
Dnase1 Family in Autoimmunity

 ,
, {kind=link}
{kind=link}
Definition
1. History and Overview of the Dnase1 Family
1.1. Discovery of Dnase1 Family Members
1.2. Evolution of Dnases
1.3. Extranuclear DNA Is Inflammatory
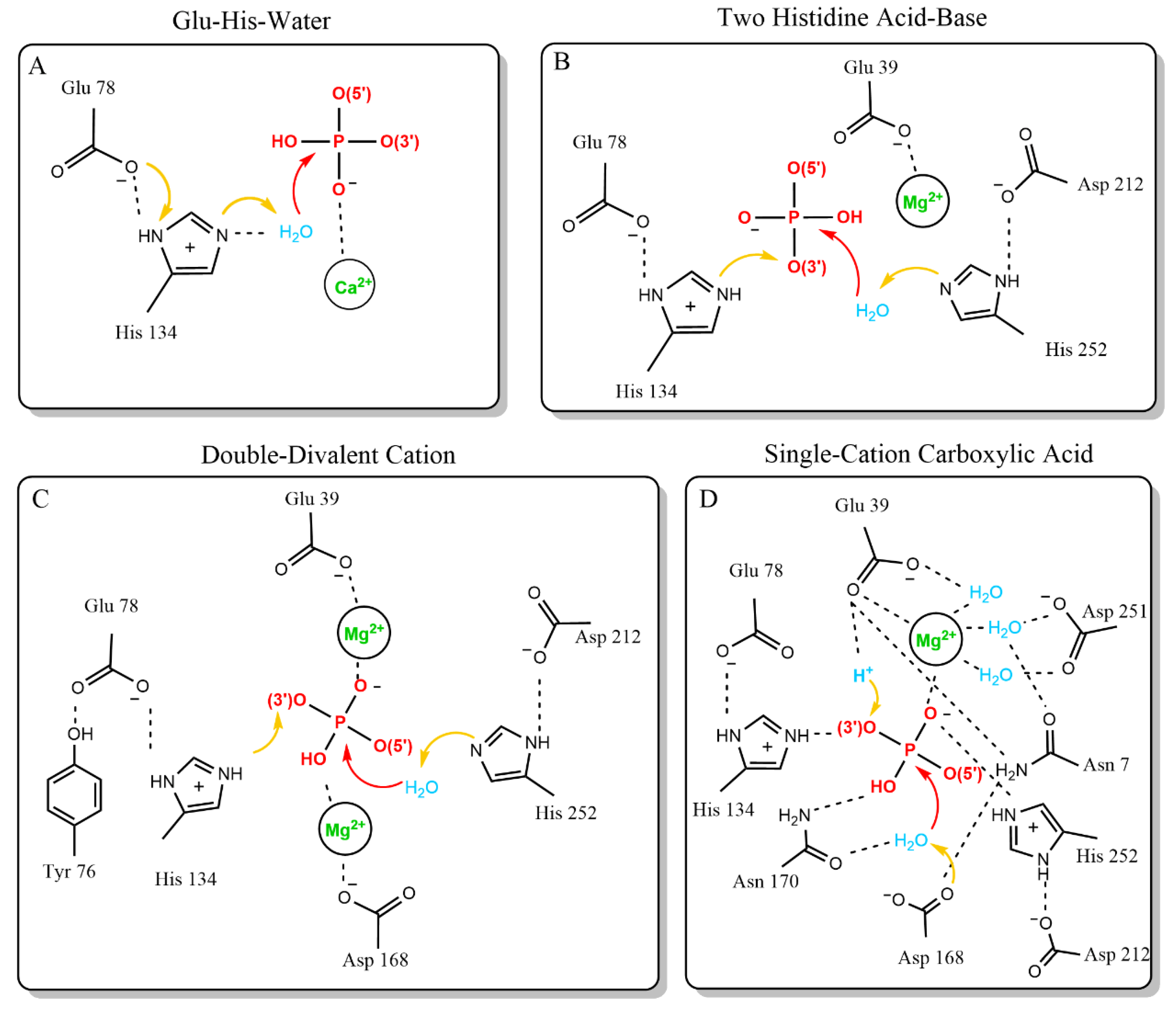
2. Mechanism of Action for the Dnase1 Family
Mechanism of Dnase1 Family Catalysis
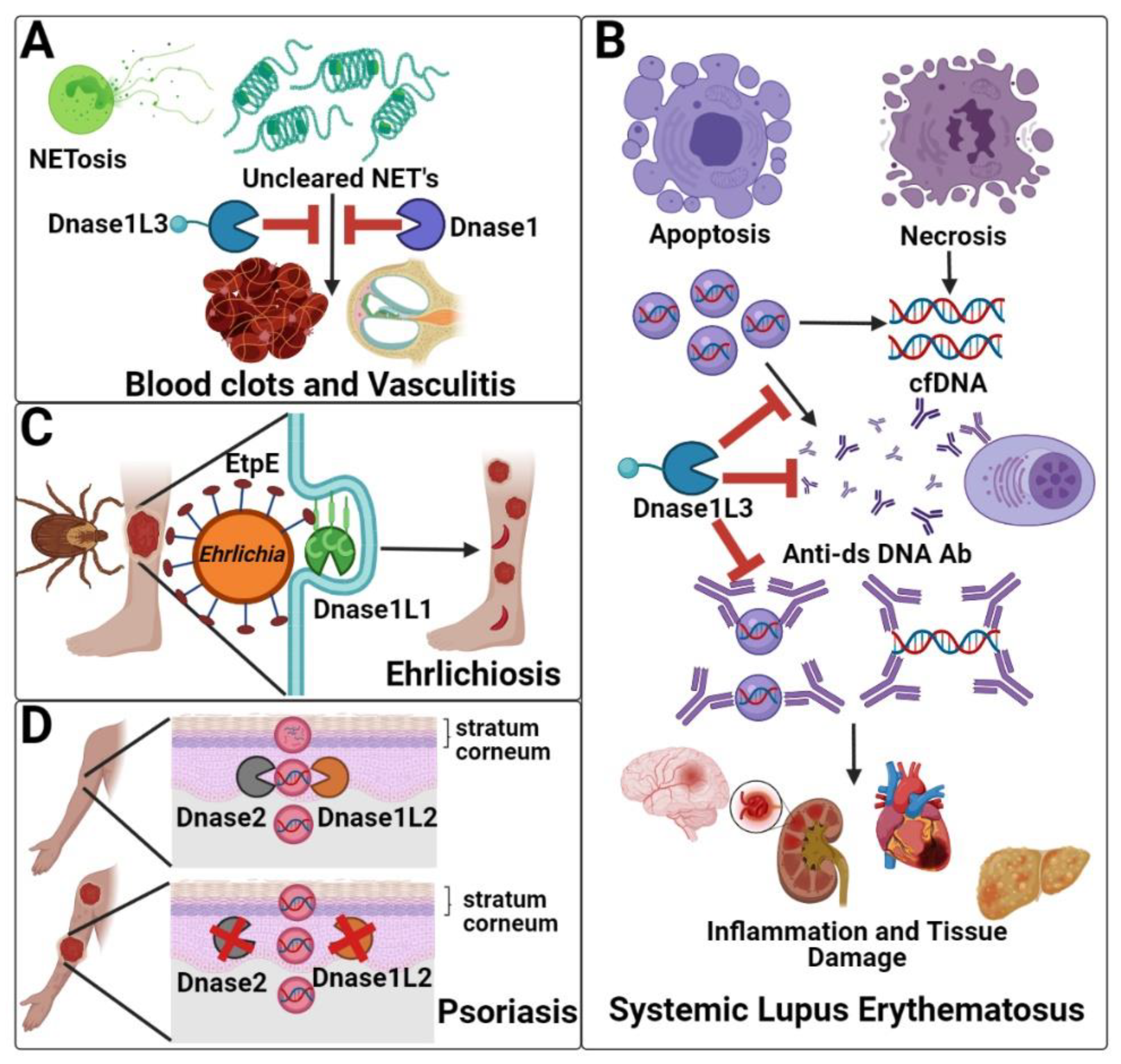
3. The Pathophysiology of the Dnase1 Family
3.1. Clearance of Neutrophil Extracellular Traps (Dnase1/Dnase1L3)
3.2. Systemic Lupus Erythematosus (Dnase1/Dnase1L3)
3.3. Hypocomplementemic Urticarial Vasculitis (Dnase1L3)
3.4. Ehrlichiosis (Dnase1L1)
3.5. Psoriasis (Dnase1L2)
4. Clinical and Experimental Applications of Dnases
4.1. Experimental Applications of Dnase1
4.2. Clinical Applications of Dnase1
4.3. Dnase1 Family Members and Cell-Free DNA as Potential Biomarkers
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Entry Link on the Encyclopedia Platform
References
- Kunitz, M. Crystalline desoxyribonuclease; isolation and general properties; spectrophotometric method for the measurement of desoxyribonuclease activity. J. Gen. Physiol. 1950, 33, 349–362. [Google Scholar] [CrossRef]
- Levene, P.A.; Medigreceanu, F. On Nucleases. J. Biol. Chem 1911, 9, 65–83. [Google Scholar] [CrossRef]
- Cunningham, L.; Laskowski, M. Presence of two different desoxyribonucleode-polymerases in veal kidney. Biochim. Biophys. Acta 1953, 11, 590–591. [Google Scholar] [CrossRef]
- Shiokawa, D.; Tanuma, S. Characterization of human DNase I family endonucleases and activation of DNase gamma during apoptosis. Biochemistry 2001, 40, 143–152. [Google Scholar] [CrossRef]
- MacLea, K.S.; Krieser, R.J.; Eastman, A. A family history of deoxyribonuclease II: Surprises from Trichinella spiralis and Burkholderia pseudomallei. Gene 2003, 305, 1–12. [Google Scholar] [CrossRef]
- Catcheside, D.G.; Holmes, B. The action of enzymes on chromosomes. Symp. Soc. Exp. Biol. 1947, 225–231. [Google Scholar]
- Keyel, P.A. Dnases in health and disease. Dev. Biol. 2017, 429, 1–11. [Google Scholar] [CrossRef]
- Parrish, J.E.; Ciccodicola, A.; Wehhert, M.; Cox, G.F.; Chen, E.; Nelson, D.L. A muscle-specific DNase I-like gene in human Xq28. Hum. Mol. Genet. 1995, 4, 1557–1564. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.M.; Rodin, D.; Nomura, H.; Morton, C.C.; Weremowicz, S.; Schneider, M.C. Identification, localization, and expression of two novel human genes similar to deoxyribonuclease I. Genomics 1997, 42, 507–513. [Google Scholar] [CrossRef]
- Shiokawa, D.; Hirai, M.; Tanuma, S. cDNA cloning of human DNase gamma: Chromosomal localization of its gene and enzymatic properties of recombinant protein. Apoptosis 1998, 3, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.Y.; Pandey, S.; Singh, R.K.; Lin, W.; Ribecco, M.; Borowy-Borowski, H.; Smith, B.; LeBlanc, J.; Walker, P.R.; Sikorska, M. DNaseY: A rat DNaseI-like gene coding for a constitutively expressed chromatin-bound endonuclease. Biochemistry 1998, 37, 10134–10143. [Google Scholar] [CrossRef]
- Shiokawa, D.; Ohyama, H.; Yamada, T.; Takahashi, K.; Tanuma, S. Identification of an endonuclease responsible for apoptosis in rat thymocytes. Eur. J. Biochem. 1994, 226, 23–30. [Google Scholar] [CrossRef]
- Zeng, Z.; Parmelee, D.; Hyaw, H.; Coleman, T.A.; Su, K.; Zhang, J.; Gentz, R.; Ruben, S.; Rosen, C.; Li, Y. Cloning and characterization of a novel human DNase. Biochem. Biophys. Res. Commun. 1997, 231, 499–504. [Google Scholar] [CrossRef]
- Baron, W.F.; Pan, C.Q.; Spencer, S.A.; Ryan, A.M.; Lazarus, R.A.; Baker, K.P. Cloning and characterization of an actin-resistant DNase I-like endonuclease secreted by macrophages. Gene 1998, 215, 291–301. [Google Scholar] [CrossRef]
- Kishi, K.; Yasuda, T.; Takeshita, H. DNase I: Structure, function, and use in medicine and forensic science. Leg. Med. 2001, 3, 69–83. [Google Scholar] [CrossRef]
- Los, M.; Neubuser, D.; Coy, J.F.; Mozoluk, M.; Poustka, A.; Schulze-Osthoff, K. Functional characterization of DNase X, a novel endonuclease expressed in muscle cells. Biochemistry 2000, 39, 7365–7373. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.; Eckhart, L.; Mildner, M.; Jaeger, K.; Buchberger, M.; Ghannadan, M.; Tschachler, E. DNase1L2 degrades nuclear DNA during corneocyte formation. J. Investig. Dermatol. 2007, 127, 24–30. [Google Scholar] [CrossRef]
- Sisirak, V.; Sally, B.; D’Agati, V.; Martinez-Ortiz, W.; Ozcakar, Z.B.; David, J.; Rashidfarrokhi, A.; Yeste, A.; Panea, C.; Chida, A.S.; et al. Digestion of Chromatin in Apoptotic Cell Microparticles Prevents Autoimmunity. Cell 2016, 166, 88–101. [Google Scholar] [CrossRef]
- Ueki, M.; Kimura-Kataoka, K.; Takeshita, H.; Fujihara, J.; Iida, R.; Sano, R.; Nakajima, T.; Kominato, Y.; Kawai, Y.; Yasuda, T. Evaluation of all non-synonymous single nucleotide polymorphisms (SNPs) in the genes encoding human deoxyribonuclease I and I-like 3 as a functional SNP potentially implicated in autoimmunity. FEBS J. 2014, 281, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Lyon, C.J.; Evans, C.J.; Bill, B.R.; Otsuka, A.J.; Aguilera, R.J. The C. elegans apoptotic nuclease NUC-1 is related in sequence and activity to mammalian DNase II. Gene 2000, 252, 147–154. [Google Scholar] [CrossRef]
- Fafandel, M.; Ravlic, S.; Smodlaka, M.; Bihari, N. Deoxyribonucleases (DNases) in the cortex and endosome from the marine sponge Tethya aurantium. Russ. J. Mar. Biol. 2010, 36, 383–389. [Google Scholar] [CrossRef][Green Version]
- Kumar, V. The Trinity of cGAS, TLR9, and ALRs Guardians of the Cellular Galaxy Against Host-Derived Self-DNA. Front. Immunol. 2020, 11, 624597. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Kaplan, M.J.; Radic, M. Neutrophil extracellular traps: Double-edged swords of innate immunity. J. Immunol. 2012, 189, 2689–2695. [Google Scholar] [CrossRef] [PubMed]
- Apel, F.; Andreeva, L.; Knackstedt, L.S.; Streeck, R.; Frese, C.K.; Goosmann, C.; Hopfner, K.P.; Zychlinsky, A. The cytosolic DNA sensor cGAS recognizes neutrophil extracellular traps. Sci. Signal. 2021, 14. [Google Scholar] [CrossRef]
- Soni, C.; Reizis, B. DNA as a self-antigen: Nature and regulation. Curr. Opin. Immunol. 2018, 55, 31–37. [Google Scholar] [CrossRef]
- Suck, D.; Oefner, C.; Kabsch, W. Three-dimensional structure of bovine pancreatic DNase I at 2.5 A resolution. EMBO J. 1984, 3, 2423–2430. [Google Scholar] [CrossRef]
- Napirei, M.; Wulf, S.; Eulitz, D.; Mannherz, H.G.; Kloeckl, T. Comparative characterization of rat deoxyribonuclease 1 (Dnase1) and murine deoxyribonuclease 1-like 3 (Dnase1l3). Biochem. J. 2005, 389, 355–364. [Google Scholar] [CrossRef]
- Napirei, M.; Ludwig, S.; Mezrhab, J.; Klockl, T.; Mannherz, H.G. Murine serum nucleases--contrasting effects of plasmin and heparin on the activities of DNase1 and DNase1-like 3 (DNase1l3). FEBS J. 2009, 276, 1059–1073. [Google Scholar] [CrossRef]
- Wilber, A.; Lu, M.; Schneider, M.C. Deoxyribonuclease I-like III is an inducible macrophage barrier to liposomal transfection. Mol. Ther. 2002, 6, 35–42. [Google Scholar] [CrossRef]
- Galburt, E.A.; Stoddard, B.L. Catalytic mechanisms of restriction and homing endonucleases. Biochemistry 2002, 41, 13851–13860. [Google Scholar] [CrossRef]
- Yang, W. Nucleases: Diversity of structure, function and mechanism. Q. Rev. Biophys. 2011, 44, 1–93. [Google Scholar] [CrossRef]
- Suck, D.; Oefner, C. Structure of DNase I at 2.0 A resolution suggests a mechanism for binding to and cutting DNA. Nature 1986, 321, 620–625. [Google Scholar] [CrossRef]
- Parsiegla, G.; Noguere, C.; Santell, L.; Lazarus, R.A.; Bourne, Y. The structure of human DNase I bound to magnesium and phosphate ions points to a catalytic mechanism common to members of the DNase I-like superfamily. Biochemistry 2012, 51, 10250–10258. [Google Scholar] [CrossRef] [PubMed]
- Weston, S.A.; Lahm, A.; Suck, D. X-ray structure of the DNase I-d(GGTATACC)2 complex at 2.3 A resolution. J. Mol. Biol. 1992, 226, 1237–1256. [Google Scholar] [CrossRef]
- Jones, S.J.; Worrall, A.F.; Connolly, B.A. Site-directed mutagenesis of the catalytic residues of bovine pancreatic deoxyribonuclease I. J. Mol. Biol. 1996, 264, 1154–1163. [Google Scholar] [CrossRef]
- Price, P.A.; Moore, S.; Stein, W.H. Alkylation of a histidine residue at the active site of bovine pancreatic deoxyribonuclease. J. Biol. Chem. 1969, 244, 924–928. [Google Scholar] [CrossRef]
- Oshima, R.G.; Price, P.A. Alkylation of an essential histidine residue in porcine spleen deoxyribonuclease. J. Biol. Chem. 1973, 248, 7522–7526. [Google Scholar] [CrossRef]
- Nielsen, J.E.; McCammon, J.A. Calculating pKa values in enzyme active sites. Protein Sci. Publ. Protein Soc. 2003, 12, 1894–1901. [Google Scholar] [CrossRef]
- Chen, W.J.; Lai, P.J.; Lai, Y.S.; Huang, P.T.; Lin, C.C.; Liao, T.H. Probing the catalytic mechanism of bovine pancreatic deoxyribonuclease I by chemical rescue. Biochem. Biophys. Res. Commun. 2007, 352, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.Q.; Ulmer, J.S.; Herzka, A.; Lazarus, R.A. Mutational analysis of human DNase I at the DNA binding interface: Implications for DNA recognition, catalysis, and metal ion dependence. Protein Sci. Publ. Protein Soc. 1998, 7, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Gueroult, M.; Picot, D.; Abi-Ghanem, J.; Hartmann, B.; Baaden, M. How cations can assist DNase I in DNA binding and hydrolysis. PLoS Comput. Biol. 2010, 6, e1001000. [Google Scholar] [CrossRef]
- Beernink, P.T.; Segelke, B.W.; Hadi, M.Z.; Erzberger, J.P.; Wilson, D.M., 3rd; Rupp, B. Two divalent metal ions in the active site of a new crystal form of human apurinic/apyrimidinic endonuclease, Ape1: Implications for the catalytic mechanism. J. Mol. Biol. 2001, 307, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Mol, C.D.; Izumi, T.; Mitra, S.; Tainer, J.A. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination [corrected]. Nature 2000, 403, 451–456. [Google Scholar] [CrossRef]
- Freudenthal, B.D.; Beard, W.A.; Cuneo, M.J.; Dyrkheeva, N.S.; Wilson, S.H. Capturing snapshots of APE1 processing DNA damage. Nat. Struct. Mol. Biol. 2015, 22, 924–931. [Google Scholar] [CrossRef]
- Shi, G.; Abbott, K.N.; Wu, W.; Salter, R.D.; Keyel, P.A. Dnase1L3 Regulates Inflammasome-Dependent Cytokine Secretion. Front. Immunol. 2017, 8, 522. [Google Scholar] [CrossRef]
- Yamada, Y.; Fujii, T.; Ishijima, R.; Tachibana, H.; Yokoue, N.; Takasawa, R.; Tanuma, S. DR396, an apoptotic DNase gamma inhibitor, attenuates high mobility group box 1 release from apoptotic cells. Bioorg. Med. Chem. 2011, 19, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Dassler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Alcazar, M.; Rangaswamy, C.; Panda, R.; Bitterling, J.; Simsek, Y.J.; Long, A.T.; Bilyy, R.; Krenn, V.; Renne, C.; Renne, T.; et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 2017, 358, 1202–1206. [Google Scholar] [CrossRef] [PubMed]
- Merza, M.; Hartman, H.; Rahman, M.; Hwaiz, R.; Zhang, E.; Renstrom, E.; Luo, L.; Morgelin, M.; Regner, S.; Thorlacius, H. Neutrophil Extracellular Traps Induce Trypsin Activation, Inflammation, and Tissue Damage in Mice With Severe Acute Pancreatitis. Gastroenterology 2015, 149, 1920–1931.e8. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef]
- Englert, H.; Rangaswamy, C.; Deppermann, C.; Sperhake, J.P.; Krisp, C.; Schreier, D.; Gordon, E.; Konrath, S.; Haddad, M.; Pula, G.; et al. Defective NET clearance contributes to sustained FXII activation in COVID-19-associated pulmonary thrombo-inflammation. EBioMedicine 2021, 67, 103382. [Google Scholar] [CrossRef]
- Hakkim, A.; Furnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [PubMed]
- Toma, A.; Darwish, C.; Taylor, M.; Harlacher, J.; Darwish, R. The Use of Dornase Alfa in the Management of COVID-19-Associated Adult Respiratory Distress Syndrome. Crit. Care Res. Pract. 2021, 2021, 8881115. [Google Scholar] [PubMed]
- Tan, E.M.; Schur, P.H.; Carr, R.I.; Kunkel, H.G. Deoxybonucleic acid (DNA) and antibodies to DNA in the serum of patients with systemic lupus erythematosus. J. Clin. Investig. 1966, 45, 1732–1740. [Google Scholar] [CrossRef]
- Rekvig, O.P.; van der Vlag, J.; Seredkina, N. Review: Antinucleosome antibodies: A critical reflection on their specificities and diagnostic impact. Arthritis Rheumatol. 2014, 66, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Relle, M.; Weinmann-Menke, J.; Scorletti, E.; Cavagna, L.; Schwarting, A. Genetics and novel aspects of therapies in systemic lupus erythematosus. Autoimmun. Rev. 2015, 14, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- Napirei, M.; Karsunky, H.; Zevnik, B.; Stephan, H.; Mannherz, H.G.; Moroy, T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat. Genet. 2000, 25, 177–181. [Google Scholar] [CrossRef]
- Al-Mayouf, S.M.; Sunker, A.; Abdwani, R.; Abrawi, S.A.; Almurshedi, F.; Alhashmi, N.; Al Sonbul, A.; Sewairi, W.; Qari, A.; Abdallah, E.; et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat. Genet. 2011, 43, 1186–1188. [Google Scholar] [CrossRef]
- Hartl, J.; Serpas, L.; Wang, Y.; Rashidfarrokhi, A.; Perez, O.A.; Sally, B.; Sisirak, V.; Soni, C.; Khodadadi-Jamayran, A.; Tsirigos, A.; et al. Autoantibody-mediated impairment of DNASE1L3 activity in sporadic systemic lupus erythematosus. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef]
- Almlof, J.C.; Nystedt, S.; Leonard, D.; Eloranta, M.L.; Grosso, G.; Sjowall, C.; Bengtsson, A.A.; Jonsen, A.; Gunnarsson, I.; Svenungsson, E.; et al. Whole-genome sequencing identifies complex contributions to genetic risk by variants in genes causing monogenic systemic lupus erythematosus. Hum. Genet. 2019, 138, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Ozcakar, Z.B.; Foster, J., 2nd; Diaz-Horta, O.; Kasapcopur, O.; Fan, Y.S.; Yalcinkaya, F.; Tekin, M. DNASE1L3 mutations in hypocomplementemic urticarial vasculitis syndrome. Arthritis Rheum. 2013, 65, 2183–2189. [Google Scholar] [CrossRef]
- Yasutomo, K.; Horiuchi, T.; Kagami, S.; Tsukamoto, H.; Hashimura, C.; Urushihara, M.; Kuroda, Y. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat. Genet. 2001, 28, 313–314. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.C., Jr.; Manzi, S.; Yarboro, C.; Rairie, J.; McInnes, I.; Averthelyi, D.; Sinicropi, D.; Hale, V.G.; Balow, J.; Austin, H.; et al. Recombinant human Dnase I (rhDNase) in patients with lupus nephritis. Lupus 1999, 8, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Macanovic, M.; Sinicropi, D.; Shak, S.; Baughman, S.; Thiru, S.; Lachmann, P.J. The treatment of systemic lupus erythematosus (SLE) in NZB/W F1 hybrid mice; studies with recombinant murine DNase and with dexamethasone. Clin. Exp. Immunol. 1996, 106, 243–252. [Google Scholar] [CrossRef]
- Lachmann, P.J. Allergic reactions, connective tissue, and disease. Sci. Basis Med. Annu. Rev. 1967, 36–58. [Google Scholar]
- Wisnieski, J.J.; Baer, A.N.; Christensen, J.; Cupps, T.R.; Flagg, D.N.; Jones, J.V.; Katzenstein, P.L.; McFadden, E.R.; McMillen, J.J.; Pick, M.A.; et al. Hypocomplementemic urticarial vasculitis syndrome. Clinical and serologic findings in 18 patients. Medicine 1995, 74, 24–41. [Google Scholar] [CrossRef]
- Hamad, A.; Jithpratuck, W.; Krishnaswamy, G. Urticarial vasculitis and associated disorders. Ann. Allergy Asthma Immunol. 2017, 118, 394–398. [Google Scholar] [CrossRef]
- Ion, O.; Obrisca, B.; Ismail, G.; Sorohan, B.; Balanica, S.; Mircescu, G.; Sinescu, I. Kidney Involvement in Hypocomplementemic Urticarial Vasculitis Syndrome-A Case-Based Review. J. Clin. Med. 2020, 9, 2131. [Google Scholar] [CrossRef]
- Ueki, M.; Takeshita, H.; Fujihara, J.; Iida, R.; Yuasa, I.; Kato, H.; Panduro, A.; Nakajima, T.; Kominato, Y.; Yasuda, T. Caucasian-specific allele in non-synonymous single nucleotide polymorphisms of the gene encoding deoxyribonuclease I-like 3, potentially relevant to autoimmunity, produces an inactive enzyme. Clin. Chim. Acta Int. J. Clin. Chem. 2009, 407, 20–24. [Google Scholar] [CrossRef]
- Stevenson, H.L.; Estes, M.D.; Thirumalapura, N.R.; Walker, D.H.; Ismail, N. Natural killer cells promote tissue injury and systemic inflammatory responses during fatal Ehrlichia-induced toxic shock-like syndrome. Am. J. Pathol. 2010, 177, 766–776. [Google Scholar] [CrossRef]
- Thomas, R.J.; Dumler, J.S.; Carlyon, J.A. Current management of human granulocytic anaplasmosis, human monocytic ehrlichiosis and Ehrlichia ewingii ehrlichiosis. Expert Rev. Anti Infect. Ther. 2009, 7, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.B.; Walker, D.H. A Tick Vector Transmission Model of Monocytotropic Ehrlichiosis. J. Infect. Dis. 2015, 212, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Rikihisa, Y. Molecular Pathogenesis of Ehrlichia chaffeensis Infection. Annu. Rev. Microbiol. 2015, 69, 283–304. [Google Scholar] [CrossRef] [PubMed]
- Shiokawa, D.; Shika, Y.; Saito, K.; Yamazaki, K.; Tanuma, S. Physical and biochemical properties of mammalian DNase X proteins: Non-AUG translation initiation of porcine and bovine mRNAs for DNase X. Biochem. J. 2005, 392, 511–517. [Google Scholar] [CrossRef][Green Version]
- Shiokawa, D.; Matsushita, T.; Shika, Y.; Shimizu, M.; Maeda, M.; Tanuma, S. DNase X is a glycosylphosphatidylinositol-anchored membrane enzyme that provides a barrier to endocytosis-mediated transfer of a foreign gene. J. Biol. Chem. 2007, 282, 17132–17140. [Google Scholar] [CrossRef]
- Teymournejad, O.; Lin, M.; Rikihisa, Y. Ehrlichia chaffeensis and Its Invasin EtpE Block Reactive Oxygen Species Generation by Macrophages in a DNase X-Dependent Manner. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Mohan Kumar, D.; Yamaguchi, M.; Miura, K.; Lin, M.; Los, M.; Coy, J.F.; Rikihisa, Y. Ehrlichia chaffeensis uses its surface protein EtpE to bind GPI-anchored protein DNase X and trigger entry into mammalian cells. PLoS Pathog. 2013, 9, e1003666. [Google Scholar] [CrossRef]
- Kallick, C.A. Diabetic retinopathy and ehrlichia: The possible relationship. Med. Hypothesis Discov. Innov. Ophthalmol. 2012, 1, 33–36. [Google Scholar]
- Brumpton, B.M.; Ferreira, M.A. Multivariate eQTL mapping uncovers functional variation on the X-chromosome associated with complex disease traits. Hum. Genet. 2016, 135, 827–839. [Google Scholar] [CrossRef]
- Zhu, B.; Gong, Y.; Chen, P.; Zhang, H.; Zhao, T.; Li, P. Increased DNase I activity in diabetes might be associated with injury of pancreas. Mol. Cell. Biochem. 2014, 393, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Brady, S.P. Parakeratosis. J. Am. Acad. Dermatol. 2004, 50, 77–84. [Google Scholar] [CrossRef]
- Liang, Y.; Sarkar, M.K.; Tsoi, L.C.; Gudjonsson, J.E. Psoriasis: A mixed autoimmune and autoinflammatory disease. Curr. Opin. Immunol. 2017, 49, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.; Szabo, S.; Scherz, J.; Jaeger, K.; Rossiter, H.; Buchberger, M.; Ghannadan, M.; Hermann, M.; Theussl, H.C.; Tobin, D.J.; et al. Essential role of the keratinocyte-specific endonuclease DNase1L2 in the removal of nuclear DNA from hair and nails. J. Investig. Dermatol. 2011, 131, 1208–1215. [Google Scholar] [CrossRef]
- Ueki, M.; Takeshita, H.; Kimura-Kataoka, K.; Fujihara, J.; Iida, R.; Yasuda, T. Identification of functional SNPs potentially served as a genetic risk factor for the pathogenesis of parakeratosis in the gene encoding human deoxyribonuclease I-like 2 (DNase 1L2) implicated in terminal differentiation of keratinocytes. Gene 2015, 561, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Manils, J.; Fischer, H.; Climent, J.; Casas, E.; Garcia-Martinez, C.; Bas, J.; Sukseree, S.; Vavouri, T.; Ciruela, F.; de Anta, J.M.; et al. Double deficiency of Trex2 and DNase1L2 nucleases leads to accumulation of DNA in lingual cornifying keratinocytes without activating inflammatory responses. Sci. Rep. 2017, 7, 11902. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.; Fumicz, J.; Rossiter, H.; Napirei, M.; Buchberger, M.; Tschachler, E.; Eckhart, L. Holocrine Secretion of Sebum Is a Unique DNase2-Dependent Mode of Programmed Cell Death. J. Investig. Dermatol. 2017, 137, 587–594. [Google Scholar] [CrossRef]
- Galas, D.J.; Schmitz, A. DNAse footprinting: A simple method for the detection of protein-DNA binding specificity. Nucleic Acids Res. 1978, 5, 3157–3170. [Google Scholar] [CrossRef]
- Shak, S. Aerosolized recombinant human DNase I for the treatment of cystic fibrosis. Chest 1995, 107, 65S–70S. [Google Scholar] [CrossRef]
- Shak, S.; Capon, D.J.; Hellmiss, R.; Marsters, S.A.; Baker, C.L. Recombinant human DNase I reduces the viscosity of cystic fibrosis sputum. Proc. Natl. Acad. Sci. USA 1990, 87, 9188–9192. [Google Scholar] [CrossRef]
- Shah, P.L.; Scott, S.F.; Knight, R.A.; Marriott, C.; Ranasinha, C.; Hodson, M.E. In vivo effects of recombinant human DNase I on sputum in patients with cystic fibrosis. Thorax 1996, 51, 119–125. [Google Scholar] [CrossRef]
- Delfino, D.; Mori, G.; Rivetti, C.; Grigoletto, A.; Bizzotto, G.; Cavozzi, C.; Malatesta, M.; Cavazzini, D.; Pasut, G.; Percudani, R. Actin-Resistant DNase1L2 as a Potential Therapeutics for CF Lung Disease. Biomolecules 2021, 11, 410. [Google Scholar] [CrossRef] [PubMed]
- Cagliani, J.; Yang, W.L.; Brenner, M.; Wang, P. Deoxyribonuclease Reduces Tissue Injury and Improves Survival after Hemorrhagic Shock. J. Surg. Res. 2020, 249, 104–113. [Google Scholar] [CrossRef] [PubMed]
- da Cunha, A.A.; Nunez, N.K.; de Souza, R.G.; Moraes Vargas, M.H.; Silveira, J.S.; Antunes, G.L.; Durante Lda, S.; Porto, B.N.; Marczak, E.S.; Jones, M.H.; et al. Recombinant human deoxyribonuclease therapy improves airway resistance and reduces DNA extracellular traps in a murine acute asthma model. Exp. Lung. Res. 2016, 42, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Boogaard, R.; de Jongste, J.C.; Merkus, P.J. Pharmacotherapy of impaired mucociliary clearance in non-CF pediatric lung disease. A review of the literature. Pediatr. Pulmonol. 2007, 42, 989–1001. [Google Scholar] [CrossRef] [PubMed]
- Laukova, L.; Konecna, B.; Janovicova, L.; Vlkova, B.; Celec, P. Deoxyribonucleases and Their Applications in Biomedicine. Biomolecules 2020, 10, 1036. [Google Scholar] [CrossRef] [PubMed]
- Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; De Meyer, S.F.; Bhandari, A.A.; Wagner, D.D. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 2012, 10, 136–144. [Google Scholar] [CrossRef]
- Vokalova, L.; Laukova, L.; Conka, J.; Meliskova, V.; Borbelyova, V.; Babickova, J.; Tothova, L.; Hodosy, J.; Vlkova, B.; Celec, P. Deoxyribonuclease partially ameliorates thioacetamide-induced hepatorenal injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G457–G463. [Google Scholar] [CrossRef]
- Takada, S.; Watanabe, T.; Mizuta, R. DNase gamma-dependent DNA fragmentation causes karyolysis in necrotic hepatocyte. J. Vet. Med. Sci. 2020, 82, 23–26. [Google Scholar] [CrossRef]
- Tetz, V.; Tetz, G. Effect of deoxyribonuclease I treatment for dementia in end-stage Alzheimer’s disease: A case report. J. Med. Case Rep. 2016, 10, 131. [Google Scholar] [CrossRef]
- Mayes, M.D.; Bossini-Castillo, L.; Gorlova, O.; Martin, J.E.; Zhou, X.; Chen, W.V.; Assassi, S.; Ying, J.; Tan, F.K.; Arnett, F.C.; et al. Immunochip analysis identifies multiple susceptibility loci for systemic sclerosis. Am. J. Hum. Genet. 2014, 94, 47–61. [Google Scholar] [CrossRef]
- Zochling, J.; Newell, F.; Charlesworth, J.C.; Leo, P.; Stankovich, J.; Cortes, A.; Zhou, Y.; Stevens, W.; Sahhar, J.; Roddy, J.; et al. An Immunochip-based interrogation of scleroderma susceptibility variants identifies a novel association at DNASE1L3. Arthritis Res. Ther. 2014, 16, 438. [Google Scholar] [CrossRef]
- Westra, H.J.; Martinez-Bonet, M.; Onengut-Gumuscu, S.; Lee, A.; Luo, Y.; Teslovich, N.; Worthington, J.; Martin, J.; Huizinga, T.; Klareskog, L.; et al. Fine-mapping and functional studies highlight potential causal variants for rheumatoid arthritis and type 1 diabetes. Nat. Genet. 2018, 50, 1366–1374. [Google Scholar] [CrossRef]
- Lenert, A.; Fardo, D.W. Detecting novel micro RNAs in rheumatoid arthritis with gene-based association testing. Clin. Exp. Rheumatol. 2017, 35, 586–592. [Google Scholar]
- Zhao, Q.; Yang, C.; Wang, J.; Li, Y.; Yang, P. Serum level of DNase1l3 in patients with dermatomyositis/polymyositis, systemic lupus erythematosus and rheumatoid arthritis, and its association with disease activity. Clin. Exp. Med. 2017, 17, 459–465. [Google Scholar] [CrossRef]
- Malickova, K.; Duricova, D.; Bortlik, M.; Hruskova, Z.; Svobodova, B.; Machkova, N.; Komarek, V.; Fucikova, T.; Janatkova, I.; Zima, T.; et al. Impaired deoxyribonuclease I activity in patients with inflammatory bowel diseases. Autoimmune Dis. 2011, 2011, 945861. [Google Scholar]
- Wang, K.; Gao, M.; Yang, M.; Meng, F.; Li, D.; Lu, R.; Wang, Y.; Zhuang, H.; Li, M.; Cheng, G.; et al. Transcriptome analysis of bronchoalveolar lavage fluid from children with severe Mycoplasma pneumoniae pneumonia reveals novel gene expression and immunodeficiency. Hum. Genom. 2017, 11, 4. [Google Scholar] [CrossRef]
- Berthon, B.S.; Gibson, P.G.; Wood, L.G.; MacDonald-Wicks, L.K.; Baines, K.J. A sputum gene expression signature predicts oral corticosteroid response in asthma. Eur. Respir. J. 2017, 49, 1700180. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Luis, E.; Lorenzo-Diaz, F.; Samedy-Bates, L.A.; Eng, C.; Villar, J.; Rodriguez-Santana, J.R.; Burchard, E.G.; Pino-Yanes, M. A deoxyribonuclease 1-like 3 genetic variant associates with asthma exacerbations. J. Allergy Clin. Immunol. 2021, 147, 1095–1097 e1010. [Google Scholar] [CrossRef] [PubMed]
- Kawai, Y.; Yoshida, M.; Arakawa, K.; Kumamoto, T.; Morikawa, N.; Masamura, K.; Tada, H.; Ito, S.; Hoshizaki, H.; Oshima, S.; et al. Diagnostic use of serum deoxyribonuclease I activity as a novel early-phase marker in acute myocardial infarction. Circulation 2004, 109, 2398–2400. [Google Scholar] [CrossRef] [PubMed]
- Kumamoto, T.; Kawai, Y.; Arakawa, K.; Morikawa, N.; Kuribara, J.; Tada, H.; Taniguchi, K.; Tatami, R.; Miyamori, I.; Kominato, Y.; et al. Association of Gln222Arg polymorphism in the deoxyribonuclease I (DNase I) gene with myocardial infarction in Japanese patients. Eur. Heart J. 2006, 27, 2081–2087. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ma, H.; Li, X.; Mo, X.; Zhang, H.; Yang, L.; Deng, Y.; Yan, Y.; Yang, G.; Liu, X.; et al. DNASE1L3 as an indicator of favorable survival in hepatocellular carcinoma patients following resection. Aging 2020, 12, 1171–1185. [Google Scholar] [CrossRef]
- Bhalla, S.; Chaudhary, K.; Kumar, R.; Sehgal, M.; Kaur, H.; Sharma, S.; Raghava, G.P. Gene expression-based biomarkers for discriminating early and late stage of clear cell renal cancer. Sci. Rep. 2017, 7, 44997. [Google Scholar] [CrossRef]
- Deng, Z.; Xiao, M.; Du, D.; Luo, N.; Liu, D.; Liu, T.; Lian, D.; Peng, J. DNASE1L3 as a Prognostic Biomarker Associated with Immune Cell Infiltration in Cancer. OncoTargets Ther. 2021, 14, 2003–2017. [Google Scholar] [CrossRef]
- Ouyang, B.; Xie, Q.Q.; Huang, W.; Wang, L.; Tang, S.; Fu, J. Diagnostic Value of Serum DNASE1L3 in Hepatitis B Virus-Related Hepatocellular Carcinoma. Clin. Lab. 2021, 67. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Xie, T.; Ding, S.C.; Zhou, Z.; Cheng, S.H.; Chan, R.W.Y.; Lee, W.S.; Peng, W.; Wong, J.; Wong, V.W.S.; et al. Detection and characterization of jagged ends of double-stranded DNA in plasma. Genome Res. 2020, 30, 1144–1153. [Google Scholar] [CrossRef]
- Chan, R.W.Y.; Serpas, L.; Ni, M.; Volpi, S.; Hiraki, L.T.; Tam, L.S.; Rashidfarrokhi, A.; Wong, P.C.H.; Tam, L.H.P.; Wang, Y.; et al. Plasma DNA Profile Associated with DNASE1L3 Gene Mutations: Clinical Observations, Relationships to Nuclease Substrate Preference, and In Vivo Correction. Am. J. Hum. Genet. 2020, 107, 882–894. [Google Scholar] [CrossRef]
- Badeau, M.; Lindsay, C.; Blais, J.; Nshimyumukiza, L.; Takwoingi, Y.; Langlois, S.; Legare, F.; Giguere, Y.; Turgeon, A.F.; Witteman, W.; et al. Genomics-based non-invasive prenatal testing for detection of fetal chromosomal aneuploidy in pregnant women. Cochrane Database Syst. Rev. 2017, 11, CD011767. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Werlang, A.; Cheng, W.; Lanes, A.; Wen, S.W.; Walker, M. Association between Levels of Total Cell-Free DNA and Development of Preeclampsia-A Literature Review. AJP Rep. 2021, 11, e38–e48. [Google Scholar] [PubMed]
- Alix-Panabieres, C.; Pantel, K. Liquid Biopsy: From Discovery to Clinical Application. Cancer Discov. 2021, 11, 858–873. [Google Scholar] [CrossRef]
- Vrablicova, Z.; Tomova, K.; Tothova, L.; Babickova, J.; Gromova, B.; Konecna, B.; Liptak, R.; Hlavaty, T.; Gardlik, R. Nuclear and Mitochondrial Circulating Cell-Free DNA Is Increased in Patients With Inflammatory Bowel Disease in Clinical Remission. Front. Med. 2020, 7, 593316. [Google Scholar] [CrossRef] [PubMed]
- Duvvuri, B.; Lood, C. Cell-Free DNA as a Biomarker in Autoimmune Rheumatic Diseases. Front Immunol 2019, 10, 502. [Google Scholar] [CrossRef] [PubMed]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res 1977, 37, 646–650. [Google Scholar]
- Volik, S.; Alcaide, M.; Morin, R.D.; Collins, C. Cell-free DNA (cfDNA): Clinical Significance and Utility in Cancer Shaped By Emerging Technologies. Mol. Cancer Res. MCR 2016, 14, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Bronkhorst, A.J.; Ungerer, V.; Holdenrieder, S. The emerging role of cell-free DNA as a molecular marker for cancer management. Biomol. Detect. Quantif. 2019, 17, 100087. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Engavale, M.; McCord, J.; Mapp, B.; Nzimulinda, N.; Bengtson, E.; Sutton, R.B.; Keyel, P.A. Dnase1 Family in Autoimmunity. Encyclopedia 2021, 1, 527-541. https://doi.org/10.3390/encyclopedia1030044
Engavale M, McCord J, Mapp B, Nzimulinda N, Bengtson E, Sutton RB, Keyel PA. Dnase1 Family in Autoimmunity. Encyclopedia. 2021; 1(3):527-541. https://doi.org/10.3390/encyclopedia1030044
Chicago/Turabian StyleEngavale, Minal, Jon McCord, Britney Mapp, Nadine Nzimulinda, Elisabeth Bengtson, R. Bryan Sutton, and Peter A. Keyel. 2021. "Dnase1 Family in Autoimmunity" Encyclopedia 1, no. 3: 527-541. https://doi.org/10.3390/encyclopedia1030044
APA StyleEngavale, M., McCord, J., Mapp, B., Nzimulinda, N., Bengtson, E., Sutton, R. B., & Keyel, P. A. (2021). Dnase1 Family in Autoimmunity. Encyclopedia, 1(3), 527-541. https://doi.org/10.3390/encyclopedia1030044