1. Introduction
Antimicrobial resistance led to over 4.95 million deaths in 2019, with projections suggesting it could result in approximately 10 million deaths annually by 2050 [
1]. Among the myriad bacterial pathogens posing a threat to public health,
Mycobacterium tuberculosis (Mtb) stands out as the causative agent of tuberculosis (TB). According to the World Health Organization (WHO), TB accounted for over 1.6 million deaths in 2021, affecting more than 10.6 million individuals, including over 187,000 HIV-positive individuals [
2].
Typically, TB is treated with a regimen comprising isoniazid, rifampicin, pyrazinamide, and ethambutol for the first two months, which constitutes the intensive treatment phase, followed by isoniazid and rifampicin for an additional four months, known as the continuation phase. However, the emergence of resistance, particularly to isoniazid and rifampicin, has necessitated alternative treatment approaches depending on the level of resistance [
3]. In 2019, approximately 14% of Mtb strains exhibited resistance to isoniazid, and an estimated 4.6% were classified as multi-drug resistant (MDR) TB strains, resistant to both isoniazid and rifampicin. Various second-line treatments such as fluoroquinolones or aminoglycosides are currently employed to combat MDR TB, but antimicrobial resistance poses a formidable challenge to TB control efforts. Extensively drug-resistant TB (XDR TB), defined as TB resistant to isoniazid, rifampicin, at least one fluoroquinolone, and one aminoglycoside, further exacerbates the situation [
3,
4].
Several targets are being explored for anti-tuberculosis therapy to address these challenges. InhA, the enoyl-ACP reductase involved in mycolic acid synthesis, is targeted by isoniazid and ethionamide. Inhibiting InhA disrupts the synthesis of mycolic acids, which are essential components of the mycobacterial cell wall [
5,
6]. RNA polymerase, targeted by rifampicin, inhibits bacterial RNA synthesis by binding to the β-subunit of RNA polymerase, preventing the transcription of DNA to RNA [
7]. ATP synthase, targeted by bedaquiline, inhibits the mycobacterial ATP synthase, leading to energy depletion and bacterial death [
8]. Additionally, topoisomerase II (DNA gyrase) is targeted by fluoroquinolones, such as levofloxacin and moxifloxacin, which inhibit the supercoiling activity of DNA gyrase that is essential for DNA replication and transcription [
9].
Moreover, the cost and adverse effects associated with conventional drugs compromise patient quality of life and impede treatment adherence [
2]. Given these challenges, urgent action is warranted to develop novel therapeutic agents. This review aims to elucidate the biosynthesis of the mycobacterial arabinogalactan cell wall and its inhibition by targeting DprE1 with small molecules. It will provide an overview of known DprE1 inhibitors and offer insights into key considerations for the development of novel therapeutics. By targeting DprE1, a critical enzyme in the biosynthesis of the mycobacterial cell wall, new treatments could potentially overcome existing drug resistance and improve patient outcomes.
2. Mycobacterial Cell Wall Composition
The bacterial cell wall is a complex structure that is essential for bacterial integrity and survival [
5]. It protects bacteria from various stresses, maintains homeostasis, and is responsible for different mechanisms of resistance to antibiotics, such as efflux and membrane impermeability. Moreover, the bacterial cell wall is the first point of contact between the bacterium and a potential drug. Therefore, the bacterial cell wall is a prime target for antibiotics [
10,
11,
12,
13,
14,
15].
While most bacteria can be classified as either “Gram-positive” or “Gram-negative”, a subset known as acid-fast bacilli does not discolor under acidic conditions or the action of alcohol. Among these acid-fast bacilli are members of the
Mycobacterium family. Prominent members of the
Mycobacterium genus include
Mycobacterium leprae (the causative agent of leprosy),
Mycobacterium avium,
Mycobacterium bovis, and Mtb, which are responsible for tuberculosis [
16].
The mycobacterial cell wall exhibits distinct characteristics that set it apart from other bacterial cell walls, including, notably, its extreme lipophilicity and complexity [
16,
17,
18,
19]. Structurally, it consists of the following:
A cytoplasmic phospholipidic membrane linked to the cytoplasm and the periplasm via phosphatidylinositol mannosides (PIMs), lipomannan (LM), and lipoarabinomannan (LAM). This membrane plays a pivotal role in maintaining inner membrane integrity, regulating cell division, septation, and cell wall permeability;
LM and LAM traverse through the cytoplasm and the peptidoglycan layer, which is rich in proteins, lipids, and glycolipids. Peptidoglycan is linked to arabinogalactan (AG) by a rhamnosyl-acetylglucoseamine-1-phosphate linker. AG, mainly composed of galactofuranose and D-arabinofuranose, acts as a transporter involved in exporting cell wall and capsular polysaccharides;
AG and LAM may be linked to various coenzymes or small molecules involved in metabolic pathways, such as galactosamine, methyl-thio-xylose, or succinate;
Mycolic acids, the first components of the outer membrane, are α-alkyl and β-hydroxy fatty acids (C60-90) that play a crucial role in cell wall fluidity and permeability. They are integral to the PG-AG–mycolyl complex, which is essential for cell wall integrity and rigidity;
Various lipids (such as sulfolipids and phenolic glycolipids), trehalose derivatives (polyacetyltrehalose, diacetyltrehalose, trehalose dimycolates, and trehaloses monomycolates), and mycolic acid derivatives (phthiocerol dimycocerosates) are present, each contributing to the structure and permeability of the cell wall;
PIMs, LM, and LAM are also found on the outer membrane, constituents of the mycobacterial capsule primarily composed of D-glucan, D-Mannan, Arabino-D-mannan, and various proteins. This lipophilic capsule forms the final barrier between Mycobacterium and the extracellular space.
The intricate composition of the mycobacterial cell wall underscores its importance in bacterial physiology and pathogenesis. Understanding these components’ role is essential, especially when considering the development of novel therapeutic agents to combat TB and other mycobacterial infections. Previous studies have highlighted the role of these complex structures in mycobacterial resistance to conventional antibiotics, emphasizing the need for targeted treatments.
2.1. Arabinan Synthesis
Arabinogalactan consists of two components. The first part is a linear chain of β-D-galactofuranose, comprising approximately 30 residues linked to each other at positions 5 and 6 of the cycle. The second part consists of three branched arabinofuranose units, each composed of around 30 residues and linked to the galactan chain [
17,
18,
20]. As previously noted, arabinogalactan biosynthesis (
Figure 1) plays a crucial role in the structure of mycobacteria cell walls. Inhibiting its synthesis has been demonstrated to be fatal for
Mycobacterium, particularly affecting the survival and pathogenicity of Mtb within a living host [
17,
18]. The role of arabinogalactan is also essential in controlled settings outside a living organism [
19].
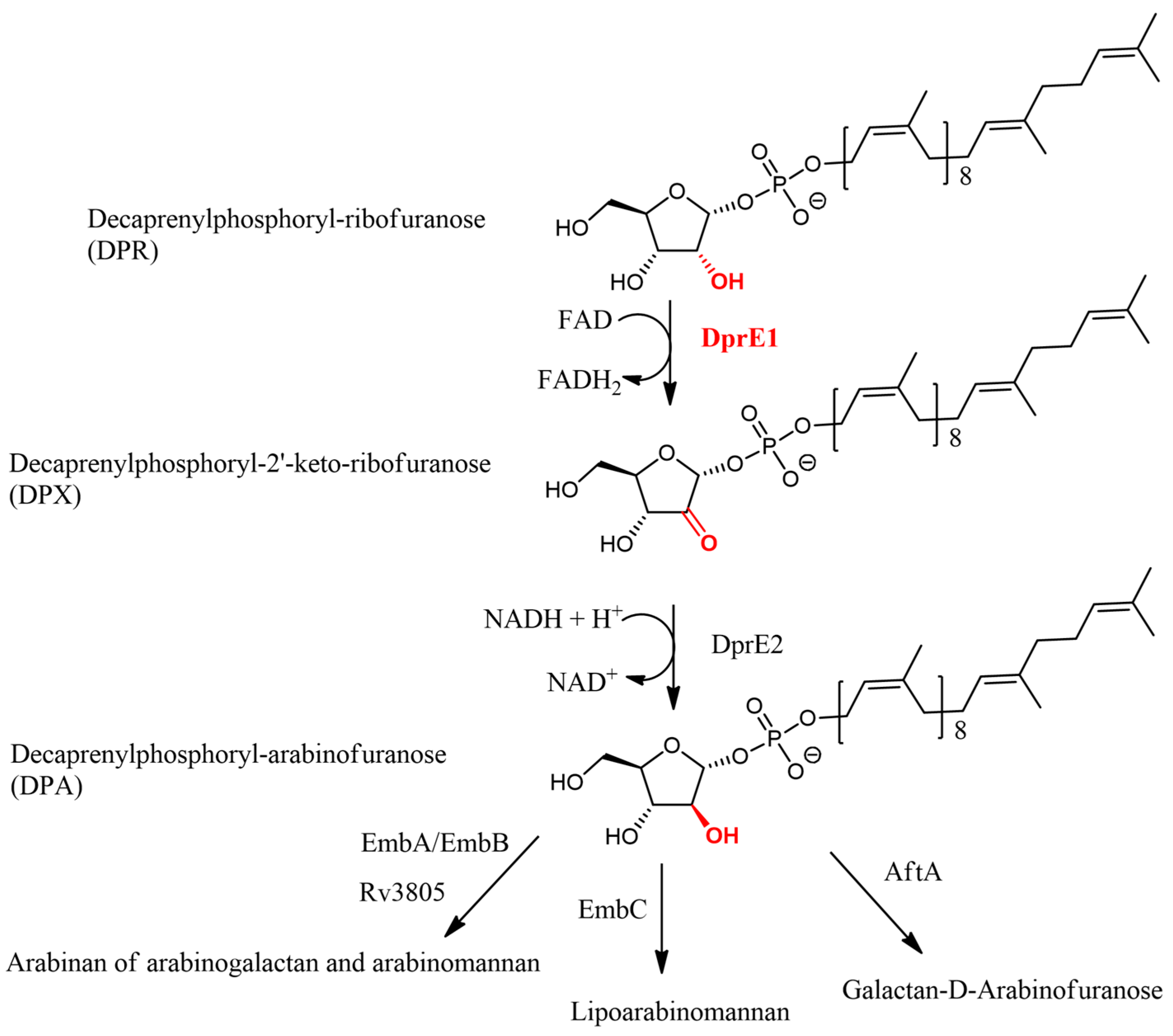
The approved drug ethambutol operates by inhibiting arabinogalactan synthesis, specifically targeting arabinan synthesis by inhibiting arabinosyltransferases A, B, and C (EmbA, EmbB, and EmbC). These enzymes are responsible for synthesizing arabinan from arabinogalactan and lipoarabinomannan (
Figure 1), starting from decaprenyl-phospho-D-arabinofuranose [
18,
21]. Another drug candidate, Macozinone, targets the biosynthesis of decaprenyl-phospho-D-arabinofuranose by inhibiting decaprenylphosphoryl-β-D-ribose-oxydase (DprE1), one of the two subunits of the decaprenylphosphoryl-β-D-ribose-2′-epimerase complex, along with the decaprenylphosphoryl-2-keto-β-D-erythropentose reductase (DprE2).
Arabinan synthesis has thus proven to be a viable target for the development of new tuberculosis treatments. The metabolic pathway of arabinan begins with D-ribose-5-phosphate, which is converted into 5-phosphoribose-1-pyrophosphate (pRpp) by ribose phosphatase pyrophosphokinase (PrsA) in an ATP-dependent reaction. Decaprenyl-phosphate phosphoribosyltransferase then replaces pRpp’s pyrophosphate with a decaprenyl phosphate substituent to form decaprenyl-P-ribofuranose-5-phosphate (DPPR). DPPR is subsequently dephosphorylated to form decaprenyl-P-ribofuranose (DPR) by a phospholipid phosphatase encoded by Rv3807c. DprE1 then oxidizes DPR to decaprenyl-P-2′keto-ribofuranose (DPX) using the FADH2/FAD coenzyme. DPX is then reduced to decaprenyl-P-arabinofuranose (DPA) by decaprenylphosphoryl-2-keto-β-D-erythro-pentose reductase (DprE2) with the NADH/NAD+ coenzyme. DPA is ultimately translocated to the periplasm via an unknown mechanism and processed by EmbA, EmbB, EmbC, or Galactan 5-O-arabinofuranosyltransferase (AftA) to produce arabinan from arabinogalactan [
22,
23,
24,
25,
26].
Future research should focus on elucidating the unknown mechanisms involved in DPA translocation and the precise roles of various arabinosyltransferases in arabinan synthesis. Additionally, exploring the development of inhibitors targeting other enzymes within this pathway could provide novel therapeutic options to address TB and other mycobacterial infections. Such advancements are crucial for overcoming the challenges posed by drug-resistant TB strains and improving treatment outcomes.
2.2. DprE1/DprE2 Complex
As previously described, the DprE1/DprE2 complex plays a pivotal role in the biosynthesis of DPA from DPR in a two-step pathway. In the initial step, DPR interacts with DprE1 bound to the coenzyme FAD (Flavin adenine dinucleotide), which oxidizes DPR to produce DPX and FADH2. Subsequently, DPX is transferred to DprE2 along with its coenzyme NADH, where it undergoes reduction to form DPA and NAD+ [
22,
23,
24,
25,
26].
To date, there are no solved crystallographic structures available for DprE2 or the DprE1/DprE2 complex. However, several crystallographic structures of DprE1 have been elucidated, co-crystallized with its coenzyme FAD, and occasionally with an inhibitor (PDB code: 4FEH co-crystallized with FAD, 4NRC co-crystallized with FAD and Macozinone). Regarding the structure of DprE2 and the DprE1/DprE2 complex, a model has been constructed based on the amino acid sequence of DprE2, employing techniques such as homology modeling, protein threading, molecular docking, and dynamic studies. Considering these insights, the DprE1/DprE2 complex appears to be a bipartite complex localized in the
Mycobacterium periplasm. Transport between DprE1 and DprE2 is speculated to occur via substrate channeling [
27].
3. DprE1 in Mtb
3.1. A Promising Target
DprE1, comprising 461 amino acid residues [
27], in complex with the FAD/FADH2 coenzyme, is currently under investigation with four inhibitors in clinical trials (Macozinone, BTZ-043, TBA-7371, and OPC-16732,
Figure 2). DprE1 stands out as one of the prime drug targets in tuberculosis (TB) for several reasons [
28,
29,
30,
31,
32,
33]:
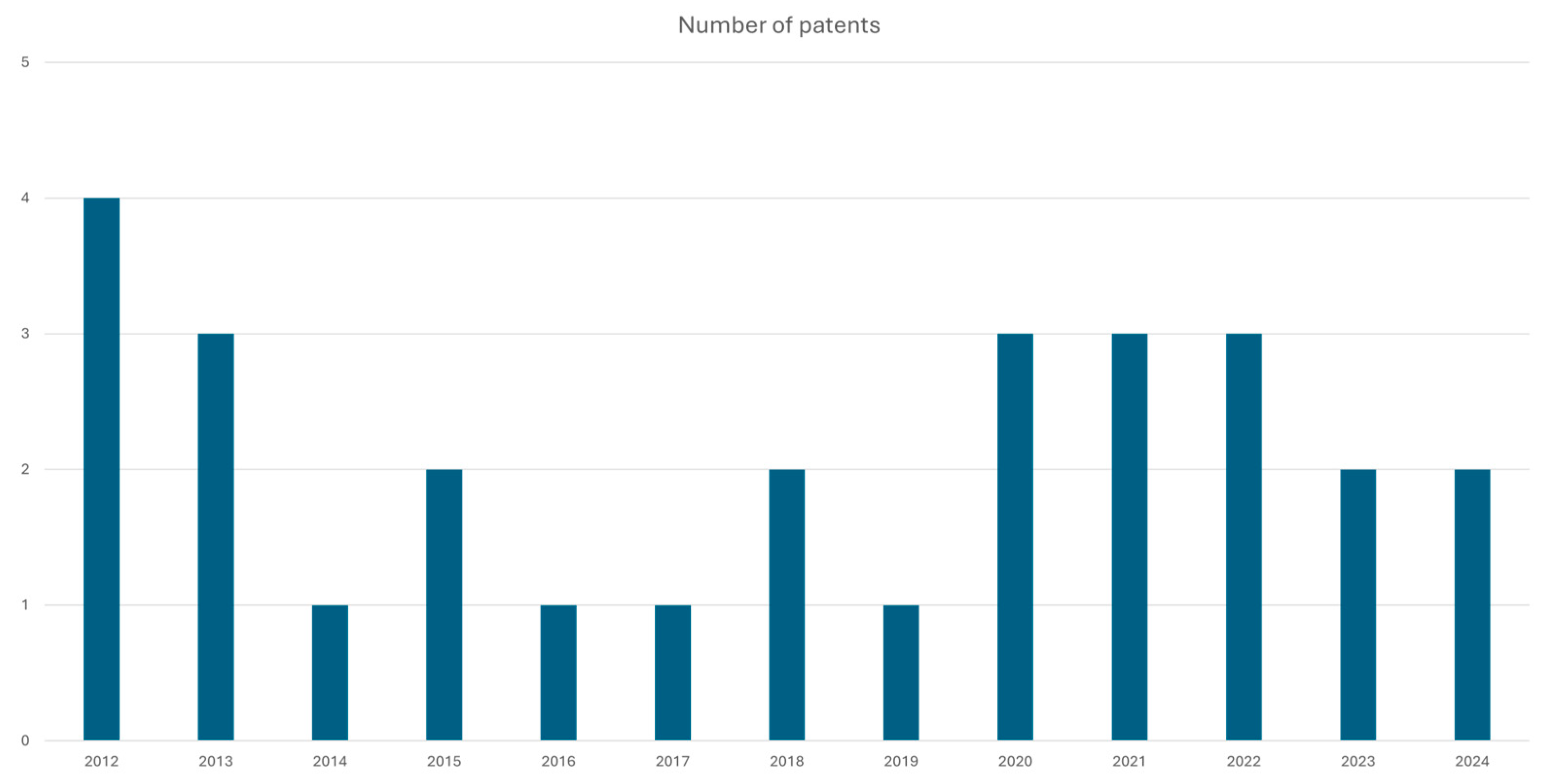
DprE1 is recognized as an established putative target for TB treatment, as evidenced by various patents, patent applications [
34,
35,
36,
37,
38,
39,
40,
41,
42,
43,
44,
45,
46,
47,
48,
49,
50,
51,
52,
53,
54,
55,
56,
57,
58,
59,
60,
61] as illustrated in
Figure 3, and ongoing clinical trials. Inhibiting DprE1 could be critical for combating Mtb;
The absence of DprE1 in the human body mitigates the risk of human toxicities arising from target similarity, enhancing the safety profile of potential inhibitors;
As a novel TB target, DprE1 holds promise in addressing current challenges posed by multi-drug-resistant (MDR) and extensively drug-resistant (XDR) TB strains;
DprE1’s localization in the Mycobacterium periplasm facilitates accessibility compared to various cytoplasmic targets, potentially improving the efficacy of inhibitors.
These factors underscore DprE1’s versatility and significance in TB treatment, warranting further research and development.
3.2. DprE1 Covalent Inhibitors
Among the inhibitors of DprE1, some form covalent bonds with Cysteine 387 in Mtb and Cys 394 in
Mycobacterium smegmatis. This covalent linkage typically occurs following the reduction in a nitro group to a nitroso group [
62]. In the case of a covalent drug with a nitro substituent, DprE1 initially recognizes the nitro group, which is then reduced to a nitroso group by the coenzyme FADH2/FAD. The ligand is subsequently released and binds to its binding pocket, forming a covalent bond with Cys387 of DprE1. This covalent bond can then be cleaved, and the nitroso group can be further reduced to a hydroxylamino group, which can non-covalently bind to the binding pocket [
63,
64,
65,
66].
While most known covalent ligands feature a nitro group, Liu et al. [
66] demonstrated the possibility of using different electrophilic warheads such as cyano, methylester, or primary amides. These findings open new avenues for designing effective DprE1 inhibitors that can overcome resistance mechanisms and enhance TB treatment.
The current understanding of the DprE1/DprE2 complex and its role in mycobacterial cell wall biosynthesis highlights its potential as a drug target. Despite the lack of crystallographic structures for DprE2 and the DprE1/DprE2 complex, advancements in modeling and structural predictions have provided significant insights. The selective targeting of DprE1, combined with the development of covalent inhibitors, represents a promising strategy against MDR and XDR TB strains. Future research should focus on elucidating the detailed mechanisms of substrate channeling between DprE1 and DprE2, as well as exploring new inhibitory compounds with diverse electrophilic warheads. This comprehensive approach could pave the way for novel therapeutics that are more effective and less prone to resistance.
3.2.1. Nitrobenzothiazinones, 2-Nitrothiochromen-ones, Nitrochromen-ones, Nitrobenzooxazinones, and Nitroquinolinones
The first family of known covalent DprE1 inhibitors is Nitrobenzothiazinone and its derivatives. The two most representative compounds in this series are Macozinone and BTZ-043 (both in Phase 2 clinical trials). The success of these compounds in preclinical and early clinical phases has sparked a wave of studies on Nitrobenzothiazinone analogs. BTZ-043 was the first benzothiazine to enter clinical trials as a DprE1 inhibitor. It exhibited a MIC of 2.3 nM on Mtb H37Rv with a toxicity of 11.5 μM (IC
50) on HepG2. Macozinone, also known as PBTZ-169, was obtained after the optimization of BTZ-043 and showed superior in vitro results with a MIC of 0.65 nM and a toxicity of 127 μM (IC
50 on HepG2), as well as better in vivo activities (Log
10 CFU/lung reduction of about 2.4 in lung for PBTZ-169 and of 1.9 for BTZ-043) [
67,
68]. After these two compounds entered clinical trials, numerous analogs have been developed in the pursuit of finding new active compounds.
Various studies have explored modifications and analogs of these compounds. For instance, attempts to add sulfone to the piperazine ring of Macozinone led to the identification of promising compounds [
69,
70]. In another study, compounds combining the structures of BTZ043 and Macozinone, replacing piperazine substituent with a spyrocycle, demonstrated low toxicity and potent antimycobacterial activity [
71,
72]. Additionally, studies have investigated analogs [
70,
73] with a cyclohexyl O-methyl oxime substituent and quinoline substituted derivatives [
74], which exhibited favorable MICs against Mtb H37Rv strains and low cytotoxicity on Vero cells.
Further efforts have explored the impact of replacing the benzothiazinone structure with benzothiopyranone, benzoxazinone, or benzopyranone. These studies have identified compounds with potent antimycobacterial activity and reduced toxicity, showcasing the potential of these structural modifications in enhancing DprE1 inhibition [
70,
75].
Dube et al. developed compounds with a nitroquinolone structure, positioning the nitro group at position 6 instead of the usual position 8. Although the activities observed were not as potent as other studies, this research highlights the potential for covalent binding to DprE1 with a nitro group at position 7 of the active skeleton. An X-ray structure with DprE1 could provide insights into the binding differences compared to inhibitors with the nitro group in position 8 [
76,
77].
These studies illustrate the extensive optimization of PBZ169 and the challenges in developing a superior DprE1 inhibitor with this structure. However, benzothiazinones and benzothiopyranones show high activity against DprE1 with minimal toxicity on HepG2 and Vero cells. The presence of a nitro group facilitates covalent binding to the target and hydrophobic groups at position 2 of the structure enhance activity. Positions 3 and 7 have been less explored and could be investigated to further improve compound activity. Nonetheless, discovering new scaffolds for covalent DprE1 inhibitors might be more beneficial to prevent future resistance rather than continually optimizing an already well-optimized structure.
Overall, these studies highlight the extensive optimization and exploration of DprE1 inhibitors based on the benzothiazinone skeleton. While significant progress has been made in developing potent inhibitors with favorable safety profiles, further research is warranted to explore new structures and optimize existing ones to address emerging challenges in TB treatment, including drug resistance.
3.2.2. Nitrobenzothiazoles
Following the success of benzothiazinones as covalent DprE1 inhibitors, various studies aimed to develop inhibitors with a Nitrobenzothiazole scaffold. Initial investigations by Landge et al. [
78] revealed the influence of different substituents on the benzothiazole ring. The presence of specific groups, such as a methyl group at position 6 or an oxygen atom on the nitrogen of the structure, significantly impacted the compound’s activity against Mtb H37Rv. Additionally, compounds incorporating piperidine or pyridine rings exhibited potent activities against Mtb.
Liu et al. [
79] further explored different substituents in position 2 of the Nitrobenzothiazole structure and identified compounds with remarkable activities (MIC = 0.01 µM) against Mtb H37Rv and low toxicities (IC
50 > 100 µM), highlighting the potential of this structural motif in DprE1 inhibition.
3.2.3. Nitrophenyltriazoles
Another structure capable of covalently binding to DprE1 is the nitrophenyl triazole. Although distinct from the benzothiazinone, compounds containing this scaffold displayed potent activities (MIC = 0.03–0.06 µM) against Mtb H37Rv with moderate toxicities on HepG2 cells (CC50 > 30 µM). While these compounds show promising activities, further exploration is needed to diversify this class of inhibitors [
80].
3.2.4. Nitrobenzene
A new class of DprE1 inhibitors was discovered by Batt et al. [
81] with the identification of the compound CT325. This compound, co-crystallized with DprE1, exhibited potent inhibitory activity against
Mycobacterium bovis. Further studies focusing on the modification of this structure could lead to the development of novel inhibitors against
Mycobacterium tuberculosis.
3.2.5. Non-Nitrated Compounds
In an attempt to explore alternative electrophilic groups for covalent binding to DprE1, Liu et al. [
66] investigated compounds replacing the nitro group of Macozinone. While these compounds exhibited inhibitory activity against Mtb H37Rv (MIC = 0.03–0.63 µM) and demonstrated covalent binding to DprE1, their activities were generally lower than those of Macozinone. Further evaluation of cytotoxicities is warranted to assess the potential of these compounds as DprE1 inhibitors. Nonetheless, this study provides insights into alternative substituents for covalent binding to DprE1, which could be valuable for researchers exploring inhibitors with different structural motifs.
3.2.6. Comparisons of Different Covalent Inhibitors
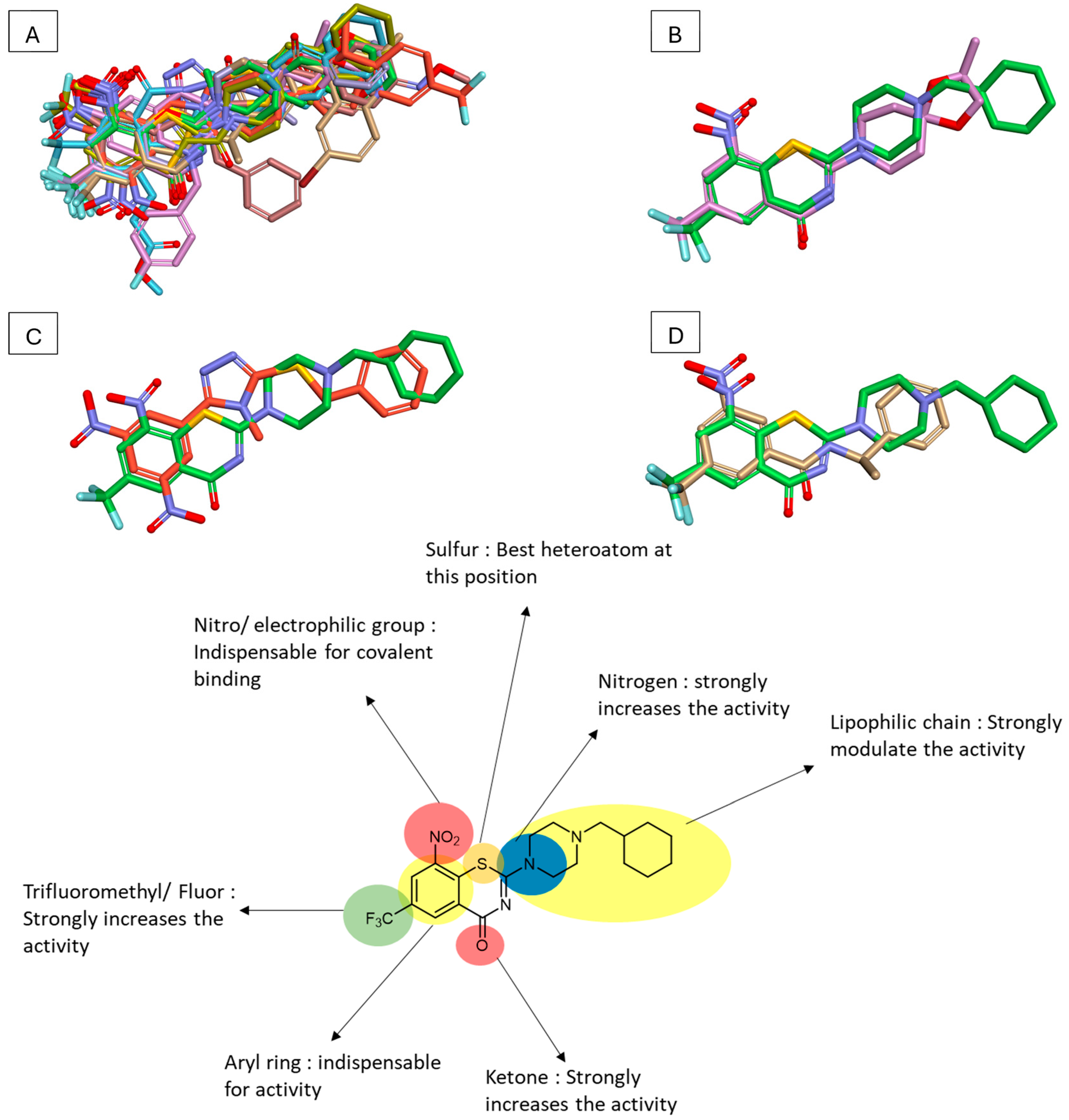
To compare various DprE1 inhibitors, representatives from each series were overlaid on the Discovery Studio Visualizer (
Figure 4). This visualization revealed significant uniformity in the presence and position of key functional groups across different inhibitor classes. Notably, a nitro group and a trifluoromethyl or fluoro group were consistently present, often directly linked to a benzene ring. Additionally, a predominantly hydrophobic moiety, composed of different rings, was observed in diverse structures such as the triazole series or CT325.
Figure 5 also illustrates different Macozinone pharmacophores, highlighting critical features for activity against DprE1. These include the presence of a phenyl ring with a nitro or electrophilic group, a trifluoromethyl or fluor group, and a heteroatom, particularly sulfur, which appears important for activity. Nitrogen presence across all ligands indicates its likely role in favorable hydrogen bonding. Lastly, the presence of a lipophilic chain, typically composed of one or several rings, strongly influences activity.
The chemical properties of compounds were studied to ascertain the important properties of a DprE1 covalent inhibitor. pKa and LogP values were computed using Chemaxon, while molecular weight and complexity were sourced from PubChem. Notably, most active inhibitors exhibited a relatively high number of hydrogen bond acceptors (between 8 and 10), with few hydrogen bond donors. The optimal number of free rotations appears to be 3 or 4, and compounds with a complexity slightly above 700 tend to be more active. LogP values indicate the compounds’ lipophilicity, with most falling between 3.5 and 5. The mean pKa of active compounds is typically around seven, while the optimal number of rings appears to be between three and four, with two or three of them being aromatic. Additionally, compounds with a molecular weight between 350 g/mol and 500 g/mol may increase the chances of observing activity against Mtb, as higher molecular weights may have difficulty crossing Mtb membranes due to their complexity.
These comparisons provide valuable insights into the structural and chemical features essential for the development of effective DprE1 inhibitors, aiding researchers in the design of novel compounds targeting tuberculosis.
3.3. Non-Covalent Inhibitors
Non-covalent inhibitors of DprE1 represent a significant family of compounds, with two currently in Phase 2 clinical trials (TBA-7371 [
82] and OPC-167832 [
68]).
3.3.1. Pyrrolopyridines, Benzimidazoles, Imidazopyridines, Pyrazolopyridines, and Thiazolopyridines
TBA-7371, a pyrrolopyridine-based compound, exhibited promising activities (CMI = 0.78 µM) against Mtb H37Rv. Additionally, compounds such as PP1 and PP2 (MIC: <0.39 µM and 0.78 µM, respectively) [
83] showed significant potencies, albeit with some toxicity concerns. Toxicities were evaluated revealing cytochrome P450 (CYP) inhibition (CYP1A2, CYP2C9, CYP2C19, CYP3A4) at doses exceeding 33 µM and hERG inhibition above 33 µM. Additionally, PDE6 IC50 (retina proteins) assessments showed toxicities (TBA7371: 4 µM; PP1: >100 µM; PP2: 1 µM). The presence of a methyl on the heteroaryl group reduced toxicities on PDE6 and enhanced compound activity. Predicted clearances on human microsomes were between 15% and 30% liver blood flow (LBF). A scaffold morphing study revealed the potency of benzimidazoles through the discovery of compounds BZ1-8, modulating the amide and pyrimidine ring substituents. This study suggested replacing the methyl group with a methoxy group to enhance solubility. Although CYP inhibition and hERG toxicities remained consistent, PDE6 toxicities were not assessed. The study presented several compounds with MICs < 0.39 µM, but structural discussion was limited due to a lack of testing at lower concentrations.
Imidazolopyridine compounds (IZP1-4) [
84] exhibited MICs ranging from 0.5 µM to 0.8 µM, highlighting their affinity for DprE1. Interestingly, despite the presence of a nitro group on these compounds, they do not appear to covalently bind to DprE1. This observation highlights the potential significance of the nitro group’s position for covalent binding to DprE1.
Pyrazolopyridone compounds [
85,
86] emphasized the importance of specific structural features for activity against Mtb. This study showed the importance of the methyl in para of the CF
3 group, the cyclopropyl group, and the presence of a phenyl group or a hydrophobic group in position 1 of the pyrazolopyridine ring.
Compounds based on benzothiazoles and thiazolopyridines [
87,
88,
89] demonstrated significant MICs (0.77–3.1 µM) on Mtb H37Rv, although with some toxicity concerns. The objective of these compounds was to target Leu317 of DprE1 with a hydrophobic binding to enhance their activity.
3.3.2. Imidazoles, Thiophene, and Thiadiazoles
Studies on the hydantoin structures [
90,
91,
92,
93] revealed promising activities (MIC = 2.5–10 µM) against Mtb H37Rv, with low cytotoxicities (IC
50 > 100 µM). Studies aimed to optimize hits through Structure Activity Relationship (SAR) analysis, synthesizing over 85 compounds tested on Mtb. Compounds showed sub-micromolar activities (MICs: 0.6–0.9 µM) without toxicity. In vivo testing revealed a favorable pharmacokinetic profile but modest activity. Notably, the S enantiomer of compound IMI8 was inactive, while the R enantiomer IMI8 showed a MIC of 0.78 µM. The racemic mixture exhibited higher activity (MIC: 0.7 µM), indicating enantiomeric synergy. Despite modest in vivo results, the study highlights the potential of the hydantoin moiety for new DprE1 inhibitors.
Thiophene [
70,
94] compounds exhibited significant activities (MIC = 0.035–0.32 µM) against Mtb H37Rv, with compound Thio1 showing promising in vivo results (1.11 reduction log
10 CFU/macrophages at 5 µg/mL). Moreover, activity was maintained (MIC: 0.062 µg/mL) against PBTZ-169-resistant strains. Various PK parameters were calculated for these inhibitors, and future studies will aim to enhance their drug-like properties.
Among 177 GSK small molecules published for TB, a thiadiazole-based inhibitor [
70,
95,
96], presented a MIC90 of 4 µM on Mtb H37Rv, contributing to the pool of potential drugs for tuberculosis treatment.
3.3.3. Pyrimidine
The previous structures underwent optimization, with the thiadiazole moiety being replaced by a pyrimidine one and the thiophene one by a morpholine [
96]. In this study, compounds were identified as potential drug candidates, with MICs on Mtb H37Rv of 0.6–1.7 µM, low toxicities on HepG2 (pIC50 4.3–4.5), and promising efficacy in vivo in a mouse model (ED99 around 30 mg/kg), despite low t
1/2 (0.45–1.0 h at 5 mg/kg). These compounds still require optimization but demonstrate the potency of a pyrimidino–morpholine structure.
In another study by Oh et al. [
97], a structure–activity relationship (SAR) study based on the pyrimidine moiety was conducted, where compounds appeared to be promising, with MIC = 2.3–4.7 µM on Mtb H37Rv and exhibited an IC50 over 100 µM on HEPG2. Compound Py2 underwent further experimentation, particularly in vivo, and demonstrated favorable pharmacokinetic properties with a T
1/2 of 4.1 h at a 10 mg/kg dose.
Additionally, a structure-based virtual screening [
98] identified another pyrimidine compound that exhibited a MIC of 3.1 µM on M. smegmatis ATCC 607 with an IC
50 on Vero cells, Lo2 cells, and L929 cells over 100 times the MIC and no damage observed in mice at doses under 2 g/kg during a 14-day period. At 30 mg/kg, this compound showed a 2.1 CFU log10 reduction in the lung and a 1.3 CFU log10 reduction in the spleen. All these compounds underscore the potency of the pyrimidine nucleus in developing in vitro and in vivo efficient DprE1 inhibitors.
These studies highlight the diversity of non-covalent inhibitors targeting DprE1 and highlight the potential for developing effective therapies against tuberculosis. Further research into optimizing these compounds and understanding their pharmacokinetic properties is warranted to enhance their clinical utility.
3.3.4. Quinoline, Quinoxaline, and Benzodioxine
Currently, in clinical trials, OPC-167832 demonstrates exceptional potency against Mtb H37Rv with a sub-nanomolar MIC of 0.5 nM. It exhibits promising in vivo results and shows potential as a new tuberculosis drug. Additionally, it displays nanomolar or sub-nanomolar MICs on various resistant Mtb strains without significant toxicities, highlighting its therapeutic potential [
99].
Quinolinone derivatives exhibit submicromolar MIC on Mtb H37Rv with minimal cytotoxicity. A scaffold morphing study led to the discovery of analogs showing improved potency [
86,
100].
Quinoxaline compounds demonstrate activity against Mtb H37Rv, albeit with relatively high toxicities on HepG2 cells [
86,
101]. X-ray structures confirmed the fact those compounds are non-covalent DprE1 inhibitors.
Benzodioxine derivatives served as a starting point for developing new DprE1 inhibitors, leading to the discovery of compounds with improved MIC = 2.5–4 µM. Further optimization of these compounds is warranted [
102].
3.3.5. Selamectin
Originally a veterinary drug for treating fleas, Selamectin inhibits DprE1 with a MIC of 5.1 µM on Mtb H37Rv [
103]. While its size may limit its in vivo efficacy, further research, especially crystallization studies, could offer insights into its binding mechanisms and potential for tuberculosis treatment.
3.3.6. Comparison of Non-Covalent Inhibitors
A physicochemical characteristics analysis of non-covalent DprE1 inhibitors reveals essential factors influencing their activity. The most active compounds typically have 6–8 hydrogen bond acceptors (HBAs) and generally one to two hydrogen bond donors (HBDs). These compounds tend to exhibit five to six free rotations. The complexity of these compounds varies, with most falling between 350 and 850. While most compounds lack asymmetric centers, their presence can impact activity. Active compounds usually have a LogP between three and five. There is considerable variability in pKa values, with no clear correlation to activity. Most active compounds have three to four rings, with two to four of these being aromatic. The optimal molar weight range for efficient membrane penetration falls between 350 g/mol and 480 g/mol.
Structural analysis highlights the importance of hydrophobic groups, phenyl rings, HBA, and specific functional groups in enhancing activity. Understanding these pharmacophores guides the design of potent DprE1 inhibitors for tuberculosis therapy.
To determine if a general structural shape is evident among different DprE1 non-covalent inhibitors, the best compounds were overlaid (50% steric/50% electrostatic) using Discovery Studio Visualizer (
Figure 4). The overlay reveals that these compounds form a general shape that closely aligns with the structure of OPC-167832, highlighting the extent to which this compound has been optimized to fit the DprE1 pocket.
The molecular overlay between TBA7371 and OPC-167832 shows that, while the pyrrolopyridine and quinoline rings are not superimposed, the overall shape of the molecules appears coherent. This suggests a similar binding conformation within the DprE1 pocket, warranting further investigation with X-ray models to compare their binding modes directly.
Additionally, while it was suggested that Selamectin might bind to a different pocket [
103], the overlay with OPC-167832 indicates that Selamectin could fit into the same binding pocket as other inhibitors. This finding enhances our understanding of the structural requirements for effective DprE1 inhibition and provides valuable guidance for the future design and optimization of inhibitors.
Figure 5 further illustrates various OPC-167832 pharmacophores, highlighting critical features essential for the design of new TB drugs.
3.4. Link between Covalent and Non-Covalent Inhibitors
3.4.1. Structural and Characteristic Differences
A comparison of HBAs reveals that covalent inhibitors generally have more HBAs compared to non-covalent inhibitors. This is attributed to the presence of nitro groups in many covalent inhibitors. Conversely, non-covalent inhibitors typically exhibit fewer HBAs, possibly due to the accessibility of certain amino acids in the binding pocket.
In terms of HBDs, non-covalent inhibitors tend to possess more HBDs than covalent inhibitors. This is likely influenced by the conformational flexibility and accessibility of functional groups in the binding pocket.
Regarding free rotations, non-covalent inhibitors show a higher number of free rotations compared to covalent inhibitors. This may be due to the absence of a conformational lock provided by covalent binding.
Interestingly, covalent inhibitors exhibit higher complexity than non-covalent inhibitors. This may be influenced by factors such as the number of HBAs, rings, and molecular weight.
Non-covalent inhibitors, particularly those influenced by Selamectin, have a higher average count of asymmetric centers. Excluding Selamectin, the difference between covalent and non-covalent inhibitors is less pronounced.
Both covalent and non-covalent inhibitors show relatively stable LogP values, reflecting the challenge of membrane penetration. Variation in pKa may be attributed to structural diversity among the inhibitors.
Covalent and non-covalent inhibitors typically possess three to four rings, with two to three aromatic rings. This indicates the importance of this structural feature for DprE1 inhibition.
Finally, covalent and non-covalent inhibitors exhibit relatively stable molar weights. This reflects the size and shape of DprE1’s pocket and the challenges of membrane penetration.
3.4.2. Structural Comparison: OPC-167832 and Macozinone
OPC-167832 and Macozinone, representing the most potent inhibitors of each class, display striking similarities in their fitting into DprE1’s pocket, suggesting potential overlaps in their binding modes.
This highlights common pharmacophores essential for DprE1 inhibition, underscoring the shared features required for effective inhibition across covalent and non-covalent inhibitor classes.
3.4.3. Insights and Future Directions
While covalent and non-covalent inhibitors exhibit differences in certain structural and characteristic aspects, their similarities underscore common features crucial for effective DprE1 inhibition.
Further structural studies, such as X-ray crystallography, could provide valuable insights into the binding modes of these inhibitors, aiding in the rational design of new DprE1 inhibitors.
4. Conclusions
In conclusion, DprE1 emerges as a pivotal protein in Mtb responsible for arabinogalactan synthesis, particularly arabinose. Its indispensable role in Mtb survival, unique presence in mycobacteria, and location in the bacterial periplasm make it an attractive target for new anti-tuberculosis (TB) drugs.
While there are currently no marketed DprE1 inhibitors as anti-TB agents, promising candidates such as Macozinone, BTZ-043, TBA-7371, and OPC-167832 are undergoing clinical trials, demonstrating sub-nanomolar minimum inhibitory concentrations (MICs). The availability of X-ray structures of DprE1 co-crystallized with various inhibitors offers valuable insights for drug development endeavors.
However, a notable lack of structural diversity among known DprE1 ligands exists. Hence, the discovery of new molecular scaffolds capable of targeting DprE1 could significantly advance TB treatment strategies. SAR studies highlighted in this review provide crucial guidelines for developing effective DprE1 inhibitors, emphasizing key physicochemical properties such as optimal LogP around 3.5, 3–4 rings, approximately 8 hydrogen bond acceptors, and a molecular weight of around 430 g/mol.
Looking ahead, leveraging the wealth of available data on known DprE1 ligands and the structural insights into the target protein will be pivotal for the development of novel and potent anti-TB drugs. By expanding the structural diversity of DprE1 inhibitors and optimizing their pharmacological properties, researchers can contribute significantly to the global effort to combat tuberculosis.
Author Contributions
Conceptualization, M.F. and S.A.; methodology, M.F. and S.A.; software, M.F.; validation, M.F. and S.A.; formal analysis, M.F. and S.A.; investigation, M.F. and S.A.; resources, M.F. and S.A.; data curation, M.F. and S.A.; writing—original draft preparation, M.F. and S.A.; writing—review and editing, M.F. and S.A.; visualization, M.F. and S.A.; supervision, S.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Acknowledgments
We would like to express our sincere gratitude to Elnur Garayev for facilitating our involvement in the SONATA project.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- WHO (World Health Organization). Global Tuberculosis Report 2022; WHO: Geneva, Switzerland, 2022. [Google Scholar]
- WHO. Consolidated Guidelines on Tuberculosis: Module 4: Treatment: Drug-Resistant Tuberculosis Treatment; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- WHO. Consolidated Guidelines on Tuberculosis: Module 3: Diagnosis: Rapid Diagnostics for Tuberculosis Detection; 2021 Update; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Banerjee, A.; Dubnau, E.; Quemard, A.; Balasubramanian, V.; Um, K.S.; Wilson, T.; Collins, D.; De Lisle, G.; Jacobs, W.R., Jr. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science 1994, 263, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Rozwarski, D.A.; Grant, G.A.; Barton, D.H.; Jacobs, W.R., Jr.; Sacchettini, J.C. Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science 1998, 279, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.A.; Korzheva, N.; Mustaev, A.; Murakami, K.; Nair, S.; Goldfarb, A.; Darst, S.A. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell 2001, 104, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Andries, K.; Verhasselt, P.; Guillemont, J.; Göhlmann, H.W.; Neefs, J.M.; Winkler, H.; Van Gestel, J.; Timmerman, P.; Zhu, M.; Lee, E.; et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Drlica, K.; Zhao, X. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol. Mol. Biol. Rev. 1997, 61, 377–392. [Google Scholar] [CrossRef] [PubMed]
- Jankute, M.; Grover, S.; Birch, H.L.; Besra, G.S. Genetics of Mycobacterial Arabinogalactan and Lipoarabinomannan Assembly. Microbiol. Spectr. 2014, 2, MGM2-0013-2013. [Google Scholar] [CrossRef] [PubMed]
- Putman, M.; van Veen, H.W.; Konings, W.N. Molecular properties of bacterial multidrug transporters. Microbiol. Mol. Biol. Rev. 2000, 64, 672–693. [Google Scholar] [CrossRef] [PubMed]
- Poole, K. Efflux pumps as antimicrobial resistance mechanisms. Ann. Med. 2007, 39, 162–176. [Google Scholar] [CrossRef]
- Romaniuk, J.A.; Cegelski, L. Bacterial cell wall composition and the influence of antibiotics by cell-wall and whole-cell NMR. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20150024. [Google Scholar] [CrossRef]
- Lambert, P.A. Enterobacteriaceae: Composition, structure and function of the cell envelope. Soc. Appl. Bacteriol. Symp. Ser. 1988, 17, 21S–34S. [Google Scholar] [CrossRef] [PubMed]
- Dörr, T.; Moynihan, P.J.; Mayer, C. Editorial: Bacterial Cell Wall Structure and Dynamics. Front. Microbiol. 2019, 10, 2051. [Google Scholar] [CrossRef] [PubMed]
- Riu, F.; Ruda, A.; Ibba, R.; Sestito, S.; Lupinu, I.; Piras, S.; Widmalm, G.; Carta, A. Antibiotics and Carbohydrate-Containing Drugs Targeting Bacterial Cell Envelopes: An Overview. Pharmaceuticals 2022, 15, 942. [Google Scholar] [CrossRef] [PubMed]
- Jankute, M.; Cox, J.A.; Harrison, J.; Besra, G.S. Assembly of the Mycobacterial Cell Wall. Annu. Rev. Microbiol. 2015, 69, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, K.A.; Besra, G.S. Mycobacterial cell wall biosynthesis: A multifaceted antibiotic target. Parasitology 2018, 145, 116–133. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.; Stevens, C.M.; Zhang, L.; Zgurskaya, H.I.; Niederweis, M. Transporters Involved in the Biogenesis and Functionalization of the Mycobacterial Cell Envelope. Chem. Rev. 2021, 121, 5124–5157. [Google Scholar] [CrossRef] [PubMed]
- Daffe, M.; Brennan, P.J.; McNeil, M. Predominant structural features of the cell wall arabinogalactan of Mycobacterium tuberculosis as revealed through characterization of oligoglycosyl alditol fragments by gas chromatography/mass spectrometry and by 1H and 13C NMR analyses. J. Biol. Chem. 1990, 265, 6734–6743. [Google Scholar] [CrossRef] [PubMed]
- Alderwick, L.J.; Radmacher, E.; Seidel, M.; Gande, R.; Hitchen, P.G.; Morris, H.R.; Dell, A.; Sahm, H.; Eggeling, L.; Besra, G.S. Deletion of Cg-emb in corynebacterianeae leads to a novel truncated cell wall arabinogalactan, whereas inactivation of Cg-ubiA results in an arabinan-deficient mutant with a cell wall galactan core. J. Biol. Chem. 2005, 280, 32362–32371. [Google Scholar] [CrossRef] [PubMed]
- Brecik, M.; Centárová, I.; Mukherjee, R.; Kolly, G.S.; Huszár, S.; Bobovská, A.; Kilacsková, E.; Mokošová, V.; Svetlíková, Z.; Šarkan, M.; et al. DprE1 Is a Vulnerable Tuberculosis Drug Target Due to Its Cell Wall Localization. ACS Chem. Biol. 2015, 10, 1631–1636. [Google Scholar] [CrossRef]
- Riccardi, G.; Pasca, M.R.; Chiarelli, L.R.; Manina, G.; Mattevi, A.; Binda, C. The DprE1 enzyme, one of the most vulnerable targets of Mycobacterium tuberculosis. Appl. Microbiol. Biotechnol. 2013, 97, 8841–8848. [Google Scholar] [CrossRef]
- Mikusová, K.; Huang, H.; Yagi, T.; Holsters, M.; Vereecke, D.; D’Haeze, W.; Scherman, M.S.; Brennan, P.J.; McNeil, M.R.; Crick, D.C. Decaprenylphosphoryl arabinofuranose, the donor of the D-arabinofuranosyl residues of mycobacterial arabinan, is formed via a two-step epimerization of decaprenylphosphoryl ribose. J. Bacteriol. 2005, 187, 8020–8025. [Google Scholar] [CrossRef] [PubMed]
- Wolucka, B.A. Biosynthesis of D-arabinose in mycobacteria—A novel bacterial pathway with implications for antimycobacterial therapy. FEBS J. 2008, 275, 2691–2711. [Google Scholar] [CrossRef] [PubMed]
- Kolly, G.S.; Boldrin, F.; Sala, C.; Dhar, N.; Hartkoorn, R.C.; Ventura, M.; Serafini, A.; McKinney, J.D.; Manganelli, R.; Cole, S.T. Assessing the essentiality of the decaprenyl-phospho-d-arabinofuranose pathway in Mycobacterium tuberculosis using conditional mutants. Mol. Microbiol. 2014, 92, 194–211. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, I.; Loharch, S.; Gupta, P.; Madathil, R.; Parkesh, R. Structure, dynamics, and interaction of Mycobacterium tuberculosis (Mtb) DprE1 and DprE2 examined by molecular modeling, simulation, and electrostatic studies. PLoS ONE 2015, 10, e0119771. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; AS, A.; Thabet, H.K.; Abida Afroz Bakht, M. Synthetic molecules as DprE1 inhibitors: A patent review. Expert Opin. Ther. Pat. 2021, 31, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Raman, K.; Yeturu, K.; Chandra, N. targetTB: A target identification pipeline for Mycobacterium tuberculosis through an interactome, reactome and genome-scale structural analysis. BMC Syst. Biol. 2008, 2, 109. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Werman, J.M.; Sampson, N.S. The pursuit of mechanism of action: Uncovering drug complexity in TB drug discovery. RSC Chem. Biol. 2021, 2, 423–440. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Abrahams, K.A.; Besra, G.S. Mycobacterial drug discovery. RSC Med. Chem. 2020, 11, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- McNeil, M.B.; Keighley, L.M.; Cook, J.R.; Cheung, C.Y.; Cook, G.M. CRISPR interference identifies vulnerable cellular pathways with bactericidal phenotypes in Mycobacterium tuberculosis. Mol. Microbiol. 2021, 116, 1033–1043. [Google Scholar] [CrossRef]
- Bosch, B.; DeJesus, M.A.; Poulton, N.C.; Zhang, W.; Engelhart, C.A.; Zaveri, A.; Lavalette, S.; Ruecker, N.; Trujillo, C.; Wallach, J.B.; et al. Genome-wide gene expression tuning reveals diverse vulnerabilities of M. tuberculosis. Cell 2021, 184, 4579–4592.e24. [Google Scholar] [CrossRef]
- Deretic, V. Screening Methods for Identifying Compounds for Diagnosis and Treatment of Autophagy-Associated Disorders. WO2012154944, 15 November 2012. [Google Scholar]
- Bennani, Y.L. Preparation of Phosphate Esters as Bacterial Gyrase and Topoisomerase IV Inhibitors Useful in Treatment of Bacterial Infections. WO2012177707, 27 December 2012. [Google Scholar]
- Shannon, D. Preparation of Solid Forms of Ethylfluorohydroxymethylethylpyrimidinyltetrahydrofuranylbenzimidazolylurea Derivatives for Use as Gyrase and Topoisomerase IV Inhibitors Useful in Treatment of Bacterial Infections. WO2012097273, 19 July 2012. [Google Scholar]
- Le Tiran, A. Preparation of Pyrimidinyltetrahydrofuranylbenzimidazolylurea Derivatives for Use as Gyrase and Topoisomerase IV Inhibitors Useful in Treatment of Bacterial Infections. WO2012097269, 19 July 2012. [Google Scholar]
- Dorsch, D. Preparation of triazolo[4,5-d]pyrimidine Derivatives as Inhibitors of GCN2. WO2013110309, 1 August 2013. [Google Scholar]
- Eickhoff, J. Preparation of Pyrazolotriazine Derivatives for Use as Selective Cyclin-Dependent Kinase Inhibitors. WO2013128028, 6 September 2013. [Google Scholar]
- Burgdorf, L. Preparation of Furopyridine Derivatives as Syk Kinase Inhibitors. WO2013124025, 29 August 2013. [Google Scholar]
- Burgdorf, L. Preparation of Pyridopyrimidine Derivatives as Syk Kinase Inhibitors. WO2014023385, 13 February 2014. [Google Scholar]
- Miller, M.J. Preparation of 1,3-benzothiazinone, Sulfoxide, and Sulfone Compounds with electrophilic Substituent as Antibacterial and Anti-tuberculosis Agents. US20150353572, 10 December 2015. [Google Scholar]
- Naik, M.N. Preparation of Azaindole Compounds that Target DprE1 for Treatment of Mycobacterium Infections. WO2015009525, 22 January 2015. [Google Scholar]
- Shirude, P.S. Preparation of Azaindole Compounds that Target DprE1 for Treatment of Mycobacterium Infections. IN2013CH03196, 31 August 2016. [Google Scholar]
- Rao, J. Fluorogenic Probes for Rapid and Specific Detection of Mycobacteria, Especially Those expressing Both a β-lactamase and DprE1. WO2017027062, 16 February 2017. [Google Scholar]
- Monkam, N. Pharmaceutical Composition including D-limonene, Lupeol and a Pharmaceutically Active Agent Selected from Cinnamaldehyde, Epicatechin, Methylhydroxychalcone Polymer, β-sitosterol, Curcumin and Mixtures Thereof for Treating Metabolic Syndrome Disorders, Infectious Diseases and Complications Thereof. FR3061658, 13 July 2018. [Google Scholar]
- Li, H. DprE1 Enzymeenzyme Inhibitor and Its Preparation. CN108623527, 9 October 2018. [Google Scholar]
- Desai, R. Preparation of Condensed Azaheteroaryl Compounds Having Antibacterial Activity against Tuberculosis Bacteria. WO2019239382, 19 December 2019. [Google Scholar]
- Ambrogelly, A. FLT3L-FC Fusion Proteins and Methods of Use. WO2020263830, 30 December 2020. [Google Scholar]
- Shao, C. Preparation of Azaindole Amide Compounds and Compositions Thereof for treatment of Tuberculosistuberculosis or Mycobacterial Infection. CN111393435, 10 July 2020. [Google Scholar]
- Shirude, P.S. Preparation of Benzimidazole Derivatives as DprE1 Inhibitors for the Treatment of Tuberculosis. WO2020188405, 24 September 2020. [Google Scholar]
- Martyn, G. Immunotherapeutic Treatment of Cancer. WO2021136933, 8 July 2021. [Google Scholar]
- Crowley, B.M. Oxazolidinone Compound and Methods of Use Thereof as an Antibacterial Agent. WO2021188606, 23 September 2021. [Google Scholar]
- Shirude, P.S. Preparation of Benzimidazole Derivatives as DprE1inhibitors for the Treatment of Tuberculosis. IN201941010942, 11 June 2021. [Google Scholar]
- Kampinga, J. A Mycobacterium for Use in Cancer Therapy. WO2022101619, 19 May 2022. [Google Scholar]
- Martyn, G. Mycobacterial Immunotherapy for Treating Cancer. WO2022096896, 12 May 2022. [Google Scholar]
- Li, H. Process for Preparation of Dpre 1 Enzyme Inhibitorinhibitor and Use Thereof. CN114516858, 20 May 2022. [Google Scholar]
- Yu, L. Preparation of aryl/heteroaryloxymethyl piperidinol/cyclohexanol Derivatives as DprE1 Enzyme Inhibitors for Treatment of Drug-Resistant Mycobacterium Tuberculosis. CN116102537, 12 May 2023. [Google Scholar]
- Xu, Y. Preparation of Pyrazolopyrimidinone Based Compound and Its Application as Mycobacterium tuberculosis DprE1enzyme Inhibitor. CN116162082, 26 May 2023. [Google Scholar]
- Vicente, C. Nitrobenzamide Compounds, Methods and Uses Thereof. EP4345092, 3 April 2024. [Google Scholar]
- Vicente, C. Benzoic Acid Derivatives, Methods and Uses Thereof in Mycobacterial Infections. EP4345091, 3 April 2024. [Google Scholar]
- Richter, A.; Rudolph, I.; Möllmann, U.; Voigt, K.; Chung, C.W.; Singh, O.M.P.; Rees, M.; Mendoza-Losana, A.; Bates, R.; Ballell, L.; et al. Novel insight into the reaction of nitro, nitroso and hydroxylamino benzothiazinones and of benzoxacinones with Mycobacterium tuberculosis DprE1. Sci. Rep. 2018, 8, 13473. [Google Scholar] [CrossRef] [PubMed]
- Trefzer, C.; Rengifo-Gonzalez, M.; Hinner, M.J.; Schneider, P.; Makarov, V.; Cole, S.T.; Johnsson, K. Benzothiazinones: Prodrugs that covalently modify the decaprenylphosphoryl-β-D-ribose 2′-epimerase DprE1 of Mycobacterium tuberculosis. J. Am. Chem. Soc. 2010, 132, 13663–13665. [Google Scholar] [CrossRef] [PubMed]
- Trefzer, C.; Škovierová, H.; Buroni, S.; Bobovská, A.; Nenci, S.; Molteni, E.; Pojer, F.; Pasca, M.R.; Makarov, V.; Cole, S.T.; et al. Benzothiazinones are suicide inhibitors of mycobacterial decaprenylphosphoryl-β-D-ribofuranose 2′-oxidase DprE1. J. Am. Chem. Soc. 2012, 134, 912–915. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, S.; Kumar, S.; Parkesh, R. Chemical Space Exploration of DprE1 Inhibitors Using Chemoinformatics and Artificial Intelligence. ACS Omega 2021, 6, 14430–14441. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Kong, C.; Fumagalli, M.; Savková, K.; Xu, Y.; Huszár, S.; Sammartino, J.C.; Fan, D.; Chiarelli, L.R.; Mikušová, K.; et al. Design, synthesis and evaluation of covalent inhibitors of DprE1 as antitubercular agents. Eur. J. Med. Chem. 2020, 208, 112773. [Google Scholar] [CrossRef] [PubMed]
- Makarov, V.; Lechartier, B.; Zhang, M.; Neres, J.; van der Sar, A.M.; Raadsen, S.A.; Hartkoorn, R.C.; Ryabova, O.B.; Vocat, A.; Decosterd, L.A.; et al. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol. Med. 2014, 6, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Angula, K.T.; Legoabe, L.J.; Beteck, R.M. Chemical Classes Presenting Novel Antituberculosis Agents Currently in Different Phases of Drug Development: A 2010–2020 Review. Pharmaceuticals 2021, 14, 461. [Google Scholar] [CrossRef] [PubMed]
- Piton, J.; Vocat, A.; Lupien, A.; Foo, C.S.; Riabova, O.; Makarov, V.; Cole, S.T. Structure-Based Drug Design and Characterization of Sulfonyl-Piperazine Benzothiazinone Inhibitors of DprE1 from Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2018, 62, e00681-18. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, G.F.S.; Thompson, A.M.; Castagnolo, D.; Denny, W.A.; Dos Santos, J.L. Tuberculosis Drug Discovery: Challenges and New Horizons. J. Med. Chem. 2022, 65, 7489–7531. [Google Scholar] [CrossRef]
- Lv, K.; You, X.; Wang, B.; Wei, Z.; Chai, Y.; Wang, B.; Wang, A.; Huang, G.; Liu, M.; Lu, Y. Identification of Better Pharmacokinetic Benzothiazinone Derivatives as New Antitubercular Agents. ACS Med. Chem. Lett. 2017, 8, 636–641. [Google Scholar] [CrossRef]
- Wang, A.; Ma, C.; Chai, Y.; Liu, X.; Lv, K.; Fu, L.; Wang, B.; Jia, X.; Liu, M.; Lu, Y. Identification of benzothiazinones containing 2-benzyl-2,7-diazaspiro[3.5]nonane moieties as new antitubercular agents. Eur. J. Med. Chem. 2020, 200, 112409. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Fu, L.; Wang, B.; Chen, X.; Zhao, J.; Liu, M.; Lu, Y. In vitro and in vivo antimicrobial activities of a novel piperazine-containing benzothiazinones candidate TZY-5-84 against Mycobacterium tuberculosis. Biomed. Pharmacother. 2020, 131, 110777. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.K.; Gajula, S.N.R.; Ahmad, M.N.; Kaul, G.; Nanduri, S.; Sonti, R.; Dasgupta, A.; Chopra, S.; Yaddanapudi, V.M. Bioevaluation of quinoline-4-carbonyl derivatives of piperazinyl-benzothiazinones as promising antimycobacterial agents. Arch. Pharm. 2022, 355, e2200168. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, B.; Zhang, X.; Batt, S.M.; Besra, G.S.; Zhang, T.; Ma, C.; Zhang, D.; Lin, Z.; Li, G.; et al. Identification of novel benzothiopyranone compounds against Mycobacterium tuberculosis through scaffold morphing from benzothiazinones. Eur. J. Med. Chem. 2018, 160, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Dube, P.S.; Legoabe, L.J.; Jordaan, A.; Jesumoroti, O.J.; Tshiwawa, T.; Warner, D.F.; Beteck, R.M. Easily accessed nitroquinolones exhibiting potent and selective anti-tubercular activity. Eur. J. Med. Chem. 2021, 213, 113207. [Google Scholar] [CrossRef] [PubMed]
- Perveen, S.; Kumari, D.; Singh, K.; Sharma, R. Tuberculosis drug discovery: Progression and future interventions in the wake of emerging resistance. Eur. J. Med. Chem. 2022, 229, 114066. [Google Scholar] [CrossRef]
- Landge, S.; Mullick, A.B.; Nagalapur, K.; Neres, J.; Subbulakshmi, V.; Murugan, K.; Ghosh, A.; Sadler, C.; Fellows, M.D.; Humnabadkar, V.; et al. Discovery of benzothiazoles as antimycobacterial agents: Synthesis, structure-activity relationships and binding studies with Mycobacterium tuberculosis decaprenylphosphoryl-β-D-ribose 2′-oxidase. Bioorg. Med. Chem. 2015, 23, 7694–7710. [Google Scholar] [CrossRef]
- Liu, R.; Markley, L.; Miller, P.A.; Franzblau, S.; Shetye, G.; Ma, R.; Savková, K.; Mikušová, K.; Lee, B.S.; Pethe, K.; et al. Hydride-induced Meisenheimer complex formation reflects activity of nitro aromatic anti-tuberculosis compounds. RSC Med. Chem. 2021, 12, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Karabanovich, G.; Dušek, J.; Savková, K.; Pavliš, O.; Pávková, I.; Korábečný, J.; Kučera, T.; Kočová Vlčková, H.; Huszár, S.; Konyariková, Z.; et al. Development of 3,5-Dinitrophenyl-Containing 1,2,4-Triazoles and Their Trifluoromethyl Analogues as Highly Efficient Antitubercular Agents Inhibiting Decaprenylphosphoryl-β-d-ribofuranose 2′-Oxidase. J. Med. Chem. 2019, 62, 8115–8139. [Google Scholar] [CrossRef]
- Batt, S.M.; Jabeen, T.; Bhowruth, V.; Quill, L.; Lund, P.A.; Eggeling, L.; Alderwick, L.J.; Fütterer, K.; Besra, G.S. Structural basis of inhibition of Mycobacterium tuberculosis DprE1 by benzothiazinone inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 11354–11359. [Google Scholar] [CrossRef]
- RM, M.; Shandil, R.; Panda, M.; Sadler, C.; Ambady, A.; Panduga, V.; Kumar, N.; Mahadevaswamy, J.; Sreenivasaiah, M.; Narayan, A.; et al. Scaffold Morphing To Identify Novel DprE1 Inhibitors with Antimycobacterial Activity. ACS Med. Chem. Lett. 2019, 10, 1480–1485. [Google Scholar] [CrossRef]
- Shirude, P.S.; Shandil, R.K.; Manjunatha, M.R.; Sadler, C.; Panda, M.; Panduga, V.; Reddy, J.; Saralaya, R.; Nanduri, R.; Ambady, A.; et al. Lead optimization of 1,4-azaindoles as antimycobacterial agents. J. Med. Chem. 2014, 57, 5728–5737. [Google Scholar] [CrossRef]
- Gawad, J.; Bonde, C. Synthesis, biological evaluation and molecular docking studies of 6-(4-nitrophenoxy)-1H-imidazo[4,5-b]pyridine derivatives as novel antitubercular agents: Future DprE1 inhibitors. Chem. Cent. J. 2018, 12, 138. [Google Scholar] [CrossRef] [PubMed]
- Panda, M.; Ramachandran, S.; Ramachandran, V.; Shirude, P.S.; Humnabadkar, V.; Nagalapur, K.; Sharma, S.; Kaur, P.; Guptha, S.; Narayan, A.; et al. Discovery of pyrazolopyridones as a novel class of noncovalent DprE1 inhibitor with potent anti-mycobacterial activity. J. Med. Chem. 2014, 57, 4761–4771. [Google Scholar] [CrossRef]
- Oh, S.; Trifonov, L.; Yadav, V.D.; Barry, C.E., 3rd; Boshoff, H.I. Tuberculosis Drug Discovery: A Decade of Hit Assessment for Defined Targets. Front. Cell. Infect. Microbiol. 2021, 11, 611304. [Google Scholar] [CrossRef]
- Wang, F.; Sambandan, D.; Halder, R.; Wang, J.; Batt, S.M.; Weinrick, B.; Ahmad, I.; Yang, P.; Zhang, Y.; Kim, J.; et al. Identification of a small molecule with activity against drug-resistant and persistent tuberculosis. Proc. Natl. Acad. Sci. USA 2013, 110, E2510–E2517. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Dai, H.; Wang, B.; Liu, H.; Tian, Z.; Zhang, Y. Exploring disordered loops in DprE1 provides a functional site to combat drug-resistance in Mycobacterium strains. Eur. J. Med. Chem. 2022, 227, 113932. [Google Scholar] [CrossRef]
- Liu, R.; Lyu, X.; Batt, S.M.; Hsu, M.H.; Harbut, M.B.; Vilchèze, C.; Cheng, B.; Ajayi, K.; Yang, B.; Yang, Y.; et al. Determinants of the Inhibition of DprE1 and CYP2C9 by Antitubercular Thiophenes. Angew. Chem. Int. Ed. Engl. 2017, 56, 13011–13015. [Google Scholar] [CrossRef] [PubMed]
- Rogacki, M.K.; Pitta, E.; Balabon, O.; Huss, S.; Lopez-Roman, E.M.; Argyrou, A.; Blanco-Ruano, D.; Cacho, M.; Vande Velde, C.M.L.; Augustyns, K.; et al. Identification and Profiling of Hydantoins—A Novel Class of Potent Antimycobacterial DprE1 Inhibitors. J. Med. Chem. 2018, 61, 11221–11249. [Google Scholar] [CrossRef]
- Makarov, V.; Salina, E.; Reynolds, R.C.; Kyaw Zin, P.P.; Ekins, S. Molecule Property Analyses of Active Compounds for Mycobacterium tuberculosis. J. Med. Chem. 2020, 63, 8917–8955. [Google Scholar] [CrossRef]
- Balabon, O.; Pitta, E.; Rogacki, M.K.; Meiler, E.; Casanueva, R.; Guijarro, L.; Huss, S.; Lopez-Roman, E.M.; Santos-Villarejo, Á.; Augustyns, K.; et al. Optimization of Hydantoins as Potent Antimycobacterial Decaprenylphosphoryl-β-d-Ribose Oxidase (DprE1) Inhibitors. J. Med. Chem. 2020, 63, 5367–5386. [Google Scholar] [CrossRef] [PubMed]
- Mali, S.N.; Pandey, A.; Bhandare, R.R.; Shaik, A.B. Identification of hydantoin based Decaprenylphosphoryl-β-D-Ribose Oxidase (DprE1) inhibitors as antimycobacterial agents using computational tools. Sci. Rep. 2022, 12, 16368. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Batt, S.M.; Wang, B.; Fu, L.; Qin, R.; Lu, Y.; Li, G.; Besra, G.S.; Huang, H. Discovery of Novel Thiophene-arylamide Derivatives as DprE1 Inhibitors with Potent Antimycobacterial Activities. J. Med. Chem. 2021, 64, 6241–6261. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, J.A.; Alemparte, C.; Wall, I.; Whitehurst, B.C.; Argyrou, A.; Burley, G.; de Dios-Anton, P.; Guijarro, L.; Monteiro, M.C.; Ortega, F.; et al. Mycobacterium tuberculosis Decaprenylphosphoryl-β-d-ribose Oxidase Inhibitors: Expeditious Reconstruction of Suboptimal Hits into a Series with Potent in Vivo Activity. J. Med. Chem. 2020, 63, 2557–2576. [Google Scholar] [CrossRef]
- Ballell, L.; Bates, R.H.; Young, R.J.; Alvarez-Gomez, D.; Alvarez-Ruiz, E.; Barroso, V.; Blanco, D.; Crespo, B.; Escribano, J.; González, R.; et al. Fueling open-source drug discovery: 177 small-molecule leads against tuberculosis. ChemMedChem 2013, 8, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Park, Y.; Engelhart, C.A.; Wallach, J.B.; Schnappinger, D.; Arora, K.; Manikkam, M.; Gac, B.; Wang, H.; Murgolo, N.; et al. Discovery and Structure-Activity-Relationship Study of N-Alkyl-5-hydroxypyrimidinone Carboxamides as Novel Antitubercular Agents Targeting Decaprenylphosphoryl-β-d-ribose 2′-Oxidase. J. Med. Chem. 2018, 61, 9952–9965. [Google Scholar] [CrossRef]
- Gao, Y.; Xie, J.; Tang, R.; Yang, K.; Zhang, Y.; Chen, L.; Li, H. Identification of a pyrimidinetrione derivative as the potent DprE1 inhibitor by structure-based virtual ligand screening. Bioorg. Chem. 2019, 85, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Hariguchi, N.; Chen, X.; Hayashi, Y.; Kawano, Y.; Fujiwara, M.; Matsuba, M.; Shimizu, H.; Ohba, Y.; Nakamura, I.; Kitamoto, R.; et al. OPC-167832, a Novel Carbostyril Derivative with Potent Antituberculosis Activity as a DprE1 Inhibitor. Antimicrob. Agents Chemother. 2020, 64, e02020-19. [Google Scholar] [CrossRef]
- Naik, M.; Humnabadkar, V.; Tantry, S.J.; Panda, M.; Narayan, A.; Guptha, S.; Panduga, V.; Manjrekar, P.; Jena, L.K.; Koushik, K.; et al. 4-aminoquinolone piperidine amides: Noncovalent inhibitors of DprE1 with long residence time and potent antimycobacterial activity. J. Med. Chem. 2014, 57, 5419–5434. [Google Scholar] [CrossRef]
- Neres, J.; Hartkoorn, R.C.; Chiarelli, L.R.; Gadupudi, R.; Pasca, M.R.; Mori, G.; Venturelli, A.; Savina, S.; Makarov, V.; Kolly, G.S.; et al. 2-Carboxyquinoxalines kill mycobacterium tuberculosis through noncovalent inhibition of DprE1. ACS Chem. Biol. 2015, 10, 705–714. [Google Scholar] [CrossRef]
- Whitehurst, B.C.; Young, R.J.; Burley, G.A.; Cacho, M.; Torres, P.; Vela-Gonzalez Del Peral, L. Identification of 2-((2,3-dihydrobenzo[b][1,4]dioxin-6-yl)amino)-N-phenylpropanamides as a novel class of potent DprE1 inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 127192. [Google Scholar] [CrossRef] [PubMed]
- Ezquerra-Aznárez, J.M.; Degiacomi, G.; Gašparovič, H.; Stelitano, G.; Sammartino, J.C.; Korduláková, J.; Governa, P.; Manetti, F.; Pasca, M.R.; Chiarelli, L.R.; et al. The Veterinary Anti-Parasitic Selamectin Is a Novel Inhibitor of the Mycobacterium tuberculosis DprE1 Enzyme. Int. J. Mol. Sci. 2022, 23, 771. [Google Scholar] [CrossRef] [PubMed]
| Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}