage-, vaccine-, monoclonal antibody-, and CRISPR-based targeted antimicrobial therapies—was discussed alongside their mechanism of action, advantages, and disadvantages. Whilst all of these technologies offer advantages over the current use of broad-spectrum antibiotics, each method has inherent difficulties and limitations. Substantially more work is required to address these limitations, to enhance the potential of these technologies to treat antibiotic-resistant bacterial infections in humans. Narrow-spectrum antibiotics and engineered probiotics are not discussed in this review, but they may also be useful to specifically target pathogens.
Author Contributions
Conceptualisation: M.J.Z. and I.E.C.; methodology: M.J.Z. and I.E.C.; validation, formal analysis, data curation: M.J.Z.; writing—original draft preparation: M.J.Z. and I.E.C.; writing—review and editing, supervision: M.J.C. and I.E.C.; project administration and funding acquisition: I.E.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data are available from the corresponding author on reasonable request.
Acknowledgments
MJZ is grateful to Griffith University for providing a postgraduate scholarship and for providing the facilities to undertake the study.
Conflicts of Interest
The authors have no conflicts of interest.
References
- Bartlett, J.G.; Mundy, L.M. Community-acquired pneumonia. N. Engl. J. Med. 1995, 333, 1618–1624. [Google Scholar] [CrossRef] [PubMed]
- Karchmer, A.W. Staphylococcus aureus and vancomycin: The sequel. Ann. Intern. Med. 1991, 115, 739–741. [Google Scholar] [CrossRef]
- Newman, W.; Torres, J.M.; Guck, J.K. Bacterial endocarditis: An analysis of fifty-two cases. Am. J. Med. 1954, 16, 535–542. [Google Scholar] [CrossRef]
- Hirsch, E.F. “The Treatment of Infected Wounds”, Alexis Carrel’s contribution to the care of wounded soldiers during World War I. J. Trauma Acute Care Surg. 2008, 64, S209–S210. [Google Scholar] [CrossRef]
- Hawkey, P. The growing burden of antimicrobial resistance. J. Antimicrob. Chemother. 2008, 62, i1–i9. [Google Scholar] [CrossRef] [PubMed]
- Akova, M. Epidemiology of antimicrobial resistance in bloodstream infections. Virulence 2016, 7, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Aslam, B.; Wang, W.; Arshad, M.I.; Khurshid, M.; Muzammil, S.; Rasool, M.H.; Nisar, M.A.; Alvi, R.F.; Aslam, M.A.; Qamar, M.U. Antibiotic resistance: A rundown of a global crisis. Infect. Drug Resist. 2018, 11, 1645–1658. [Google Scholar] [CrossRef] [PubMed]
- Rossolini, G.M.; Arena, F.; Pecile, P.; Pollini, S. Update on the antibiotic resistance crisis. Curr. Opin. Pharmacol. 2014, 18, 56–60. [Google Scholar] [CrossRef]
- Laxminarayan, R.; Duse, A.; Wattal, C.; Zaidi, A.K.; Wertheim, H.F.; Sumpradit, N.; Vlieghe, E.; Hara, G.L.; Gould, I.M.; Goossens, H. Antibiotic resistance—The need for global solutions. Lancet Infect. Dis. 2013, 13, 1057–1098. [Google Scholar] [CrossRef]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef]
- Jernberg, C.; Löfmark, S.; Edlund, C.; Jansson, J.K. Long-term ecological impacts of antibiotic administration on the human intestinal microbiota. ISME J. 2007, 1, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Raymann, K.; Shaffer, Z.; Moran, N.A. Antibiotic exposure perturbs the gut microbiota and elevates mortality in honeybees. PLoS Biol. 2017, 15, e2001861. [Google Scholar] [CrossRef] [PubMed]
- Langdon, A.; Crook, N.; Dantas, G. The effects of antibiotics on the microbiome throughout development and alternative approaches for therapeutic modulation. Genome Med. 2016, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Ubeda, C.; Pamer, E.G. Antibiotics, microbiota, and immune defense. Trends Immunol. 2012, 33, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Schulfer, A.; Blaser, M.J. Risks of antibiotic exposures early in life on the developing microbiome. PLoS Pathog. 2015, 11, e1004903. [Google Scholar] [CrossRef]
- Stone, V.N.; Xu, P. Targeted antimicrobial therapy in the microbiome era. Mol. Oral Microbiol. 2017, 32, 446–454. [Google Scholar] [CrossRef]
- Villa, T.G.; Feijoo-Siota, L.; Sánchez-Pérez, A.; Rama, J.R.; Sieiro, C. Horizontal gene transfer in bacteria, an overview of the mechanisms involved. In Horizontal Gene Transfer; Villa, T., Viñas, M., Eds.; Springer: Cham, Switzerland, 2019; pp. 3–76. [Google Scholar]
- Blair, J.M.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef]
- Casadevall, A. The case for pathogen-specific therapy. Expert Opin. Pharmacother. 2009, 10, 1699–1703. [Google Scholar] [CrossRef]
- Kumar, A.; Roberts, D.; Wood, K.E.; Light, B.; Parrillo, J.E.; Sharma, S.; Suppes, R.; Feinstein, D.; Zanotti, S.; Taiberg, L. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit. Care Med. 2006, 34, 1589–1596. [Google Scholar] [CrossRef]
- Faro, J.; Mitchell, M.; Chen, Y.-J.; Kamal, S.; Riddle, G.; Faro, S. Development of a novel test for simultaneous bacterial identification and antibiotic susceptibility. Infect. Dis. Obstet. Gynecol. 2016, 2016, 5293034. [Google Scholar] [CrossRef]
- She, R.C.; Alrabaa, S.; Lee, S.H.; Norvell, M.; Wilson, A.; Petti, C.A. Survey of physicians’ perspectives and knowledge about diagnostic tests for bloodstream infections. PLoS ONE 2015, 10, e0121493. [Google Scholar] [CrossRef] [PubMed]
- Maurer, F.P.; Christner, M.; Hentschke, M.; Rohde, H. Advances in rapid identification and susceptibility testing of bacteria in the clinical microbiology laboratory: Implications for patient care and antimicrobial stewardship programs. Infect. Dis. Rep. 2017, 9, 6839. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.; Chen, S.; Zhong, Q. Digital PCR for accurate quantification of pathogens: Principles, applications, challenges and future prospects. Int. J. Biol. Macromol. 2021, 184, 750–759. [Google Scholar] [CrossRef]
- Schmitz, J.E.; Stratton, C.W.; Persing, D.H.; Tang, Y.W. Forty years of molecular diagnostics for infectious diseases. J. Clin. Microbiol. 2022, 60, e02446-21. [Google Scholar] [CrossRef] [PubMed]
- Britt, N.S.; Khader, K.; He, T.; Willson, T.M.; Effiong, A.; Timbrook, T.T.; Potter, E.M.; Lodise, T.P. Examining the clinical impact of rapid multiplex polymerase chain reaction-based diagnostic testing for bloodstream infections in a national cohort of the Veterans Health Administration. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2023, 43, 24–34. [Google Scholar] [CrossRef]
- Ndraha, N.; Lin, H.Y.; Wang, C.Y.; Hsiao, H.I.; Lin, H.J. Rapid detection methods for foodborne pathogens based on nucleic acid amplification: Recent advances, remaining challenges, and possible opportunities. Food Chem. Mol. Sci. 2023, 7, 100183. [Google Scholar] [CrossRef]
- Haider, A.; Ringer, M.; Kotroczó, Z.; Mohácsi-Farkas, C.; Kocsis, T. The current level of MALDI-TOF MS applications in the detection of microorganisms: A short review of benefits and limitations. Microbiol. Res. 2023, 14, 80–90. [Google Scholar] [CrossRef]
- Singhal, N.; Kumar, M.; Kanaujia, P.K.; Virdi, J.S. MALDI-TOF mass spectrometry: An emerging technology for microbial identification and diagnosis. Front. Microbiol. 2015, 6, 791. [Google Scholar] [CrossRef]
- Jelinkova, P.; Mazumdar, A.; Sur, V.P.; Kociova, S.; Dolezelikova, K.; Jimenez, A.M.J.; Koudelkova, Z.; Mishra, P.K.; Smerkova, K.; Heger, Z. Nanoparticle-drug conjugates treating bacterial infections. J. Control. Release 2019, 307, 166–185. [Google Scholar] [CrossRef]
- Wang, L.; Hu, C.; Shao, L. The antimicrobial activity of nanoparticles: Present situation and prospects for the future. Int. J. Nanomed. 2017, 12, 1227–1249. [Google Scholar] [CrossRef]
- Li, C.; Ye, R.; Bouckaert, J.; Zurutuza, A.; Drider, D.; Dumych, T.; Paryzhak, S.; Vovk, V.; Bilyy, R.O.; Melinte, S. Flexible nanoholey patches for antibiotic-free treatments of skin infections. ACS Appl. Mater. Interfaces 2017, 9, 36665–36674. [Google Scholar] [CrossRef] [PubMed]
- Smerkova, K.; Dolezelikova, K.; Bozdechova, L.; Heger, Z.; Zurek, L.; Adam, V. Nanomaterials with active targeting as advanced antimicrobials. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020, 12, e1636. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, N.; Wu, J.; Xu, X.; Kamaly, N.; Farokhzad, O.C. Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv. Drug Deliv. Rev. 2014, 66, 2–25. [Google Scholar] [CrossRef] [PubMed]
- Gordillo Altamirano, F.L.; Barr, J.J. Phage therapy in the postantibiotic era. Clin. Microbiol. Rev. 2019, 32, e00066-18. [Google Scholar] [CrossRef] [PubMed]
- Guzman, J.; Téné, N.; Touchard, A.; Castillo, D.; Belkhelfa, H.; Haddioui-Hbabi, L.; Treilhou, M.; Sauvain, M. Anti-Helicobacter pylori properties of the ant-venom peptide bicarinalin. Toxins 2017, 10, 21. [Google Scholar] [CrossRef]
- López-Igual, R.; Bernal-Bayard, J.; Rodríguez-Patón, A.; Ghigo, J.-M.; Mazel, D. Engineered toxin–intein antimicrobials can selectively target and kill antibiotic-resistant bacteria in mixed populations. Nat. Biotechnol. 2019, 37, 755–760. [Google Scholar] [CrossRef]
- Ram, G.; Ross, H.F.; Novick, R.P.; Rodriguez-Pagan, I.; Jiang, D. Conversion of staphylococcal pathogenicity islands to CRISPR-carrying antibacterial agents that cure infections in mice. Nat. Biotechnol. 2018, 36, 971–976. [Google Scholar] [CrossRef]
- Sabu, C.; Rejo, C.; Kotta, S.; Pramod, K. Bioinspired and biomimetic systems for advanced drug and gene delivery. J. Control. Release 2018, 287, 142–155. [Google Scholar] [CrossRef]
- Mocan, L.; Matea, C.; Tabaran, F.A.; Mosteanu, O.; Pop, T.; Puia, C.; Agoston-Coldea, L.; Gonciar, D.; Kalman, E.; Zaharie, G. Selective in vitro photothermal nano-therapy of MRSA infections mediated by IgG conjugated gold nanoparticles. Sci. Rep. 2016, 6, 39466. [Google Scholar] [CrossRef]
- Knox, K.; Vesk, M.; Work, E. Relation between excreted lipopolysaccharide complexes and surface structures of a lysine-limited culture of Escherichia coli. J. Bacteriol. 1966, 92, 1206–1217. [Google Scholar] [CrossRef]
- Lee, E.Y.; Choi, D.Y.; Kim, D.K.; Kim, J.W.; Park, J.O.; Kim, S.; Kim, S.H.; Desiderio, D.M.; Kim, Y.K.; Kim, K.P. Gram-positive bacteria produce membrane vesicles: Proteomics-based characterization of Staphylococcus aureus-derived membrane vesicles. Proteomics 2009, 9, 5425–5436. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Y.; Bang, J.Y.; Park, G.W.; Choi, D.S.; Kang, J.S.; Kim, H.J.; Park, K.S.; Lee, J.O.; Kim, Y.K.; Kwon, K.H. Global proteomic profiling of native outer membrane vesicles derived from Escherichia coli. Proteomics 2007, 7, 3143–3153. [Google Scholar] [CrossRef]
- Kuehn, M.J.; Kesty, N.C. Bacterial outer membrane vesicles and the host–pathogen interaction. Genes Dev. 2005, 19, 2645–2655. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Fang, R.H.; Thamphiwatana, S.; Luk, B.T.; Li, J.; Angsantikul, P.; Zhang, Q.; Hu, C.-M.J.; Zhang, L. Modulating antibacterial immunity via bacterial membrane-coated nanoparticles. Nano Lett. 2015, 15, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Xu, L.; Yang, B.; Fan, F.; Yang, L. Kill the real with the fake: Eliminate intracellular Staphylococcus aureus using nanoparticle coated with its extracellular vesicle membrane as active-targeting drug carrier. ACS Infect. Dis. 2018, 5, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Umamaheshwari, R.; Jain, N. Receptor mediated targeting of lectin conjugated gliadin nanoparticles in the treatment of Helicobacter pylori. J. Drug Target. 2003, 11, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Angsantikul, P.; Thamphiwatana, S.; Zhang, Q.; Spiekermann, K.; Zhuang, J.; Fang, R.H.; Gao, W.; Obonyo, M.; Zhang, L. Coating nanoparticles with gastric epithelial cell membrane for targeted antibiotic delivery against Helicobacter pylori infection. Adv. Ther. 2018, 1, 1800016. [Google Scholar] [CrossRef]
- Da Silva-Candal, A.; Brown, T.; Krishnan, V.; Lopez-Loureiro, I.; Ávila-Gómez, P.; Pusuluri, A.; Pérez-Díaz, A.; Correa-Paz, C.; Hervella, P.; Castillo, J. Shape effect in active targeting of nanoparticles to inflamed cerebral endothelium under static and flow conditions. J. Control. Release 2019, 309, 94–105. [Google Scholar] [CrossRef]
- Monopoli, M.P.; Åberg, C.; Salvati, A.; Dawson, K.A. Biomolecular coronas provide the biological identity of nanosized materials. Nat. Nanotechnol. 2012, 7, 779–786. [Google Scholar] [CrossRef]
- Siemer, S.; Westmeier, D.; Barz, M.; Eckrich, J.; Wünsch, D.; Seckert, C.; Thyssen, C.; Schilling, O.; Hasenberg, M.; Pang, C. Biomolecule-corona formation confers resistance of bacteria to nanoparticle-induced killing: Implications for the design of improved nanoantibiotics. Biomaterials 2019, 192, 551–559. [Google Scholar] [CrossRef]
- Fernández-Iglesias, N.; Bettmer, J. Complementary mass spectrometric techniques for the quantification of the protein corona: A case study on gold nanoparticles and human serum proteins. Nanoscale 2015, 7, 14324–14331. [Google Scholar] [CrossRef]
- García-Álvarez, R.; Hadjidemetriou, M.; Sánchez-Iglesias, A.; Liz-Marzán, L.M.; Kostarelos, K. In vivo formation of protein corona on gold nanoparticles. The effect of their size and shape. Nanoscale 2018, 10, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Tenzer, S.; Docter, D.; Kuharev, J.; Musyanovych, A.; Fetz, V.; Hecht, R.; Schlenk, F.; Fischer, D.; Kiouptsi, K.; Reinhardt, C.; et al. Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nat. Nanotechnol. 2013, 8, 772–781. [Google Scholar] [CrossRef]
- Digiacomo, L.; Pozzi, D.; Palchetti, S.; Zingoni, A.; Caracciolo, G. Impact of the protein corona on nanomaterial immune response and targeting ability. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020, 12, e1615. [Google Scholar] [CrossRef] [PubMed]
- Subramaniyan, S.B.; Vijayakumar, S.; Megarajan, S.; Kamlekar, R.K.; Anbazhagan, V. Remarkable effect of jacalin in diminishing the protein corona interference in the antibacterial activity of pectin-capped copper sulfide nanoparticles. ACS Omega 2019, 4, 14049–14056. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.-Q.; Shu, G.-F.; Jiang, S.-P.; Xu, X.-L.; Qi, J.; Jin, F.-Y.; Liu, D.; Xiao, Y.-H.; Lu, X.-Y.; Du, Y.-Z. Effective targeted therapy for drug-resistant infection by ICAM-1 antibody-conjugated TPGS modified β-Ga2O3: Cr3+ nanoparticles. Theranostics 2019, 9, 2739. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Carmona, M.; Gun’ko, Y.K.; Vallet-Regí, M. Mesoporous silica materials as drug delivery: “The Nightmare” of bacterial infection. Pharmaceutics 2018, 10, 279. [Google Scholar] [CrossRef]
- Pang, X.; Xiao, Q.; Cheng, Y.; Ren, E.; Lian, L.; Zhang, Y.; Gao, H.; Wang, X.; Leung, W.; Chen, X. Bacteria-responsive nanoliposomes as smart sonotheranostics for multidrug resistant bacterial infections. ACS Nano 2019, 13, 2427–2438. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiu, W.; Gan, S.; Shan, J.; Ren, S.; Yuwen, L.; Weng, L.; Teng, Z.; Wang, L. Antibody-functionalized mos2 nanosheets for targeted photothermal therapy of staphylococcus aureus focal infection. Front. Bioeng. Biotechnol. 2019, 7, 218. [Google Scholar] [CrossRef]
- Santos, R.S.; Figueiredo, C.; Azevedo, N.F.; Braeckmans, K.; De Smedt, S.C. Nanomaterials and molecular transporters to overcome the bacterial envelope barrier: Towards advanced delivery of antibiotics. Adv. Drug Deliv. Rev. 2018, 136, 28–48. [Google Scholar] [CrossRef]
- Capeletti, L.B.; Oliveira, J.F.A.; Loiola, L.M.D.; Galdino, F.E.; Silva Santos, D.E.; Soares, T.A.; Oliveira Freitas, R.; Cardoso, M.B. Biomedical applications: Gram-negative bacteria targeting mediated by carbohydrate–carbohydrate interactions induced by surface-modified nanoparticles. Adv. Funct. Mater. 2019, 29, 1904216. [Google Scholar] [CrossRef]
- Lehman, S.M.; Mearns, G.; Rankin, D.; Cole, R.A.; Smrekar, F.; Branston, S.D.; Morales, S. Design and preclinical development of a phage product for the treatment of antibiotic-resistant Staphylococcus aureus infections. Viruses 2019, 11, 88. [Google Scholar] [CrossRef]
- Nilsson, A.S. Phage therapy—Constraints and possibilities. Upsala J. Med. Sci. 2014, 119, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Skurnik, M.; Pajunen, M.; Kiljunen, S. Biotechnological challenges of phage therapy. Biotechnol. Lett. 2007, 29, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Sulakvelidze, A.; Alavidze, Z.; Morris, J.G., Jr. Bacteriophage therapy. Antimicrob. Agents Chemother. 2001, 45, 649–659. [Google Scholar] [CrossRef]
- Smith, H.W.; Huggins, M. Effectiveness of phages in treating experimental Escherichia coli diarrhoea in calves, piglets and lambs. Microbiology 1983, 129, 2659–2675. [Google Scholar] [CrossRef]
- Smith, H.W.; Huggins, M.B.; Shaw, K.M. The control of experimental Escherichia coli diarrhoea in calves by means of bacteriophages. Microbiology 1987, 133, 1111–1126. [Google Scholar] [CrossRef]
- Smith, H.W.; Huggins, M.B.; Shaw, K.M. Factors influencing the survival and multiplication of bacteriophages in calves and in their environment. Microbiology 1987, 133, 1127–1135. [Google Scholar] [CrossRef]
- Soothill, J.; Lawrence, J.; Ayliffe, G. The efficacy of phages in the prevention of the destruction of pig skin in vitro by Pseudomonas aeruginosa. Med. Sci. Res. 1988, 16, 1287–1288. [Google Scholar]
- Bogovazova, G.; Voroshilova, N.; Bondarenko, V. The efficacy of Klebsiella pneumoniae bacteriophage in the therapy of experimental Klebsiella infection. Zh. Mikrobiol. Epidemiol. Immunobiol. 1991, 4, 5–8. [Google Scholar]
- Loc-Carrillo, C.; Abedon, S.T. Pros and cons of phage therapy. Bacteriophage 2011, 1, 111–114. [Google Scholar] [CrossRef]
- Krylov, V. Phagotherapy in terms of bacteriophage genetics: Hopes, perspectives, safety, limitations. Genetika 2001, 37, 869–887. [Google Scholar] [PubMed]
- Abedon, S.T.; Thomas-Abedon, C. Phage therapy pharmacology. Curr. Pharm. Biotechnol. 2010, 11, 28–47. [Google Scholar] [CrossRef]
- Cromwell, G.L. Why and how antibiotics are used in swine production. Anim. Biotechnol. 2002, 13, 7–27. [Google Scholar] [CrossRef] [PubMed]
- Morrow, C.; Fehler, F. Marek’s disease: A worldwide problem. In Marek’s Disease; Davison, F., Nair, V., Eds.; Academic Press: Cambridge, MA, USA, 2004; pp. 49–61. [Google Scholar]
- Kennedy, D.A.; Read, A.F. Why the evolution of vaccine resistance is less of a concern than the evolution of drug resistance. Proc. Natl. Acad. Sci. USA 2018, 115, 12878–12886. [Google Scholar] [CrossRef]
- Plotkin, S.A. Correlates of protection induced by vaccination. Clin. Vaccine Immunol. 2010, 17, 1055–1065. [Google Scholar] [CrossRef]
- Ghaffar, F.; Friedland, I.R.; McCracken, G.H., Jr. Dynamics of nasopharyngeal colonization by Streptococcus pneumoniae. Pediatr. Infect. Dis. J. 1999, 18, 638–646. [Google Scholar] [CrossRef]
- Lipsitch, M. Bacterial vaccines and serotype replacement: Lessons from Haemophilus influenzae and prospects for Streptococcus pneumoniae. Emerg. Infect. Dis. 1999, 5, 336. [Google Scholar] [CrossRef] [PubMed]
- Obaro, S.K.; Adegbola, R.; Banya, W.; Greenwood, B. Carriage of pneumococci after pneumococcal vaccination. Lancet 1996, 348, 271–272. [Google Scholar] [CrossRef]
- Moore, M.R.; Link-Gelles, R.; Schaffner, W.; Lynfield, R.; Lexau, C.; Bennett, N.M.; Petit, S.; Zansky, S.M.; Harrison, L.H.; Reingold, A. Effect of use of 13-valent pneumococcal conjugate vaccine in children on invasive pneumococcal disease in children and adults in the USA: Analysis of multisite, population-based surveillance. Lancet Infect. Dis. 2015, 15, 301–309. [Google Scholar] [CrossRef]
- Klugman, K.P.; Black, S. Impact of existing vaccines in reducing antibiotic resistance: Primary and secondary effects. Proc. Natl. Acad. Sci. USA 2018, 115, 12896–12901. [Google Scholar] [CrossRef] [PubMed]
- US National Library of Medicine. ClinicalTrials.gov. 2013. Available online: https://clinicaltrials.gov/ct2/show/NCT01887912 (accessed on 1 September 2024).
- US National Library of Medicine. ClinicalTrials.gov. 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03090191?term=vaccine&cond=clostridium+difficile&rank=7 (accessed on 1 September 2024).
- Péchiné, S.; Bruxelle, J.F.; Janoir, C.; Collignon, A. Targeting Clostridium difficile surface components to develop immunotherapeutic strategies against Clostridium difficile infection. Front. Microbiol. 2018, 9, 1009. [Google Scholar] [CrossRef]
- Vincent, L.R.; Jerse, A.E. Biological feasibility and importance of a gonorrhea vaccine for global public health. Vaccine 2019, 37, 7419–7426. [Google Scholar] [CrossRef] [PubMed]
- Priebe, G.P.; Goldberg, J.B. Vaccines for Pseudomonas aeruginosa: A long and winding road. Expert Rev. Vaccines 2014, 13, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Döring, G.; Meisner, C.; Stern, M.; Flagella Vaccine Trial Study Group. A double-blind randomized placebo-controlled phase III study of a Pseudomonas aeruginosa flagella vaccine in cystic fibrosis patients. Proc. Natl. Acad. Sci. USA 2007, 104, 11020–11025. [Google Scholar] [CrossRef] [PubMed]
- Rello, J.; Krenn, C.-G.; Locker, G.; Pilger, E.; Madl, C.; Balica, L.; Dugernier, T.; Laterre, P.-F.; Spapen, H.; Depuydt, P. A randomized placebo-controlled phase II study of a Pseudomonas vaccine in ventilated ICU patients. Crit. Care 2017, 21, 22. [Google Scholar] [CrossRef]
- Choi, M.; Tennant, S.M.; Simon, R.; Cross, A.S. Progress towards the development of Klebsiella vaccines. Expert Rev. Vaccines 2019, 18, 681–691. [Google Scholar] [CrossRef]
- Feldman, M.F.; Mayer Bridwell, A.E.; Scott, N.E.; Vinogradov, E.; McKee, S.R.; Chavez, S.M.; Twentyman, J.; Stallings, C.L.; Rosen, D.A.; Harding, C.M. A promising bioconjugate vaccine against hypervirulent Klebsiella pneumoniae. Proc. Natl. Acad. Sci. USA 2019, 116, 18655–18663. [Google Scholar] [CrossRef]
- Follador, R.; Heinz, E.; Wyres, K.L.; Ellington, M.J.; Kowarik, M.; Holt, K.E.; Thomson, N.R. The diversity of Klebsiella pneumoniae surface polysaccharides. Microb. Genom. 2016, 2, e000073. [Google Scholar] [CrossRef]
- MacLennan, C.A.; Martin, L.B.; Micoli, F. Vaccines against invasive Salmonella disease: Current status and future directions. Hum. Vaccin. Immunother. 2014, 10, 1478–1493. [Google Scholar] [CrossRef]
- Lin, F.Y.C.; Ho, V.A.; Khiem, H.B.; Trach, D.D.; Bay, P.V.; Thanh, T.C.; Kossaczka, Z.; Bryla, D.A.; Shiloach, J.; Robbins, J.B. The efficacy of a Salmonella typhi Vi conjugate vaccine in two-to-five-year-old children. N. Engl. J. Med. 2001, 344, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Puzari, M.; Sharma, M.; Chetia, P. Emergence of antibiotic resistant Shigella species: A matter of concern. J. Infect. Public Health 2018, 11, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.; Wierzba, T.; Walker, R.I. Status of vaccine research and development for Shigella. Vaccine 2016, 34, 2887–2894. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Becerra, F.J.; Kissmann, J.M.; Diaz-McNair, J.; Choudhari, S.P.; Quick, A.M.; Mellado-Sanchez, G.; Clements, J.D.; Pasetti, M.F.; Picking, W.L. Broadly protective Shigella vaccine based on type III secretion apparatus proteins. Infect. Immun. 2012, 80, 1222–1231. [Google Scholar] [CrossRef]
- Frost, I.; Sati, H.; Garcia-Vello, P.; Hasso-Agopsowicz, M.; Lienhardt, C.; Gigante, V.; Beyer, P. The role of bacterial vaccines in the fight against antimicrobial resistance: An analysis of the preclinical and clinical development pipeline. Lancet Microbe 2023, 4, e113–e125. [Google Scholar] [CrossRef]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Robert-Guroff, M.; Brown, M.; Gallo, R.C. HTLV-III-neutralizing antibodies in patients with AIDS and AIDS-related complex. Nature 1985, 316, 72–74. [Google Scholar] [CrossRef]
- Hey, A. History and practice: Antibodies in infectious diseases. Microbiol. Spectr. 2015, 3, 1–21. [Google Scholar] [CrossRef]
- Glanville, J.; Zhai, W.; Berka, J.; Telman, D.; Huerta, G.; Mehta, G.R.; Ni, I.; Mei, L.; Sundar, P.D.; Day, G.M. Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc. Natl. Acad. Sci. USA 2009, 106, 20216–20221. [Google Scholar] [CrossRef]
- Storek, K.M.; Auerbach, M.R.; Shi, H.; Garcia, N.K.; Sun, D.; Nickerson, N.N.; Vij, R.; Lin, Z.; Chiang, N.; Schneider, K. Monoclonal antibody targeting the β-barrel assembly machine of Escherichia coli is bactericidal. Proc. Natl. Acad. Sci. USA 2018, 115, 3692–3697. [Google Scholar] [CrossRef]
- Novotny, L.A.; Jurcisek, J.A.; Goodman, S.D.; Bakaletz, L.O. Monoclonal antibodies against DNA-binding tips of DNABII proteins disrupt biofilms in vitro and induce bacterial clearance in vivo. eBioMedicine 2016, 10, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Aye, R.; Weldearegay, Y.B.; Lutta, H.O.; Chuma, F.; Pich, A.; Jores, J.; Meens, J.; Naessens, J. Identification of targets of monoclonal antibodies that inhibit adhesion and growth in Mycoplasma mycoides subspecies mycoides. Vet. Immunol. Immunopathol. 2018, 204, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Moreira, G.M.; Gronow, S.; Dübel, S.; Mendonça, M.; Moreira, Â.N.; Conceição, F.R.; Hust, M. Phage display-derived monoclonal antibodies against internalins A and B allow specific detection of Listeria monocytogenes. Front. Public Health 2022, 10, 712657. [Google Scholar] [CrossRef]
- Nielsen, T.B.; Pantapalangkoor, P.; Luna, B.M.; Bruhn, K.W.; Yan, J.; Dekitani, K.; Hsieh, S.; Yeshoua, B.; Pascual, B.; Vinogradov, E. Monoclonal antibody protects against Acinetobacter baumannii infection by enhancing bacterial clearance and evading sepsis. J. Infect. Dis. 2017, 216, 489–501. [Google Scholar] [CrossRef]
- Visan, L.; Rouleau, N.; Proust, E.; Peyrot, L.; Donadieu, A.; Ochs, M. Antibodies to PcpA and PhtD protect mice against Streptococcus pneumoniae by a macrophage-and complement-dependent mechanism. Hum. Vaccines Immunother. 2018, 14, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Nagy, C.F.; Leach, T.S.; Hoffman, J.H.; Czech, A.; Carpenter, S.E.; Guttendorf, R. Pharmacokinetics and tolerability of obiltoxaximab: A report of 5 healthy volunteer studies. Clin. Ther. 2016, 38, 2083–2097.e7. [Google Scholar] [CrossRef]
- Henning, L.N.; Carpenter, S.; Stark, G.V.; Serbina, N.V. Development of protective immunity in New Zealand white rabbits challenged with Bacillus anthracis spores and treated with antibiotics and obiltoxaximab, a monoclonal antibody against protective antigen. Antimicrob. Agents Chemother. 2018, 62, e01590-17. [Google Scholar] [CrossRef]
- Wilcox, M.H.; Gerding, D.N.; Poxton, I.R.; Kelly, C.; Nathan, R.; Birch, T.; Cornely, O.A.; Rahav, G.; Bouza, E.; Lee, C. Bezlotoxumab for prevention of recurrent Clostridium difficile infection. N. Engl. J. Med. 2017, 376, 305–317. [Google Scholar] [CrossRef]
- Zurawski, D.V.; McLendon, M.K. Monoclonal antibodies as an antibacterial approach against bacterial pathogens. Antibiotics 2020, 9, 155. [Google Scholar] [CrossRef]
- Sause, W.E.; Buckley, P.T.; Strohl, W.R.; Lynch, A.S.; Torres, V.J. Antibody-based biologics and their promise to combat Staphylococcus aureus infections. Trends Pharmacol. Sci. 2016, 37, 231–241. [Google Scholar] [CrossRef]
- Palacios Araya, D.; Palmer, K.L.; Duerkop, B.A. CRISPR-based antimicrobials to obstruct antibiotic-resistant and pathogenic bacteria. PLoS Pathog. 2021, 17, e1009672. [Google Scholar] [CrossRef]
- Wu, Y.; Battalapalli, D.; Hakeem, M.J.; Selamneni, V.; Zhang, P.; Draz, M.S.; Ruan, Z. Engineered CRISPR-Cas systems for the detection and control of antibiotic-resistant infections. J. Nanobiotechnol. 2021, 19, 401. [Google Scholar] [CrossRef] [PubMed]
- Hille, F.; Richter, H.; Wong, S.P.; Bratovič, M.; Ressel, S.; Charpentier, E. The biology of CRISPR-Cas: Backward and forward. Cell 2018, 172, 1239–1259. [Google Scholar] [CrossRef]
- Kiga, K.; Tan, X.-E.; Ibarra-Chávez, R.; Watanabe, S.; Aiba, Y.; Sato’o, Y.; Li, F.-Y.; Sasahara, T.; Cui, B.; Kawauchi, M. Development of CRISPR-Cas13a-based antimicrobials capable of sequence-specific killing of target bacteria. Nat. Commun. 2020, 11, 2934. [Google Scholar] [CrossRef]
- Greene, A.C. CRISPR-based antibacterials: Transforming bacterial defense into offense. Trends Biotechnol. 2018, 36, 127–130. [Google Scholar] [CrossRef]
- Gründling, A.; Salipante, S.J. Oligonucleotide design and construction of a gene-targeting CRISPR–Cas9 plasmid in Escherichia coli for generating a gene-deletion strain in Staphylococcus aureus. Cold Spring Harb. Protoc. 2024, 2024, pdb-rot107920. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.; Mukhopadhyay, P. Antimicrobial resistance (AMR) management using CRISPR-Cas based genome editing. Gene Genome Ed. 2024, 7, 100031. [Google Scholar] [CrossRef]
- Mota, D.S.; Marques, J.M.; Guimarães, J.M.; Mariúba, L.A.M. CRISPR/Cas Class 2 systems and their applications in biotechnological processes. Genet Mol. Res. 2020, 19, gmr18478. [Google Scholar]
- Gomaa, A.A.; Klumpe, H.E.; Luo, M.L.; Selle, K.; Barrangou, R.; Beisel, C.L. Programmable removal of bacterial strains by use of genome-targeting CRISPR-Cas systems. mBio 2014, 5, e00928-13. [Google Scholar] [CrossRef]
- Hullahalli, K.; Rodrigues, M.; Nguyen, U.; Palmer, K. An attenuated CRISPR-Cas system in Enterococcus faecalis permits DNA acquisition. mBio 2018, 9, e00414-8. [Google Scholar] [CrossRef]
- Basavegowda, N.; Baek, K.-H. Multimetallic nanoparticles as alternative antimicrobial agents: Challenges and perspectives. Molecules 2021, 26, 912. [Google Scholar] [CrossRef] [PubMed]
- Oechslin, F. Resistance development to bacteriophages occurring during bacteriophage therapy. Viruses 2018, 10, 351. [Google Scholar] [CrossRef] [PubMed]
- Pursey, E.; Sünderhauf, D.; Gaze, W.H.; Westra, E.R.; van Houte, S. CRISPR-Cas antimicrobials: Challenges and future prospects. PLoS Pathog. 2018, 14, e1006990. [Google Scholar] [CrossRef] [PubMed]
- Gautam, V.; Maurya, P.K. Emerging trends in vaccine delivery systems. In System Vaccinology: The History, the Translational Challenges and the Future; Academic Press: Cambridge, MA, USA, 2022; pp. 361–386. [Google Scholar]
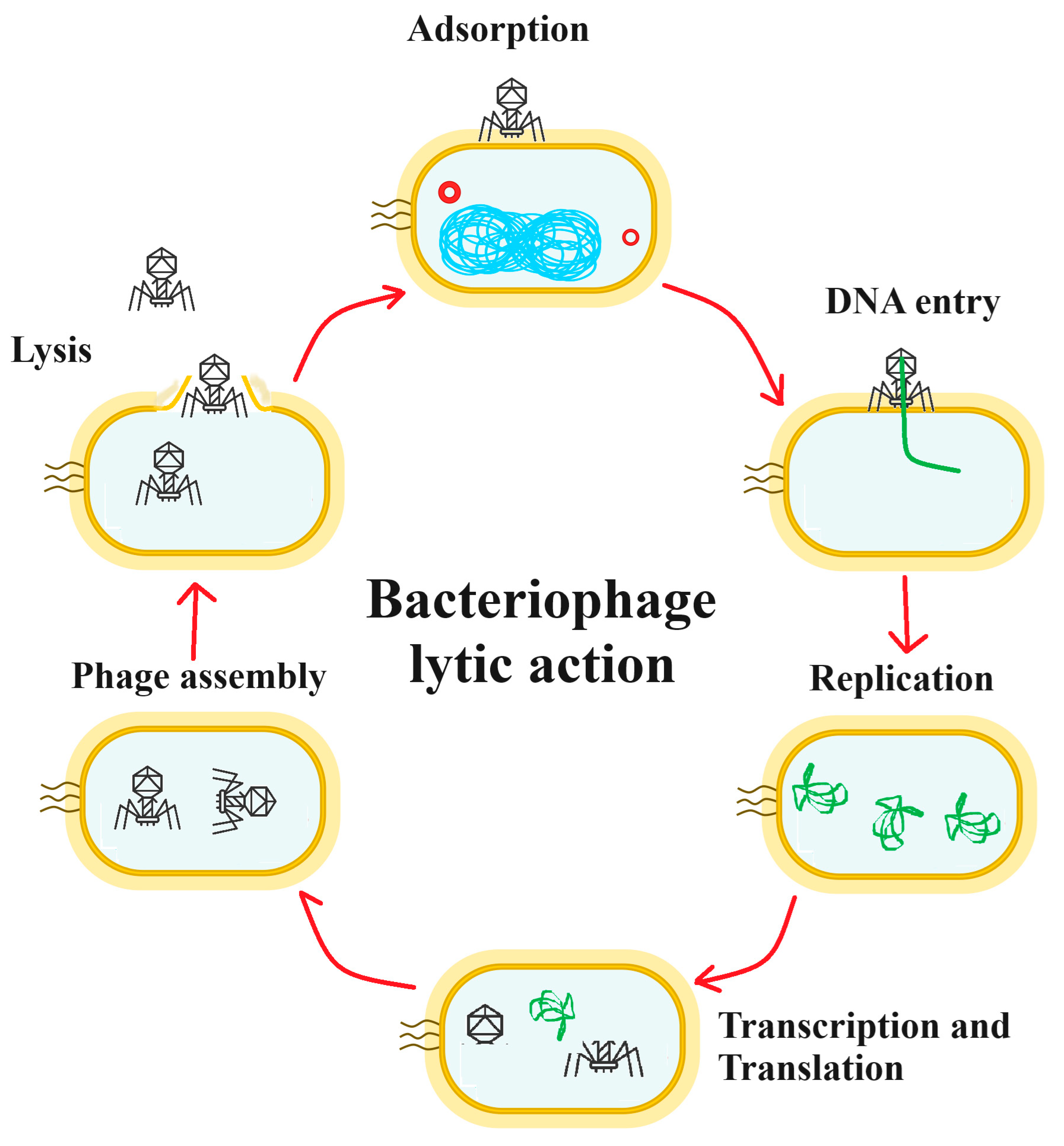
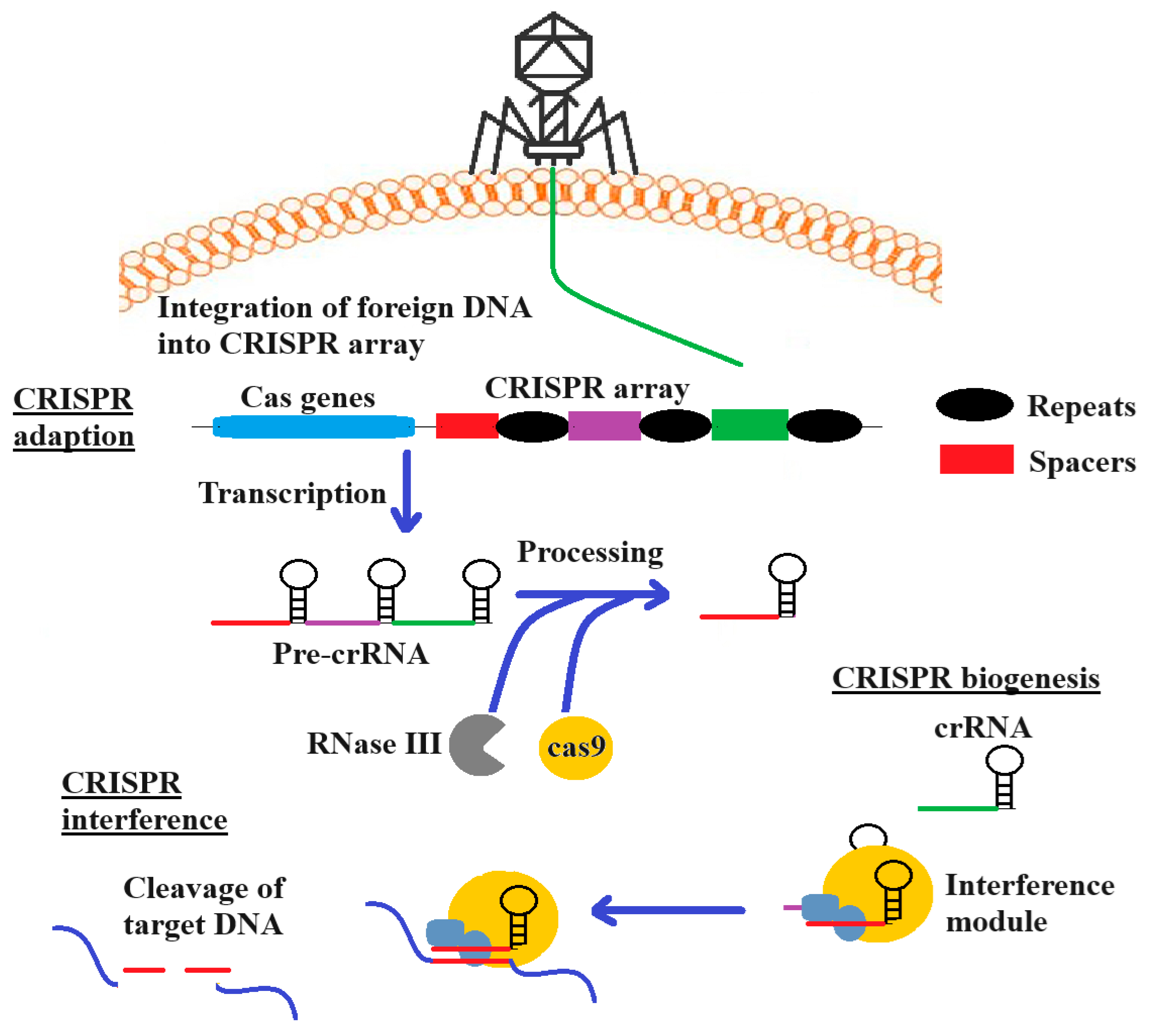
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).