
ESCRT Protein VPS4A Is Required for the Formation of Replication Centers and Replication of Human Coronavirus 229E (HCoV-229E)

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. 229E Virus Infections, Growth Curves, and Virus Titrations
2.3. TCID50 Titer Assays
2.4. Lentivirus Production and Cell Selection
2.5. RNA Isolation and RT-qPCR
2.6. Antibodies and Reagents
2.7. Immunofluorescence Staining and Imaging
2.8. Transmission Electron Microscopy
2.9. smFISH of Viral RNA, Imaging, and Analysis
2.10. Statistical Analysis
3. Results
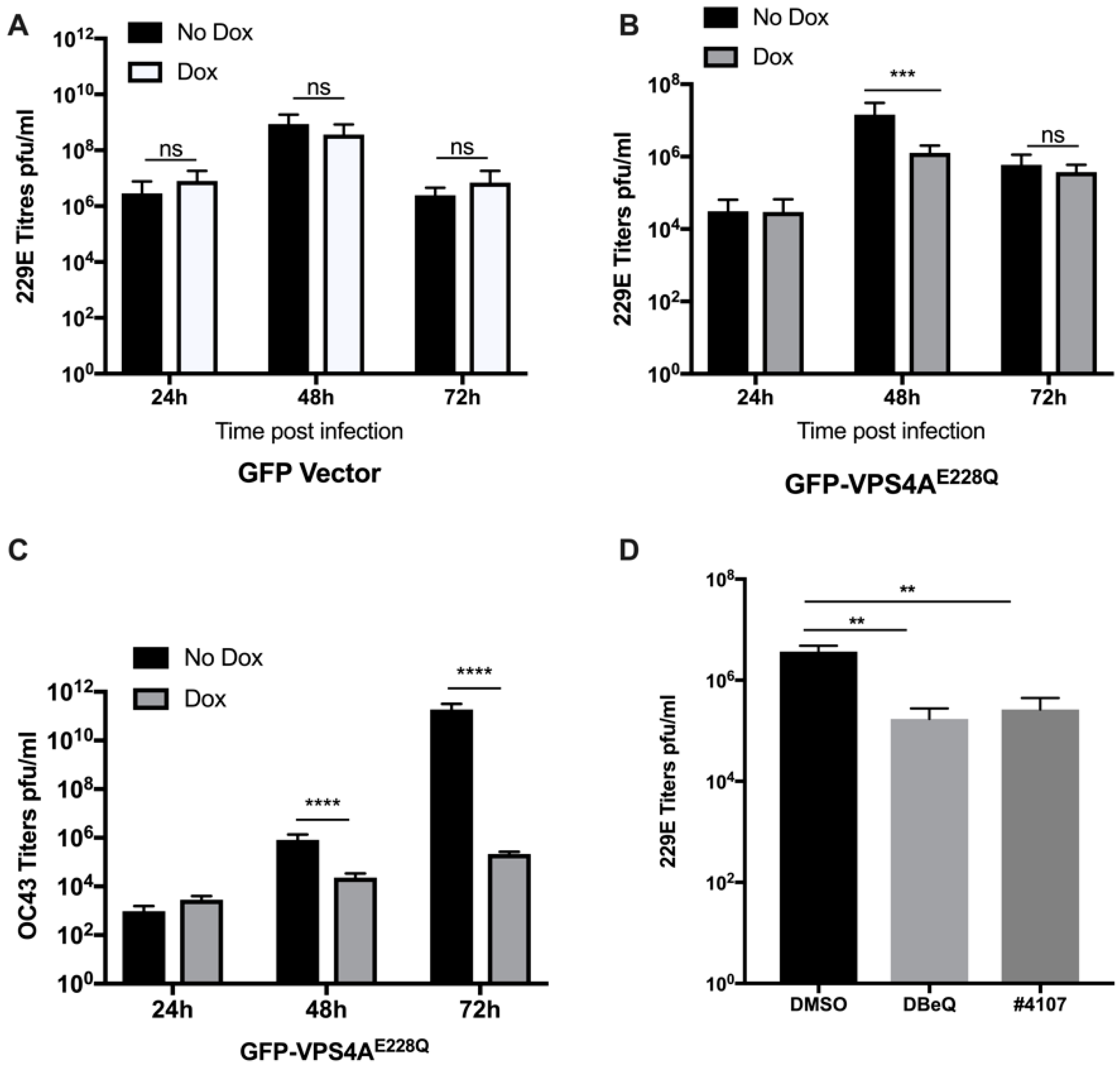
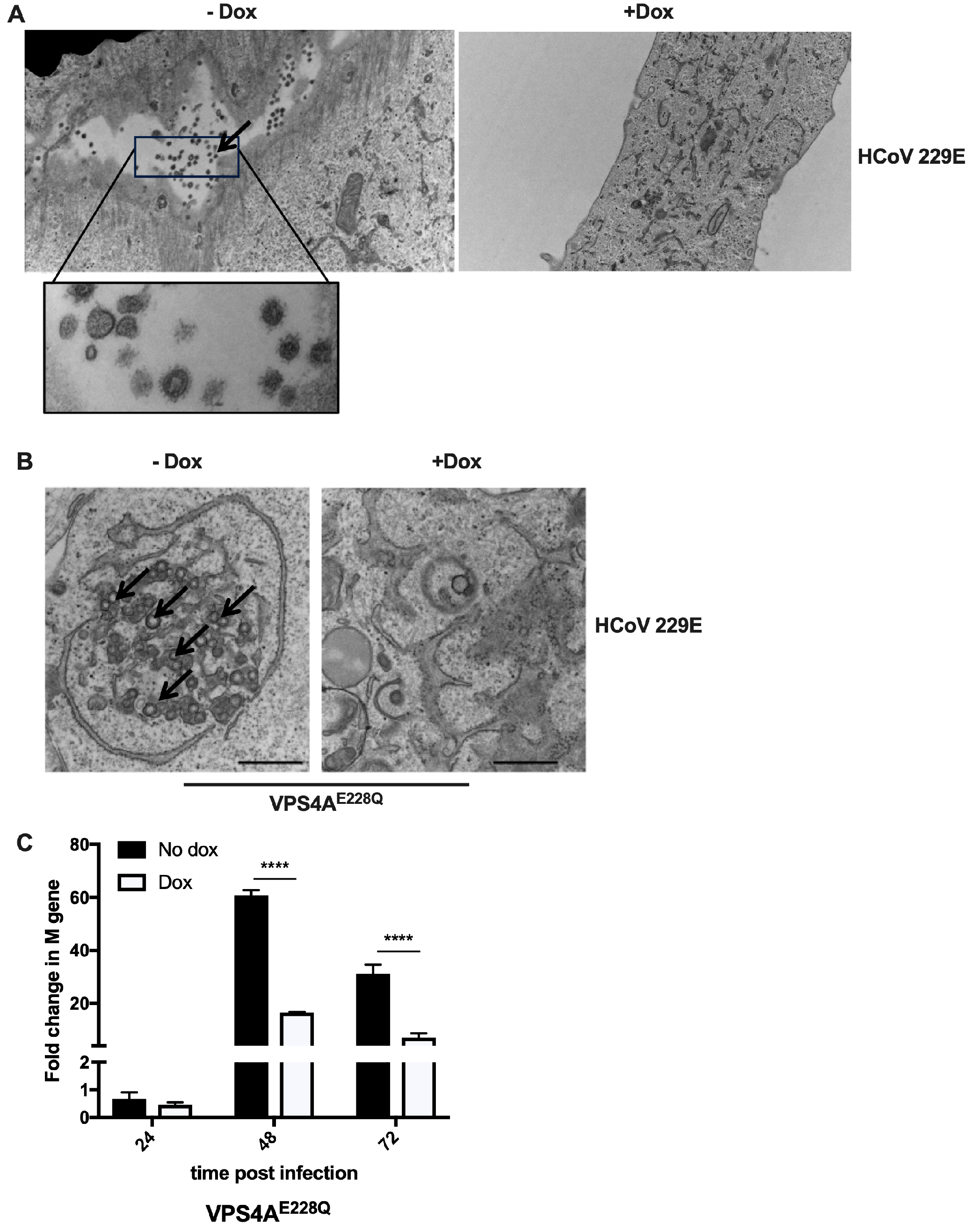
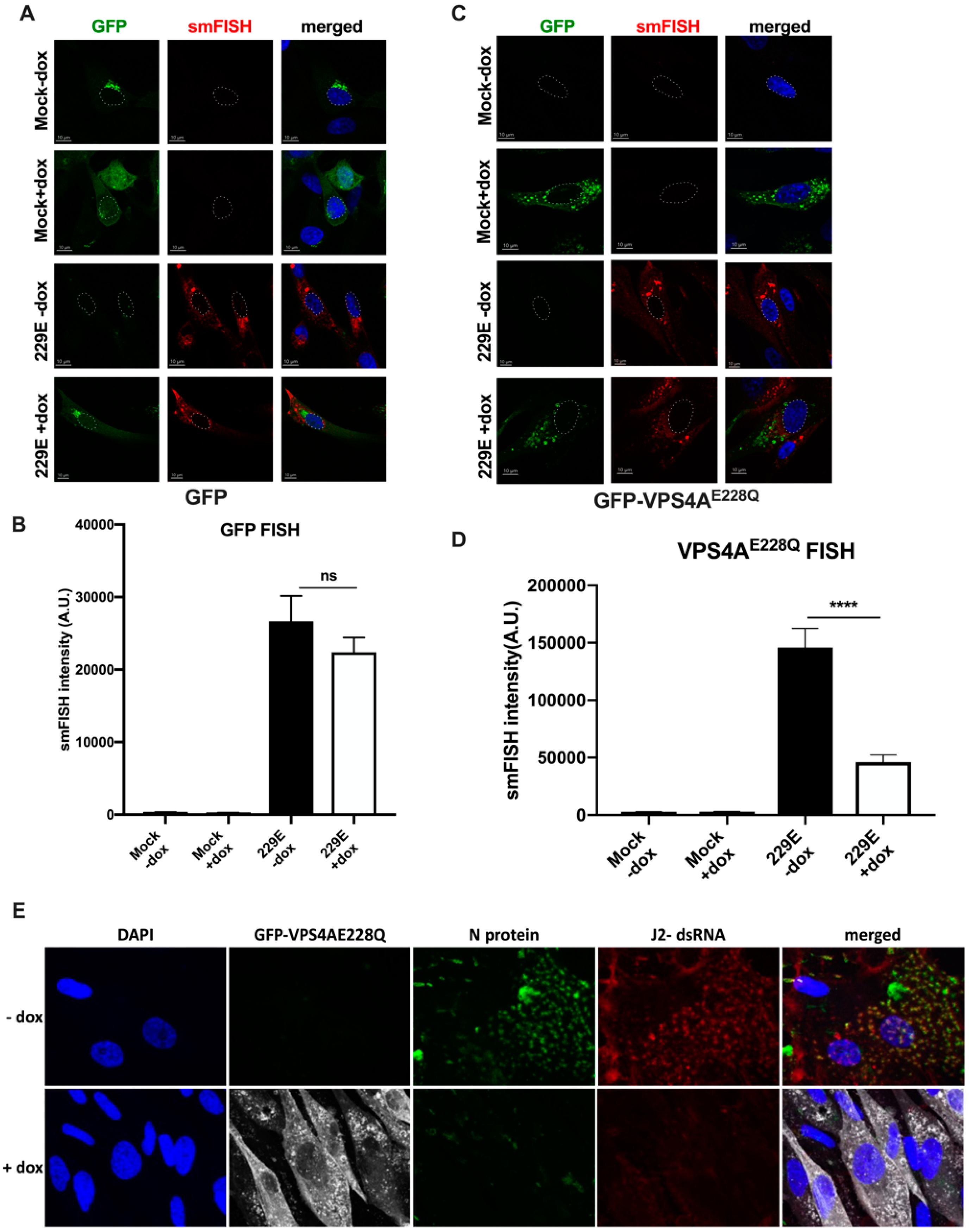
3.1. Functional VPS4A Was Required for the Formation of HCoV-229E Replication Centers
3.2. HCoV-229E Infection Triggers an Inflammatory Response in the Absence of a Functional VPS4A
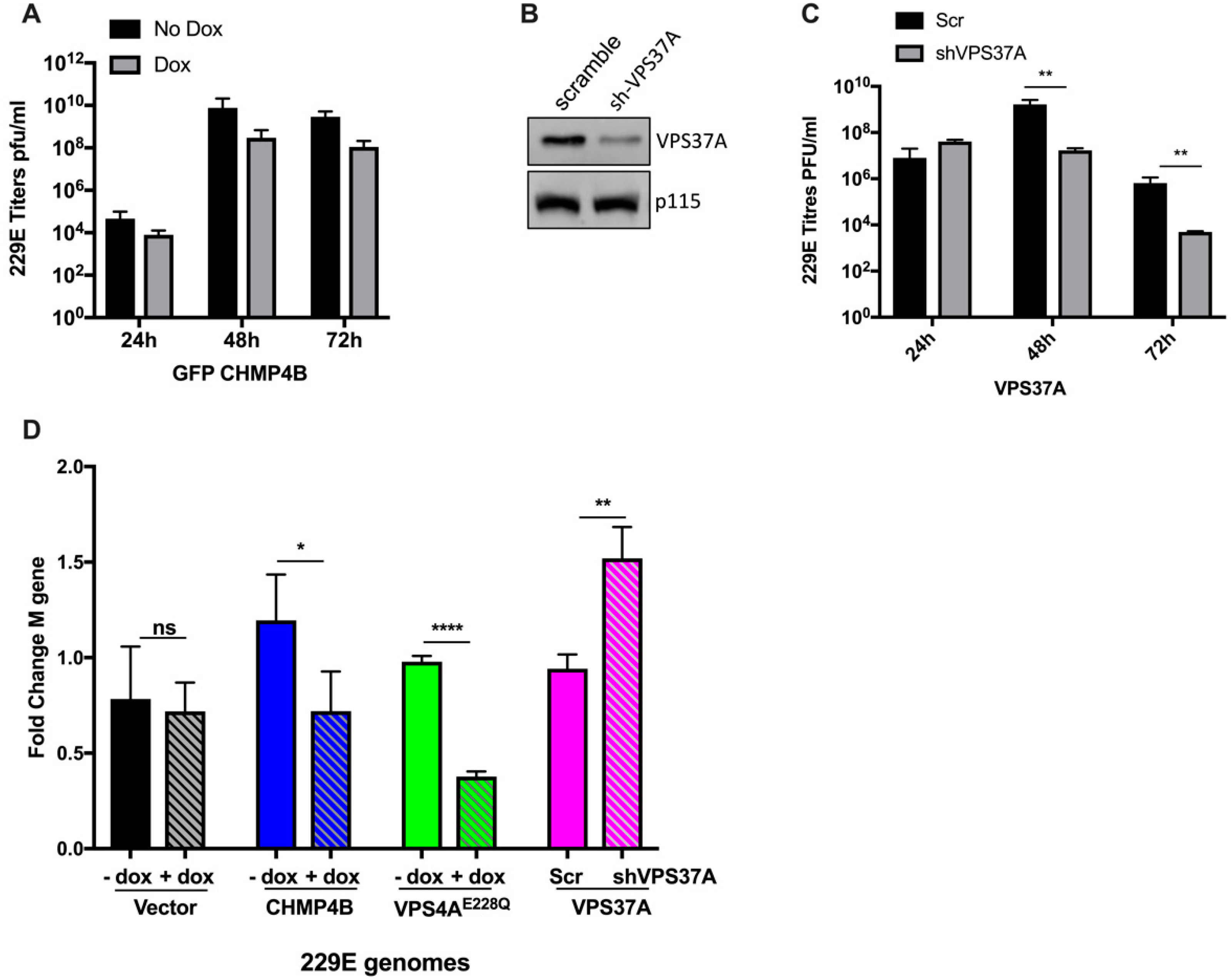
3.3. Effect of Other ESCRT Proteins on 229E Virus Growth
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pawliczek, T.; Crump, C.M. Herpes simplex virus type 1 production requires a functional ESCRT-III complex but is independent of TSG101 and ALIX expression. J. Virol. 2009, 83, 11254–11264. [Google Scholar] [CrossRef] [PubMed]
- Demirov, D.G.; Ono, A.; Orenstein, J.M.; Freed, E.O. Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc. Natl. Acad. Sci. USA 2002, 99, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Goila-Gaur, R.; Demirov, D.G.; Orenstein, J.M.; Ono, A.; Freed, E.O. Defects in human immunodeficiency virus budding and endosomal sorting induced by TSG101 overexpression. J. Virol. 2003, 77, 6507–6519. [Google Scholar] [CrossRef]
- Watanabe, T.; Sorensen, E.M.; Naito, A.; Schott, M.; Kim, S.; Ahlquist, P. Involvement of host cellular multivesicular body functions in hepatitis B virus budding. Proc. Natl. Acad. Sci. USA 2007, 104, 10205–10210. [Google Scholar] [CrossRef]
- Prange, R. Host factors involved in hepatitis B virus maturation, assembly, and egress. Med. Microbiol. Immunol. 2012, 201, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Honeychurch, K.M.; Yang, G.; Jordan, R.; Hruby, D.E. The vaccinia virus F13L YPPL motif is required for efficient release of extracellular enveloped virus. J. Virol. 2007, 81, 7310–7315. [Google Scholar] [CrossRef]
- Streck, N.T.; Carmichael, J.; Buchkovich, N.J. Nonenvelopment Role for the ESCRT-III Complex during Human Cytomegalovirus Infection. J. Virol. 2018, 92, e02096-17. [Google Scholar] [CrossRef]
- Tandon, R.; AuCoin, D.P.; Mocarski, E.S. Human cytomegalovirus exploits ESCRT machinery in the process of virion maturation. J. Virol. 2009, 83, 10797–10807. [Google Scholar] [CrossRef]
- Timmins, J.; Schoehn, G.; Ricard-Blum, S.; Scianimanico, S.; Vernet, T.; Ruigrok, R.W.; Weissenhorn, W. Ebola virus matrix protein VP40 interaction with human cellular factors Tsg101 and Nedd4. J. Mol. Biol. 2003, 326, 493–502. [Google Scholar] [CrossRef]
- Urata, S.; Noda, T.; Kawaoka, Y.; Morikawa, S.; Yokosawa, H.; Yasuda, J. Interaction of Tsg101 with Marburg virus VP40 depends on the PPPY motif, but not the PT/SAP motif as in the case of Ebola virus, and Tsg101 plays a critical role in the budding of Marburg virus-like particles induced by VP40, NP, and GP. J. Virol. 2007, 81, 4895–4899. [Google Scholar] [CrossRef]
- Itakura, Y.; Tabata, K.; Saito, T.; Intaruck, K.; Kawaguchi, N.; Kishimoto, M.; Torii, S.; Kobayashi, S.; Ito, N.; Harada, M.; et al. Morphogenesis of Bullet-Shaped Rabies Virus Particles Regulated by TSG101. J. Virol. 2023, 97, e0043823. [Google Scholar] [CrossRef]
- Finke, S.; Conzelmann, K.K. Replication strategies of rabies virus. Virus Res. 2005, 111, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Katzmann, D.J.; Babst, M.; Emr, S.D. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell 2001, 106, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H. ESCRTs are everywhere. EMBO J. 2015, 34, 2398–2407. [Google Scholar] [CrossRef] [PubMed]
- Christ, L.; Raiborg, C.; Wenzel, E.M.; Campsteijn, C.; Stenmark, H. Cellular Functions and Molecular Mechanisms of the ESCRT Membrane-Scission Machinery. Trends Biochem. Sci. 2017, 42, 42–56. [Google Scholar] [CrossRef]
- Skowyra, M.L.; Schlesinger, P.H.; Naismith, T.V.; Hanson, P.I. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 2018, 360, eaar5078. [Google Scholar] [CrossRef]
- Takahashi, Y.; Liang, X.; Hattori, T.; Tang, Z.; He, H.; Chen, H.; Liu, X.; Abraham, T.; Imamura-Kawasawa, Y.; Buchkovich, N.J.; et al. VPS37A directs ESCRT recruitment for phagophore closure. J. Cell Biol. 2019, 218, 3336–3354. [Google Scholar] [CrossRef]
- Calistri, A.; Reale, A.; Palù, G.; Parolin, C. Why Cells and Viruses Cannot Survive without an ESCRT. Cells 2021, 10, 483. [Google Scholar] [CrossRef]
- Schöneberg, J.; Lee, I.H.; Iwasa, J.H.; Hurley, J.H. Reverse-topology membrane scission by the ESCRT proteins. Nat. Rev. Mol. Cell Biol. 2017, 18, 5–17. [Google Scholar] [CrossRef]
- Schöneberg, J.; Pavlin, M.R.; Yan, S.; Righini, M.; Lee, I.H.; Carlson, L.A.; Bahrami, A.H.; Goldman, D.H.; Ren, X.; Hummer, G.; et al. ATP-dependent force generation and membrane scission by ESCRT-III and Vps4. Science 2018, 362, 1423–1428. [Google Scholar] [CrossRef]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Sergio, M.C.; Ricciardi, S.; Guarino, A.M.; Giaquinto, L.; De Matteis, M.A. Membrane remodeling and trafficking piloted by SARS-CoV-2. Trends Cell Biol. 2024. [Google Scholar] [CrossRef]
- Hoffmann, H.H.; Sánchez-Rivera, F.J.; Schneider, W.M.; Luna, J.M.; Soto-Feliciano, Y.M.; Ashbrook, A.W.; Le Pen, J.; Leal, A.A.; Ricardo-Lax, I.; Michailidis, E.; et al. Functional interrogation of a SARS-CoV-2 host protein interactome identifies unique and shared coronavirus host factors. Cell Host Microbe 2021, 29, 267–280.e5. [Google Scholar] [CrossRef] [PubMed]
- Rebendenne, A.; Roy, P.; Bonaventure, B.; Chaves Valadão, A.L.; Desmarets, L.; Arnaud-Arnould, M.; Rouillé, Y.; Tauziet, M.; Giovannini, D.; Touhami, J.; et al. Bidirectional genome-wide CRISPR screens reveal host factors regulating SARS-CoV-2, MERS-CoV and seasonal HCoVs. Nat. Genet. 2022, 54, 1090–1102. [Google Scholar] [CrossRef]
- Schneider, W.M.; Luna, J.M.; Hoffmann, H.H.; Sánchez-Rivera, F.J.; Leal, A.A.; Ashbrook, A.W.; Le Pen, J.; Ricardo-Lax, I.; Michailidis, E.; Peace, A.; et al. Genome-Scale Identification of SARS-CoV-2 and Pan-coronavirus Host Factor Networks. Cell 2021, 184, 120–132.e14. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.; Radulovic, M.; Stenmark, H. The many functions of ESCRTs. Nat. Rev. Mol. Cell Biol. 2020, 21, 25–42. [Google Scholar] [CrossRef]
- Friedman, N.; Jacob-Hirsch, J.; Drori, Y.; Eran, E.; Kol, N.; Nayshool, O.; Mendelson, E.; Rechavi, G.; Mandelboim, M. Transcriptomic profiling and genomic mutational analysis of Human coronavirus (HCoV)-229E -infected human cells. PLoS ONE 2021, 16, e0247128. [Google Scholar] [CrossRef]
- Kumar, R.; Cruz, L.; Sandhu, P.K.; Buchkovich, N.J. UL88 Mediates the Incorporation of a Subset of Proteins into the Virion Tegument. J. Virol. 2020, 94, e00474-20. [Google Scholar] [CrossRef]
- Kumar, R.; Christensen, N.D.; Kaddis Maldonado, R.J.; Bewley, M.C.; Ostman, A.; Sudol, M.; Chen, E.C.; Buchkovich, N.W.; Gontu, A.; Surendran Nair, M.; et al. Monoclonal Antibodies to S and N SARS-CoV-2 Proteins as Probes to Assess Structural and Antigenic Properties of Coronaviruses. Viruses 2021, 13, 1899. [Google Scholar] [CrossRef]
- Gui, L.; Zhang, X.; Li, K.; Frankowski, K.J.; Li, S.; Wong, D.E.; Moen, D.R.; Porubsky, P.R.; Lin, H.J.; Schoenen, F.J.; et al. Evaluating p97 Inhibitor Analogues for Potency against p97-p37 and p97-Npl4-Ufd1 Complexes. ChemMedChem 2016, 11, 953–957. [Google Scholar] [CrossRef]
- Martin, M.; Sandhu, P.; Kumar, R.; Buchkovich, N.J. The Immune-Specific E3 Ubiquitin Ligase MARCH1 Is Upregulated during Human Cytomegalovirus Infection to Regulate Iron Levels. J. Virol. 2022, 96, e0180621. [Google Scholar] [CrossRef] [PubMed]
- Dahal, S.; Clayton, K.; Cabral, T.; Cheng, R.; Jahanshahi, S.; Ahmed, C.; Koirala, A.; Villasmil Ocando, A.; Malty, R.; Been, T.; et al. On a path toward a broad-spectrum anti-viral: Inhibition of HIV-1 and coronavirus replication by SR kinase inhibitor harmine. J. Virol. 2023, 97, e0039623. [Google Scholar] [CrossRef] [PubMed]
- Jahanshahi, S.; Ouyang, H.; Ahmed, C.; Zahedi Amiri, A.; Dahal, S.; Mao, Y.Q.; Van Ommen, D.A.J.; Malty, R.; Duan, W.; Been, T.; et al. Broad spectrum post-entry inhibitors of coronavirus replication: Cardiotonic steroids and monensin. Virology 2024, 589, 109915. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Morita, E.; Sandrin, V.; McCullough, J.; Katsuyama, A.; Baci Hamilton, I.; Sundquist, W.I. ESCRT-III protein requirements for HIV-1 budding. Cell Host Microbe 2011, 9, 235–242. [Google Scholar] [CrossRef]
- den Boon, J.A.; Nishikiori, M.; Zhan, H.; Ahlquist, P. Positive-strand RNA virus genome replication organelles: Structure, assembly, control. Trends Genet. 2024, 40, 681–693. [Google Scholar] [CrossRef]
- Elia, N.; Sougrat, R.; Spurlin, T.A.; Hurley, J.H.; Lippincott-Schwartz, J. Dynamics of endosomal sorting complex required for transport (ESCRT) machinery during cytokinesis and its role in abscission. Proc. Natl. Acad. Sci. USA 2011, 108, 4846–4851. [Google Scholar] [CrossRef]
- Ye, Y.; Liang, X.; Wang, G.; Bewley, M.C.; Hamamoto, K.; Liu, X.; Flanagan, J.M.; Wang, H.G.; Takahashi, Y.; Tian, F. Identification of membrane curvature sensing motifs essential for VPS37A phagophore recruitment and autophagosome closure. Commun. Biol. 2024, 7, 334. [Google Scholar] [CrossRef]
- Mdkhana, B.; Saheb Sharif-Askari, N.; Ramakrishnan, R.K.; Goel, S.; Hamid, Q.; Halwani, R. Nucleic Acid-Sensing Pathways During SARS-CoV-2 Infection: Expectations versus Reality. J. Inflamm. Res. 2021, 14, 199–216. [Google Scholar] [CrossRef]
- Munnur, D.; Banducci-Karp, A.; Sanyal, S. ISG15 driven cellular responses to virus infection. Biochem. Soc. Trans. 2022, 50, 1837–1846. [Google Scholar] [CrossRef]
- Hurley, J.H.; Hanson, P.I. Membrane budding and scission by the ESCRT machinery: It’s all in the neck. Nat. Rev. Mol. Cell Biol. 2010, 11, 556–566. [Google Scholar] [CrossRef]
- Hurley, J.H.; Cada, A.K. Inside job: How the ESCRTs release HIV-1 from infected cells. Biochem. Soc. Trans. 2018, 46, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Votteler, J.; Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Meng, B.; Lever, A.M.L. The Interplay between ESCRT and Viral Factors in the Enveloped Virus Life Cycle. Viruses 2021, 13, 324. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Wei, Y.; Zou, J.; Jaffery, R.; Liang, S.; Zheng, C.; Chen, K.; Shi, P.Y.; Chen, Y.; Xie, X.; et al. Integrated multi-omics analyses identify key anti-viral host factors and pathways controlling SARS-CoV-2 infection. Res. Sq. 2022; preprint. [Google Scholar] [CrossRef]
- Stoten, C.L.; Carlton, J.G. ESCRT-dependent control of membrane remodelling during cell division. Semin. Cell Dev. Biol. 2018, 74, 50–65. [Google Scholar] [CrossRef]
- Lv, Y.; Zhou, S.; Gao, S.; Deng, H. Remodeling of host membranes during herpesvirus assembly and egress. Protein Cell 2019, 10, 315–326. [Google Scholar] [CrossRef]
- Zhang, J.; Lan, Y.; Sanyal, S. Membrane heist: Coronavirus host membrane remodeling during replication. Biochimie 2020, 179, 229–236. [Google Scholar] [CrossRef]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef]
- Wang, C.; Chen, Y.; Hu, S.; Liu, X. Insights into the function of ESCRT and its role in enveloped virus infection. Front. Microbiol. 2023, 14, 1261651. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Primer | Reverse Primer |
|---|---|---|
| 229E M 1 | 5′ CATACTATCAACCCATTCAACAAG 3′ | 5′ CACGGCAACTGTCATGTATT 3′ |
| IL-6 | 5′ CTGCGCAGCTTTAAGGAGTT 3′ | 5′ TAAGTTCTGTGCCCAGTGGA 3′ |
| ISG15 | 5′ GCTCCATGTCGGTGTCAGAG-3′ | 5′-CTCGAAGGTCAGCCAGAACAG 3′ |
| MX1 | 5′-CTGTGCAGCCAGTATGAGGAG 3′ | 5′ CAGGGTGATTAGCTCATGACTG 3′ |
| GAPDH | 5′ ACCCACTCCTCCACCTTTGAC 3′ | 5′ CTGTTGCTGTAGCCAAATTCGT 3′ |
| IFN-β | 5′ CTCTCCTGTTGTGCTTCTCCAC 3′ | 5′ TAGTCTCATTCCAGCCAGTGCT 3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, R.; Maldonado, R.K.; Christensen, N.D.; Bewley, M.C.; Flanagan, J.M.; Buchkovich, N.J.; Parent, L.J. ESCRT Protein VPS4A Is Required for the Formation of Replication Centers and Replication of Human Coronavirus 229E (HCoV-229E). COVID 2024, 4, 1338-1354. https://doi.org/10.3390/covid4090095
Kumar R, Maldonado RK, Christensen ND, Bewley MC, Flanagan JM, Buchkovich NJ, Parent LJ. ESCRT Protein VPS4A Is Required for the Formation of Replication Centers and Replication of Human Coronavirus 229E (HCoV-229E). COVID. 2024; 4(9):1338-1354. https://doi.org/10.3390/covid4090095
Chicago/Turabian StyleKumar, Rinki, Rebecca Kaddis Maldonado, Neil D. Christensen, Maria C. Bewley, John M. Flanagan, Nicholas J. Buchkovich, and Leslie J. Parent. 2024. "ESCRT Protein VPS4A Is Required for the Formation of Replication Centers and Replication of Human Coronavirus 229E (HCoV-229E)" COVID 4, no. 9: 1338-1354. https://doi.org/10.3390/covid4090095
APA StyleKumar, R., Maldonado, R. K., Christensen, N. D., Bewley, M. C., Flanagan, J. M., Buchkovich, N. J., & Parent, L. J. (2024). ESCRT Protein VPS4A Is Required for the Formation of Replication Centers and Replication of Human Coronavirus 229E (HCoV-229E). COVID, 4(9), 1338-1354. https://doi.org/10.3390/covid4090095