
Computational Discovery of Novel Imidazole Derivatives as Inhibitors of SARS-CoV-2 Main Protease: An Integrated Approach Combining Molecular Dynamics and Binding Affinity Analysis


Abstract
1. Introduction
2. Methodology
2.1. Imidazole Derivatives
2.2. Ligand Preparation
2.3. Preparation of Protein
2.4. Molecular Docking
2.5. Molecular Dynamics Simulation
2.6. ADMET Predictions
3. Results
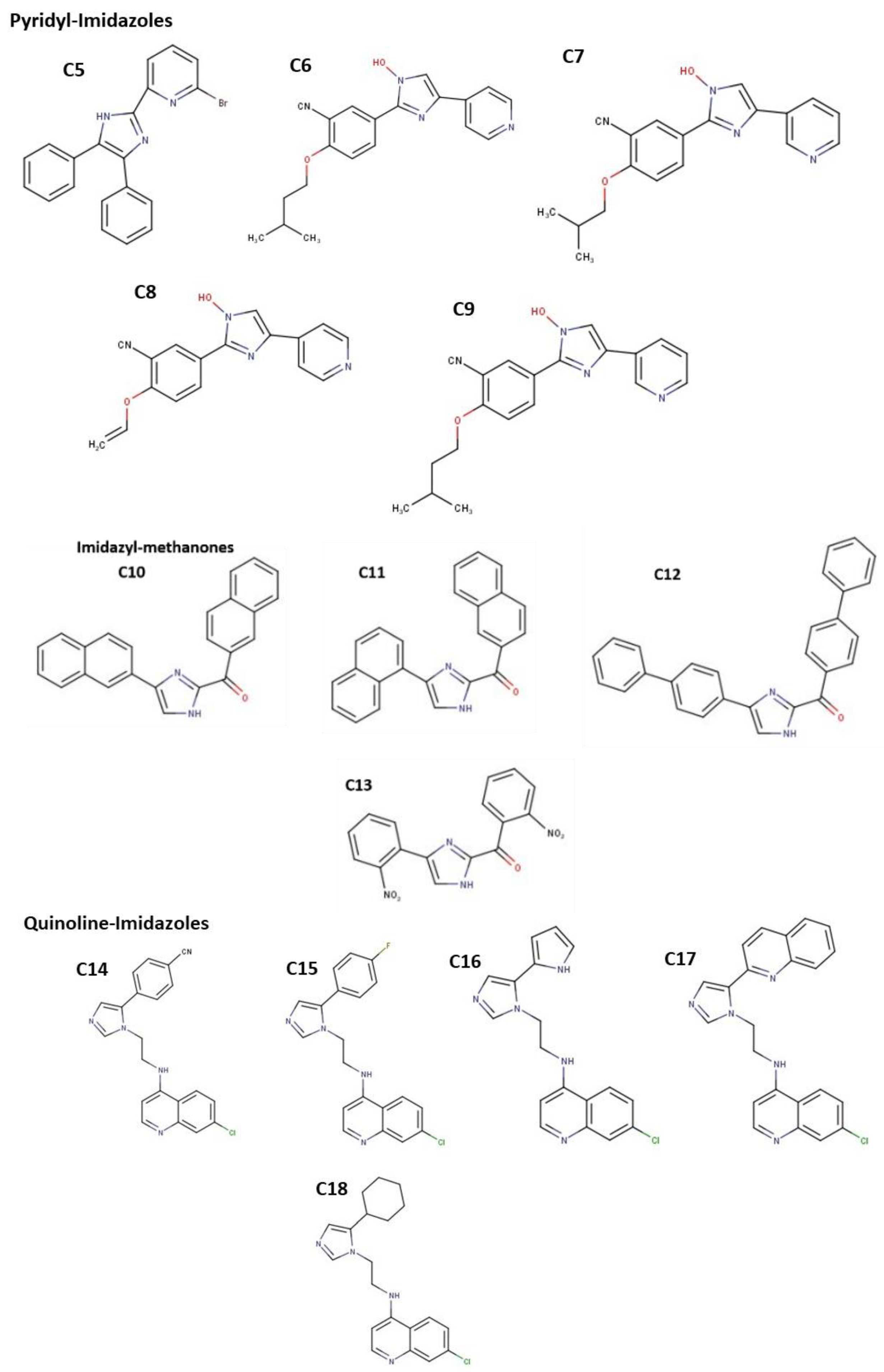
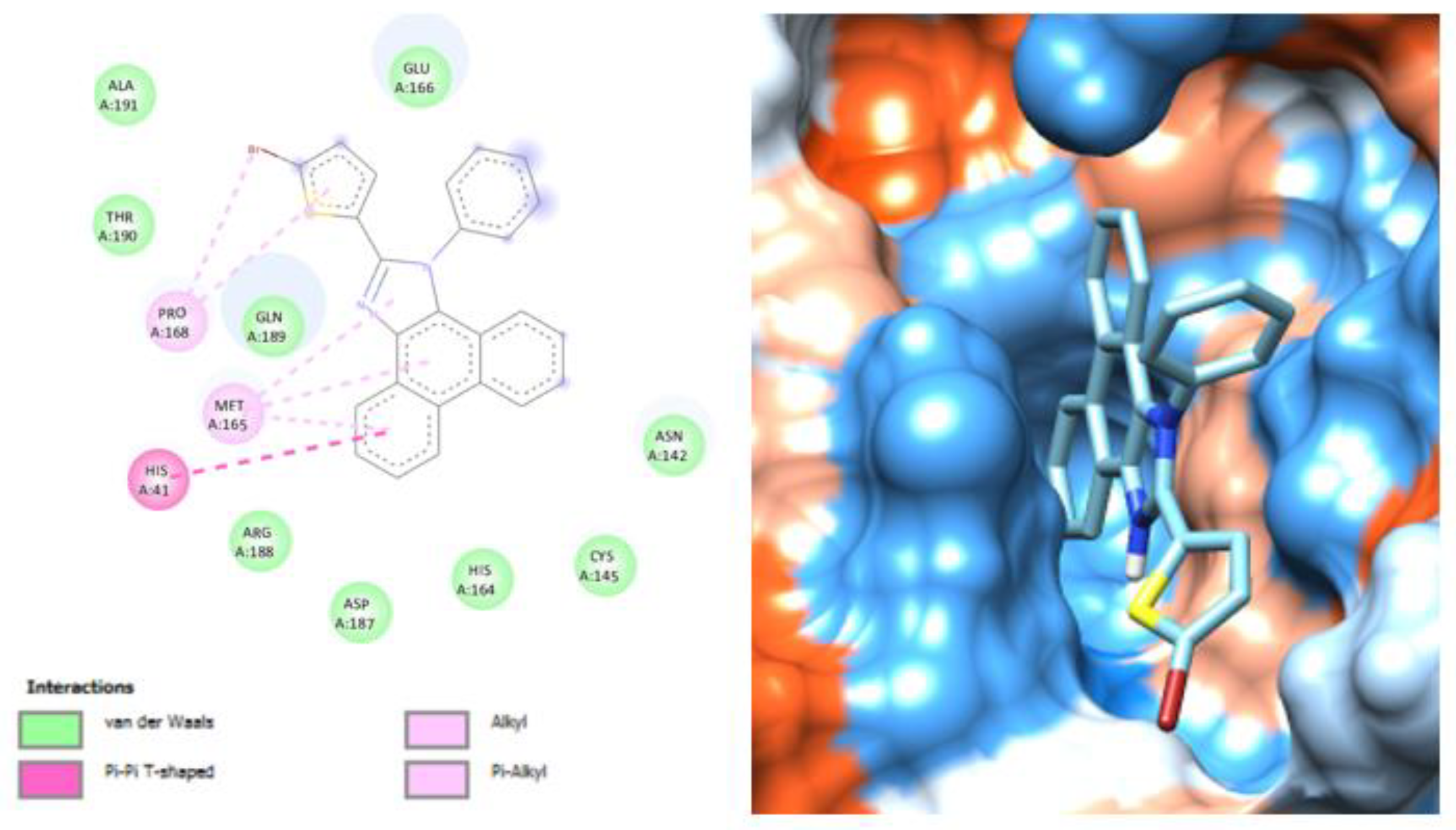
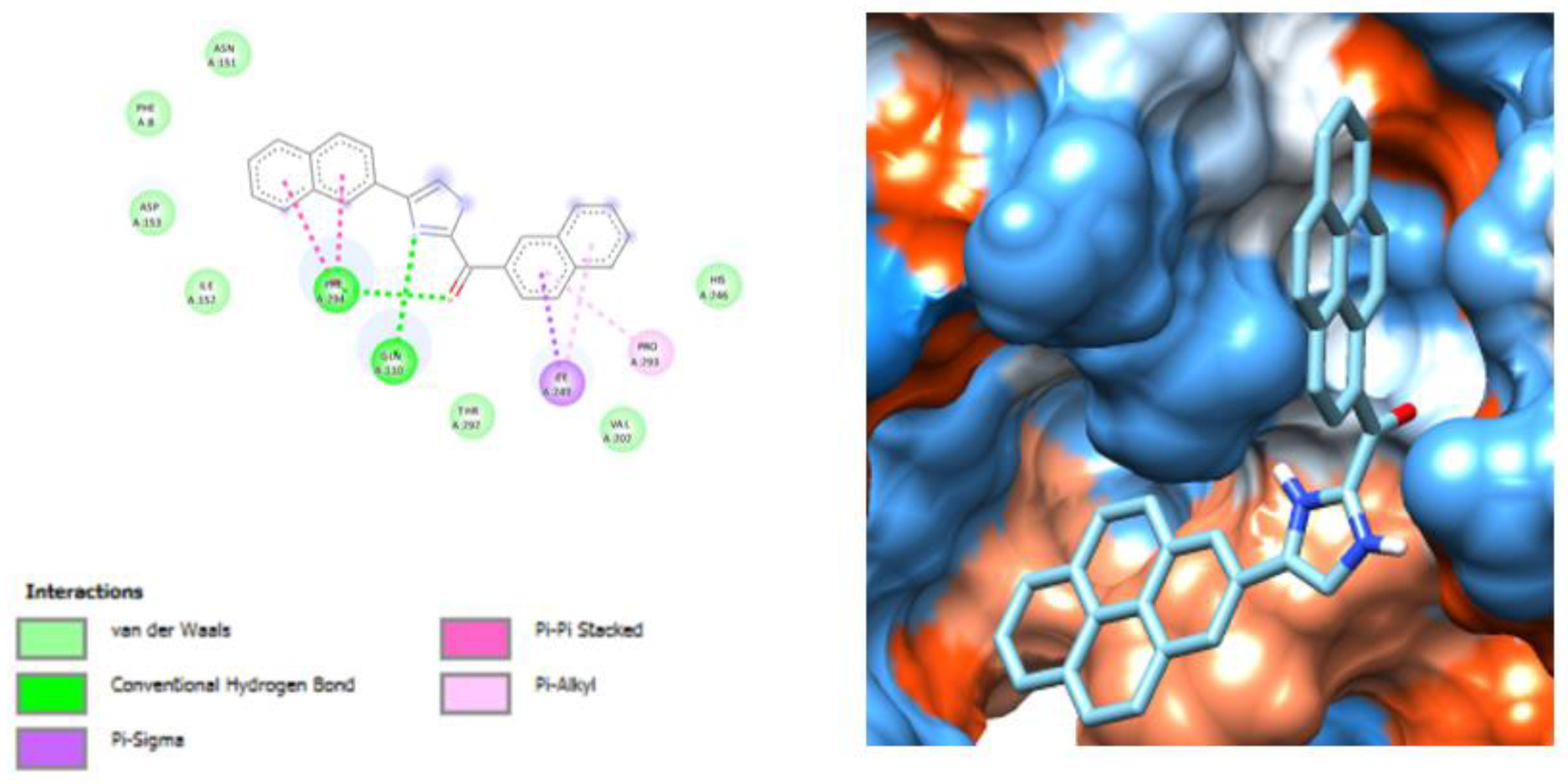
3.1. Binding Affinities and Stability of Test Compounds with SARS-CoV-2 Drug Targets
3.2. Molecular Docking Analysis of Selected Test Compounds
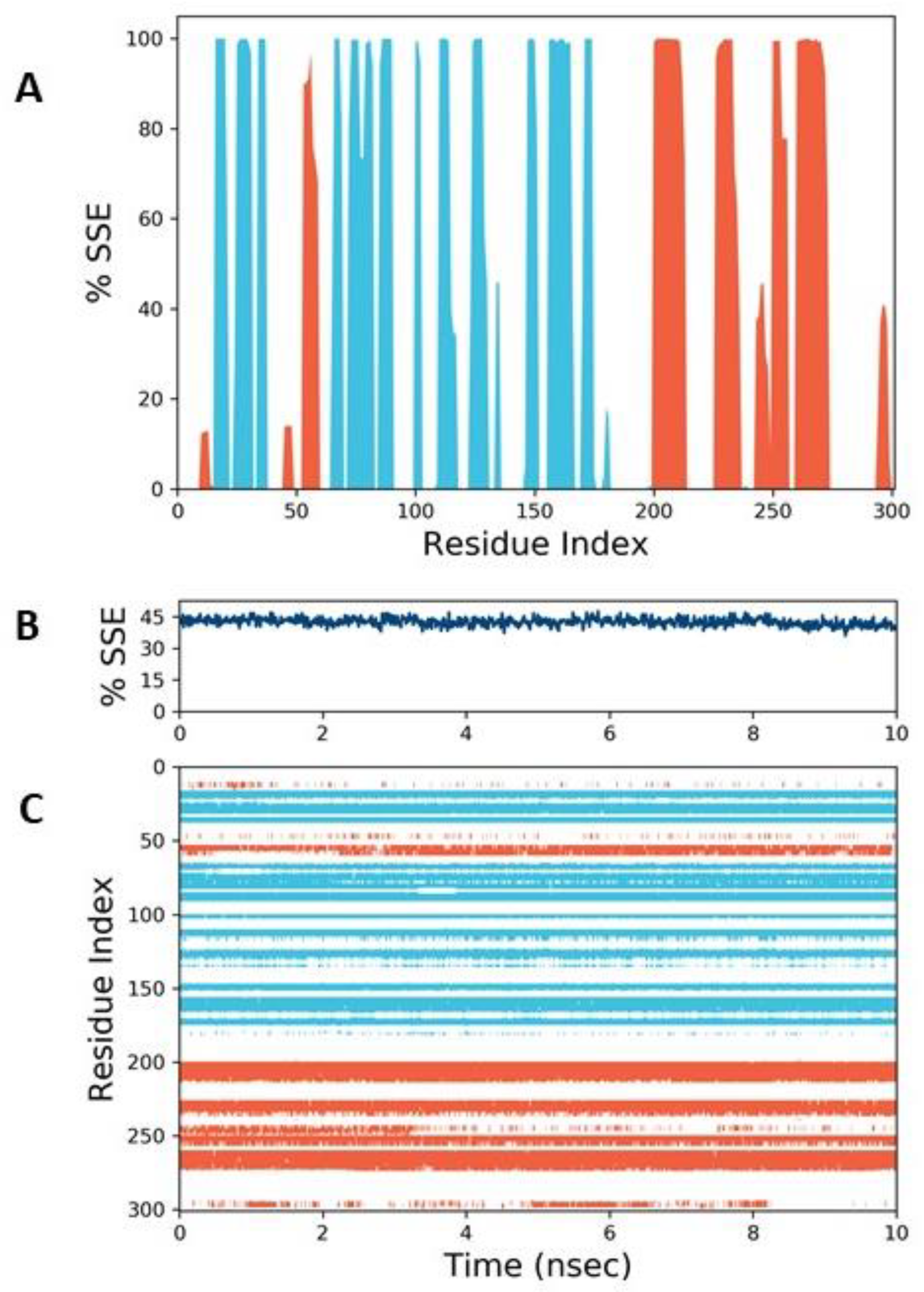
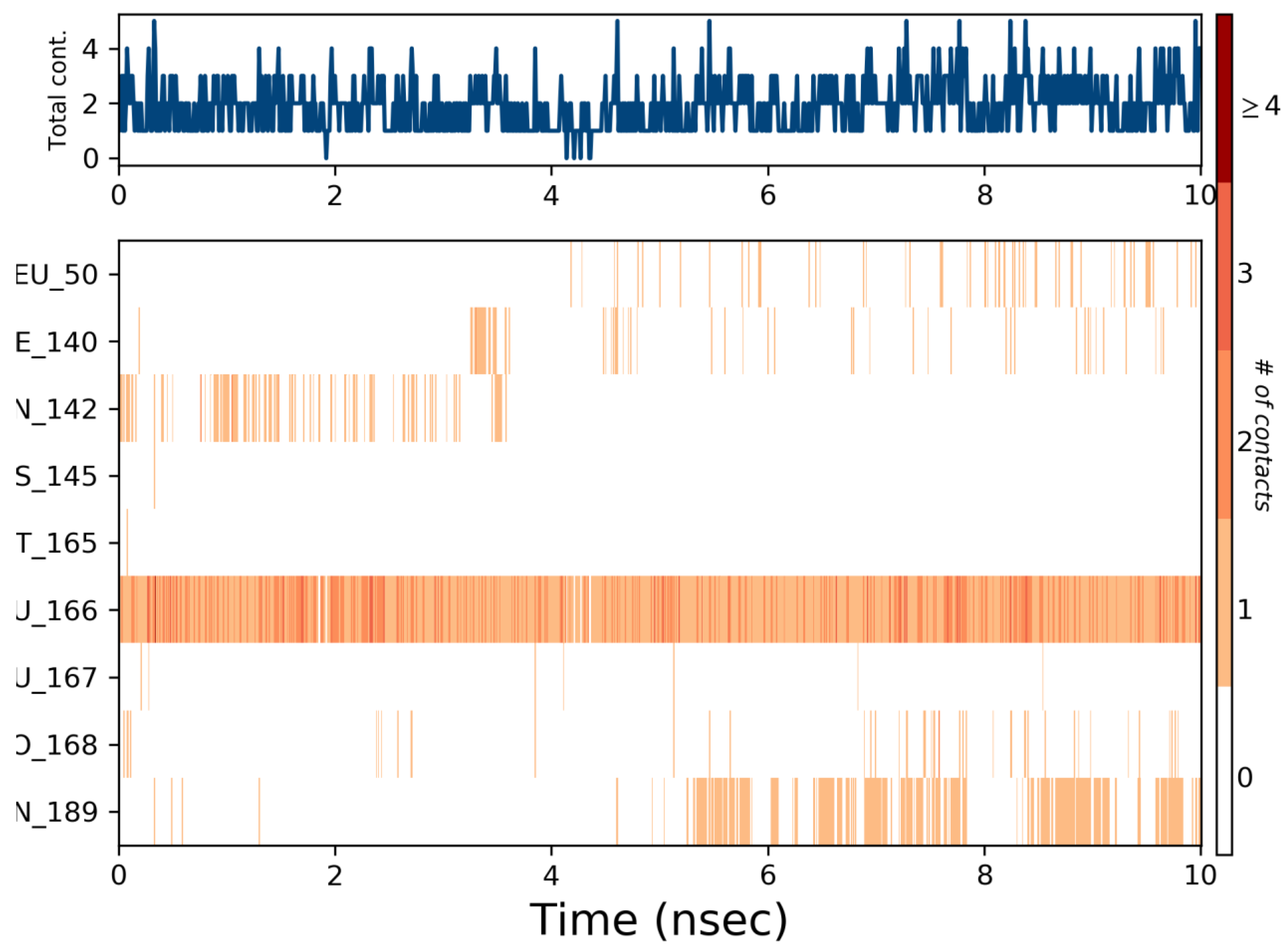
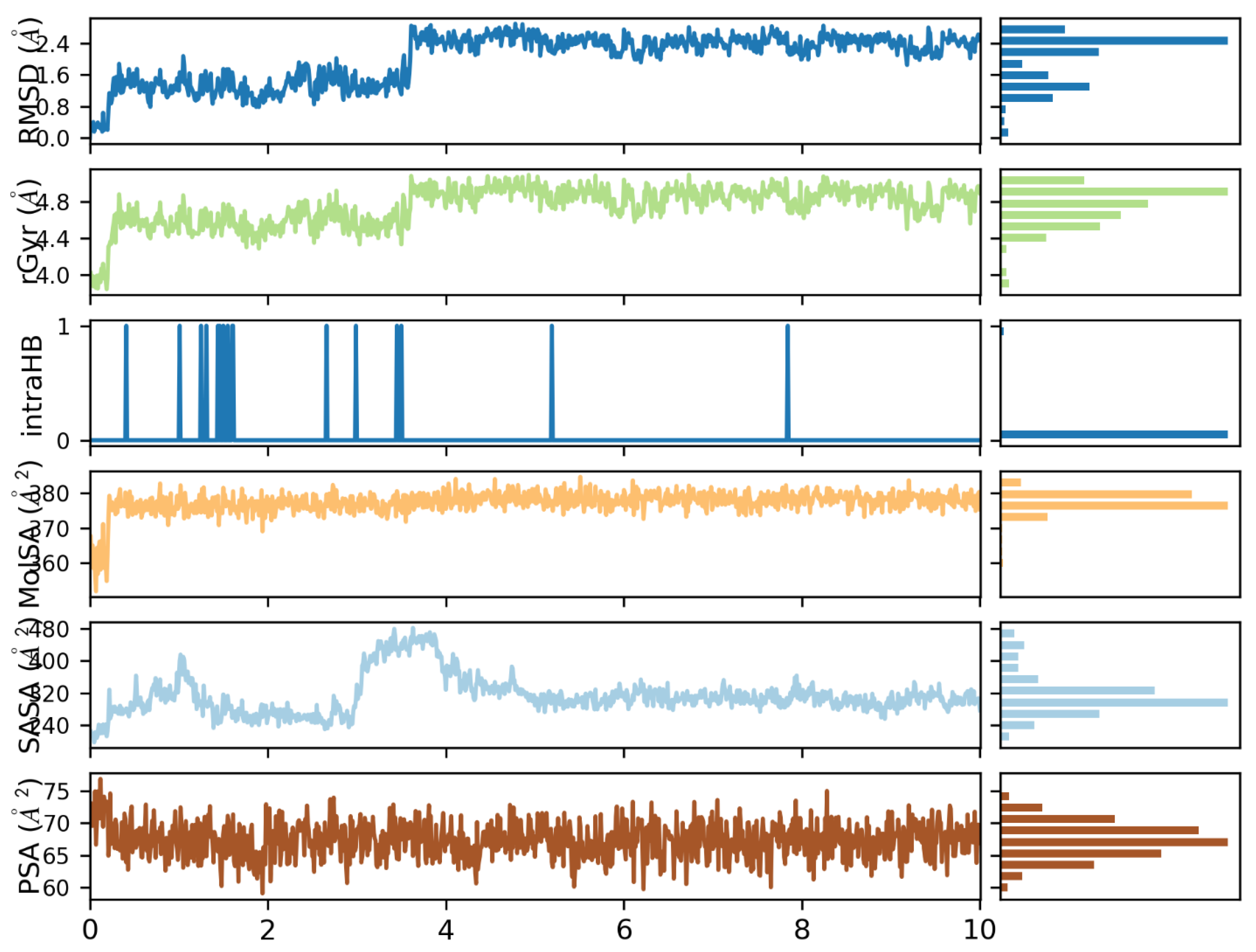
3.3. Molecular Dynamics Simulation
3.4. ADMET Profile
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization—WHO. Severe Acute Respiratory Syndrome (SARS). 2024. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 6 May 2024).
- Centers for Disease Control and Prevention—CDC. SARS Basics Fact Sheet. Available online: https://www.cdc.gov/sars/about/fs-sars.html (accessed on 13 July 2022).
- Babalola, B.; Akinsuyi, O.S.; Folajimi, E.M.; Olujimi, F.; Otunba, A.A.; Chikere, B.; Adewumagun, I.A.; Adetobi, T.E. Exploring the future of SARS-CoV-2 treatment after the first two years of the pandemic: A comparative study of alternative therapeutics. Biomed. Pharmacotherapy 2023, 165, 115099. [Google Scholar] [CrossRef] [PubMed]
- Babalola, B.A.; Adetobi, T.E.; Akinsuyi, O.S.; Adebisi, O.A.; Folajimi, E.O. Computational Study of the Therapeutic Potential of Novel Heterocyclic Derivatives against SARS-CoV-2. Covid 2021, 1, 757–774. [Google Scholar] [CrossRef]
- Pang, X.; Xu, W.; Liu, Y.; Li, H.; Chen, L. The research progress of SARS-CoV-2 main protease inhibitors from 2020 to 2022. Eur. J. Med. Chem. 2023, 257, 115491. [Google Scholar] [CrossRef]
- Li, Q.; Kang, C. Progress in Developing Inhibitors of SARS-CoV-2 3C-Like Protease. Microorganisms. 2020, 8, 1250. [Google Scholar] [CrossRef]
- Hu, Q.; Xiong, Y.; Zhu, G.H.; Zhang, Y.N.; Zhang, Y.W.; Huang, P.; Ge, G.B. The SARS-CoV-2 main protease (Mpro): Structure, function, and emerging therapies for COVID-19. MedComm 2022, 3, e151. [Google Scholar] [CrossRef]
- Adetobi, E.T.; Akinsuyi, S.O.; Ahmed, O.A.; Folajimi, E.O.; Babalola, B.A. In silico Evaluation of the Inhibitory Potential of Cymbopogonol from Cymbopogon citratus Towards Falcipain-2 (FP2) Cysteine Protease of Plasmodium falciparum. Trop. J. Nat. Prod. Res. 2022, 6, 1687–1694. [Google Scholar]
- Babalola, B.A.; Akinwande, A.I.; Gboyega, A.E.; Otunba, A.A. Extraction, purification and characterization of papain cysteine-proteases from the leaves of Carica papaya. Sci. Afr. 2023, 19, e01538. [Google Scholar] [CrossRef]
- Slassi, S.; Aarjane, M.; Amine, A. Synthesis, spectroscopic characterization (FT-IR, NMR, UV-Vis), DFT study, antibacterial and antioxidant in vitro investigations of 4, 6-bis ((E)-1-((3-(1H-imidazol-1-yl) propyl) imino) ethyl) benzene-1, 3-diol. J. Mol. Struct. 2022, 1255, 132457. [Google Scholar] [CrossRef]
- Seck, I.; Nguemo, F. Triazole, imidazole, and thiazole-based compounds as potential agents against coronavirus. Results Chem. 2021, 3, 100132. [Google Scholar] [CrossRef] [PubMed]
- Batiha, G.E.S.; Teibo, J.O.; Shaheen, H.M.; Babalola, B.A.; Teibo, T.K.A.; Al-Kuraishy, H.M.; Al-Garbeeb, A.I.; Alexiou, A.; Papadakis, M. Therapeutic potential of Lawsonia inermis Linn: A comprehensive overview. Naunyn Schmiedeberg Arch. Pharmacol. 2024, 397, 3525–3540. [Google Scholar] [CrossRef]
- Zhang, Q.; Guo, L.; Huang, Y.; Zhang, R.; Ritacca, A.G.; Leng, S.; Zheng, X.; Yang, Y.; Singh, A. Influence of an imidazole-based ionic liquid as electrolyte additive on the performance of alkaline Al-air battery. J. Power Sources 2023, 564, 232901. [Google Scholar] [CrossRef]
- Çetiner, G.; Çevik, U.A.; Celik, I.; Bostancı, H.E.; Özkay, Y.; Kaplancıklı, Z.A. New Imidazole Derivatives as Aromatase Inhibitor: Design, Synthesis, Biological Activity, Molecular Docking, and Computational ADME-Tox Studies. J. Mol. Struct. 2023, 1278, 134920. [Google Scholar] [CrossRef]
- Poyraz, S.; Döndaş, H.A.; Sansano, J.M.; Belveren, S.; Yamali, C.; Ülger, M.; Döndaş, N.Y.; Sağlık, B.N.; Pask, C.M. N-Benzoylthiourea-pyrrolidine carboxylic acid derivatives bearing an imidazole moiety: Synthesis, characterization, crystal structure, in vitro ChEs inhibition, and antituberculosis, antibacterial, antifungal studies. J. Mol. Struct. 2023, 1273, 134303. [Google Scholar] [CrossRef]
- Sadula, A.; Gaddhe, L. Synthesis, computational studies and biological evaluation of novel Acenaphthoquinone-imidazole derivatives as dual inhibitors of HSP90 and Topo II in cancer therapy. Results Chem. 2023, 5, 100796. [Google Scholar] [CrossRef]
- Muhammed, M.T.; Mustafa, E.R.; Akkoc, S. Molecular modeling and in vitro antiproliferative activity studies of some imidazole and isoxazole derivatives. J. Mol. Struct. 2023, 1282, 135066. [Google Scholar] [CrossRef]
- Chhetri, A.; Chettri, S.; Rai, P.; Sinha, B.; Brahman, D. Exploration of inhibitory action of Azo imidazole derivatives against COVID-19 main protease (Mpro): A computational study. J. Mol. Struct. 2021, 1224, 129178. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J Cheminform 2011, 3, 33. [Google Scholar] [CrossRef]
- Dong, J.; Cao, D.S.; Miao, H.Y.; Liu, S.; Deng, B.C.; Yun, Y.H.; Wang, N.N.; Lu, A.P.; Zeng, W.B.; Chen, A.F. ChemDes: An integrated web-based platform for molecular descriptor and fingerprint computation. J. Cheminform. 2015, 7, 60. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Dong, J.; Wang, N.N.; Yao, Z.J.; Zhang, L.; Cheng, Y.; Ouyang, D.; Lu, A.P.; Cao, D.S. ADMETlab: A platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. J. Cheminform. 2018, 10, 29. [Google Scholar] [CrossRef]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorganic Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Hu, Y.; Shen, Y.F.; Wang, G.X.; Zhu, B. Evaluation on antiviral activity of coumarin derivatives against spring viraemia of carp virus in epithelioma papulosum cyprini cells. Antivir. Res. 2017, 144, 173–185. [Google Scholar] [CrossRef]
- Kanwal, A.; Ahmad, M.; Aslam, S.; Naqvi, S.A.R.; Saif, M.J. Recent advances in antiviral benzimidazole derivatives: A mini review. Pharm. Chem. J. 2019, 53, 179–187. [Google Scholar] [CrossRef]
- Kneller, D.W.; Phillips, G.; Weiss, K.L.; Pant, S.; Zhang, Q.; O’Neill, H.M.; Coates, L.; Kovalevsky, A. Unusual zwitterionic catalytic site of SARS–CoV-2 main protease revealed by neutron crystallography. J. Biol. Chem. 2020, 295, 17365–17373. [Google Scholar] [CrossRef] [PubMed]
- Novak, J.; Rimac, H.; Kandagalla, S.; Pathak, P.; Naumovich, V.; Grishina, M.; Potemkin, V. Proposition of a new allosteric binding site for potential SARS-CoV-2 3CL protease inhibitors by utilizing molecular dynamics simulations and ensemble docking. J. Biomol. Struct. Dyn. 2022, 40, 9347–9360. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Sargsyan, K.; Grauffel, C.; Lim, C. How molecular size impacts RMSD applications in molecular dynamics simulations. J. Chem. Theory Comput. 2017, 13, 1518–1524. [Google Scholar] [CrossRef]
- Monhemi, H.; Housaindokht, M.R.; Moosavi-Movahedi, A.A.; Bozorgmehr, M.R. How a protein can remain stable in a solvent with high content of urea: Insights from molecular dynamics simulation of Candida antarctica lipase B in urea: Choline chloride deep eutectic solvent. Phys. Chem. Chem. Phys. 2014, 16, 14882–14893. [Google Scholar] [CrossRef]
- Uttarkar, A.; Niranjan, V. Re-profiling of natural inhibitor via combinatorial drug screening: Brefeldin A variant design as an effective antagonist leading to EPAC2 structure modification and antibody design for identification. bioRxiv 2021. [Google Scholar] [CrossRef]
- Durdagi, S.; ul Qamar, M.T.; Salmas, R.E.; Tariq, Q.; Anwar, F.; Ashfaq, U.A. Investigating the molecular mechanism of staphylococcal DNA gyrase inhibitors: A combined ligand-based and structure-based resources pipeline. J. Mol. Graph. Model. 2018, 85, 122–129. [Google Scholar] [CrossRef]
- Oum, Y.H.; Kell, S.A.; Yoon, Y.; Liang, Z.; Burger, P.; Shim, H. Discovery of novel aminopiperidinyl amide CXCR4 modulators through virtual screening and rational drug design. Eur. J. Med. Chem. 2020, 201, 112479. [Google Scholar] [CrossRef] [PubMed]
- Rai, B.K.; Sresht, V.; Yang, Q.; Unwalla, R.; Tu, M.; Mathiowetz, A.M.; Bakken, G.A. Comprehensive assessment of torsional strain in crystal structures of small molecules and protein–ligand complexes using ab initio calculations. J. Chem. Inf. Model. 2019, 59, 4195–4208. [Google Scholar] [CrossRef]
- Denning, E.J.; Priyakumar, U.D.; Nilsson, L.; Mackerell Jr, A.D. Impact of 2′-hydroxyl sampling on the conformational properties of RNA: Update of the CHARMM all-atom additive force field for, R.N.A. J. Comput. Chem. 2011, 32, 1929–1943. [Google Scholar] [CrossRef]
- Baby, K.; Maity, S.; Mehta, C.H.; Nayak, U.Y.; Shenoy, G.G.; Pai, K.S.R.; Harikumar, K.B.; Nayak, Y. Computational drug repurposing of Akt-1 allosteric inhibitors for non-small cell lung cancer. Sci. Rep. 2023, 13, 7947. [Google Scholar] [CrossRef]
- Fischer, A.; Smiesko, M.; Sellner, M.; Lill, M.A. Decision making in structure-based drug discovery: Visual inspection of docking results. J. Med. Chem. 2021, 64, 2489–2500. [Google Scholar] [CrossRef]
- Wu, J.; Hu, B.; Sun, X.; Wang, H.; Huang, Y.; Zhang, Y.; Liu, M.; Liu, Y.; Zhao, Y.; Wang, J.; et al. In silico study reveals existing drugs as α-glucosidase inhibitors: Structure-based virtual screening validated by experimental investigation. J. Mol. Struct. 2020, 1218, 128532. [Google Scholar] [CrossRef]
- Gyebi, G.A.; Ogunyemi, O.M.; Adefolalu, A.A.; Rodríguez-Martínez, A.; López-Pastor, J.F.; Banegas-Luna, A.J.; Pérez-Sánchez, H.; Adegunloye, A.P.; Ogunro, O.B.; Afolabi, S.O. African derived phytocompounds may interfere with SARS-CoV-2 RNA capping machinery via inhibition of 2′-O-ribose methyltransferase: An in silico perspective. J. Mol. Struct. 2022, 1262, 133019. [Google Scholar] [CrossRef]
- Chen, H.; Panagiotopoulos, A.Z. Molecular modeling of surfactant micellization using solvent-accessible surface area. Langmuir 2019, 35, 2443–2450. [Google Scholar] [CrossRef]
- Whitty, A.; Zhong, M.; Viarengo, L.; Beglov, D.; Hall, D.R.; Vajda, S. Quantifying the chameleonic properties of macrocycles and other high-molecular-weight drugs. Drug Discov. Today 2016, 21, 712–717. [Google Scholar] [CrossRef]
- Babalola, B.A.; Akinwande, A.I.; Otunba, A.A.; Adebami, G.E.; Babalola, O.; Nwufo, C. Therapeutic benefits of Carica papaya: A review on its pharmacological activities and characterization of papain. Arab. J. Chem. 2024, 17, 105369. [Google Scholar] [CrossRef]
- Gao, Y.; Gesenberg, C.; Zheng, W. Oral formulations for preclinical studies: Principle, design, and development considerations. In Developing Solid Oral Dosage Forms; Academic Press: Cambridge, MA, USA, 2017; pp. 455–495. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Testa, B.; Kraemer, S.D. The biochemistry of drug metabolism—An introduction: Part 5. Metabolism and bioactivity. Chem. Biodivers. 2009, 6, 591–684. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Ursu, O.; Rayan, A.; Goldblum, A.; Oprea, T.I. Understanding drug-likeness. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 760–781. [Google Scholar] [CrossRef]
- Lazzaro, S.; West, M.A.; Eatemadpour, S.; Feng, B.; Varma, M.V.; Rodrigues, A.D.; Temesszentandrási-Ambrus, C.; Kovács-Hajdu, P.; Nerada, Z.; Gáborik, Z.; et al. Translatability of in vitro inhibition potency to in vivo P-glycoprotein mediated drug interaction risk. J. Pharm. Sci. 2023, 112, 1715–1723. [Google Scholar] [CrossRef]
- Cox, B.; Nicolaï, J.; Williamson, B. The role of the efflux transporter, P-glycoprotein, at the blood–brain barrier in drug discovery. Biopharm. Drug Dispos. 2023, 44, 113–126. [Google Scholar] [CrossRef]
- Yoshitomo, A.; Asano, S.; Hozuki, S.; Tamemoto, Y.; Shibata, Y.; Hashimoto, N.; Takahashi, K.; Sasaki, Y.; Ozawa, N.; Kageyama, M.; et al. Significance of basal membrane permeability of epithelial cells in predicting intestinal drug absorption. Drug Metab. Dispos. 2023, 51, 318–328. [Google Scholar] [CrossRef]
- Danner, L.; Malard, F.; Valdes, R.; Olivier-Van Stichelen, S. Non-Nutritive Sweeteners Acesulfame Potassium and Sucralose Are Competitive Inhibitors of the Human P-glycoprotein/Multidrug Resistance Protein 1 (PGP/MDR1). Nutrients 2023, 15, 1118. [Google Scholar] [CrossRef]
- Femi-Olabisi, F.J.; Ishola, A.A.; Faokunla, O.; Agboola, A.O.; Babalola, B.A. Evaluation of the inhibitory potentials of selected compounds from Costus spicatus (Jacq.) rhizome towards enzymes associated with insulin resistance in polycystic ovarian syndrome: An in silico study. J. Genet. Eng. Biotechnol. 2021, 19, 176. [Google Scholar] [CrossRef]
- Otunba, A.A.; Osuntoki, A.A.; Okunowo, W.; Olukoya, D.K.; Babalola, B.A. Characterization of novel bacteriocin PB2 and comprehensive detection of the pediocin gene ped-A1 from Pediococcus pentosaceus PB2 strain isolated from a sorghum-based fermented beverage in Nigeria. Biotechnol. Rep. 2022, 36, e00772. [Google Scholar] [CrossRef] [PubMed]
- Otunba, A.A.; Osuntoki, A.A.; Olukoya, D.K.; Babalola, B.A. Genomic, biochemical and microbial evaluation of probiotic potentials of bacterial isolates from fermented sorghum products. Heliyon 2021, 7, e08536. [Google Scholar] [CrossRef]
- Adebami, G.E.; Fasiku, S.A.; Babalola, B.A. Physicochemical and microbial evaluations of different fish ponds’waste waters and the antibiotics profiles of isolated bacteria. Ethiop. J. Environ. Stud. Manag. 2020, 13. [Google Scholar]
- Babalola, B.A.; Adebami, G.E.; Akinsuyi, S.E. Mechanistic basis for Cancer Immune Evasion role of immune checkpoint blockades in Immuno-Oncology Glob. J. Cancer Ther. 2021, 7, 35–42. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | ∆G Energy (Kcal/mol) |
|---|---|
| Thiophenyl–imidazole | |
| C1 | −8.2 |
| C2 | −8.1 |
| C3 | −8.0 |
| C4 | −7.9 |
| Pyridyl–imidazole | |
| C5 | −8.3 |
| C6 | −7.6 |
| C7 | −7.3 |
| C8 | −7.2 |
| C9 | −7.2 |
| Imidazolyl–methanones | |
| C10 | −9.2 |
| C11 | −8.9 |
| C12 | −8.9 |
| C13 | −8.2 |
| Quinoline–imidazole | |
| C14 | −7.7 |
| C15 | −7.6 |
| C16 | −7.4 |
| C17 | −7.6 |
| C18 | −7.2 |
| Standard ligand | |
| K36 | −7.1 |
| Molecule | MW | Consensus Log p | Silicos-IT LogSw | log Kp (cm/s) | Lipinski #violations | Veber #violations | Bioavailability Score |
|---|---|---|---|---|---|---|---|
| C1 | 457.38 | 6.06 | −8.66 | −4.31 | 1 | 0 | 0.55 |
| C2 | 436.52 | 5.86 | −9.88 | −4.05 | 1 | 0 | 0.55 |
| C3 | 376.47 | 5.94 | −9.68 | −3.64 | 1 | 0 | 0.55 |
| C4 | 316.42 | 4.9 | −8.42 | −4.57 | 0 | 0 | 0.55 |
| C5 | 376.25 | 4.64 | −9.21 | −5.02 | 0 | 0 | 0.55 |
| C6 | 348.4 | 3.33 | −5.92 | −5.78 | 0 | 0 | 0.55 |
| C7 | 304.3 | 2.39 | −4.76 | −6.37 | 0 | 0 | 0.55 |
| C8 | 334.37 | 2.94 | −5.52 | −5.95 | 0 | 0 | 0.55 |
| C9 | 348.4 | 3.25 | −5.92 | −5.78 | 0 | 0 | 0.55 |
| C10 | 348.4 | 4.72 | −9.53 | −4.24 | 0 | 0 | 0.55 |
| C11 | 348.4 | 4.76 | −9.53 | −4.24 | 0 | 0 | 0.55 |
| C12 | 338.27 | 1.29 | −4.95 | −6.19 | 0 | 0 | 0.55 |
| C13 | 351.83 | 3.21 | −7.78 | −6.05 | 0 | 0 | 0.55 |
| C14 | 440.92 | 4.87 | −10.37 | −4.94 | 0 | 0 | 0.55 |
| C15 | 378.85 | 3.61 | −8.29 | −5.66 | 0 | 0 | 0.55 |
| C16 | 398.89 | 4.64 | −9.82 | −4.88 | 0 | 0 | 0.55 |
| C17 | 364.83 | 3.3 | −7.6 | −5.81 | 0 | 0 | 0.55 |
| C18 | 485.55 | 0.63 | −3.75 | −8.76 | 1 | 2 | 0.11 |
| Molecule | GI Absorption | BBB Permeant | Pgp Substrate | CYP1A2 Inhibitor | CYP2C19 Inhibitor | CYP2C9 Inhibitor | CYP2D6 Inhibitor | CYP3A4 Inhibitor |
|---|---|---|---|---|---|---|---|---|
| C1 | Low | No | No | Yes | Yes | No | No | No |
| C2 | Low | No | Yes | Yes | Yes | No | No | Yes |
| C3 | Low | No | Yes | No | Yes | No | No | No |
| C4 | High | No | Yes | Yes | Yes | Yes | No | Yes |
| C5 | High | Yes | Yes | Yes | Yes | No | Yes | Yes |
| C6 | High | No | No | Yes | Yes | Yes | Yes | Yes |
| C7 | High | No | No | Yes | No | Yes | Yes | Yes |
| C8 | High | No | No | Yes | Yes | Yes | Yes | Yes |
| C9 | High | Yes | Yes | Yes | Yes | No | No | No |
| C10 | High | Yes | Yes | Yes | Yes | No | No | No |
| C11 | Low | No | No | Yes | Yes | Yes | Yes | Yes |
| C12 | High | No | Yes | Yes | Yes | No | No | Yes |
| C13 | High | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| C14 | High | No | Yes | Yes | Yes | Yes | Yes | Yes |
| C15 | High | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| C16 | High | Yes | Yes | Yes | Yes | No | Yes | Yes |
| C17 | High | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| C18 | Low | No | Yes | No | No | No | No | No |
| Compound | LD50 (mg/kg) | Toxicity Class | Carcinogenicity | Cytotoxicity | Eye Irritation | Eye Corrosion | Skin Sensitization | Nephrotoxicity |
|---|---|---|---|---|---|---|---|---|
| C1 | 5000 | 5 | Inactive | Inactive | Inactive | Inactive | Inactive | Active |
| C2 | 3420 | 5 | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| C3 | 770 | 4 | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| C4 | 300 | 3 | Inactive | Inactive | Active | Inactive | Inactive | Active |
| C5 | 2000 | 4 | Inactive | Inactive | Active | Active | Inactive | Active |
| C6 | 2800 | 5 | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| C7 | 2000 | 4 | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| C8 | 2000 | 4 | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| C9 | 2800 | 5 | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| C10 | 300 | 3 | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| C11 | 300 | 3 | Inactive | Inactive | Active | Inactive | Inactive | Inactive |
| C12 | 720 | 4 | Inactive | Inactive | Active | Inactive | Inactive | Inactive |
| C13 | 292 | 3 | Active | Inactive | Inactive | Inactive | Inactive | Active |
| C14 | 640 | 4 | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| C15 | 780 | 4 | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| C16 | 1150 | 4 | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| C17 | 590 | 4 | Inactive | Inactive | Inactive | Inactive | Inactive | Active |
| C18 | 640 | 4 | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babalola, B.A.; Adegboyega, A.E. Computational Discovery of Novel Imidazole Derivatives as Inhibitors of SARS-CoV-2 Main Protease: An Integrated Approach Combining Molecular Dynamics and Binding Affinity Analysis. COVID 2024, 4, 672-695. https://doi.org/10.3390/covid4060046
Babalola BA, Adegboyega AE. Computational Discovery of Novel Imidazole Derivatives as Inhibitors of SARS-CoV-2 Main Protease: An Integrated Approach Combining Molecular Dynamics and Binding Affinity Analysis. COVID. 2024; 4(6):672-695. https://doi.org/10.3390/covid4060046
Chicago/Turabian StyleBabalola, Benjamin Ayodipupo, and Abayomi Emmanuel Adegboyega. 2024. "Computational Discovery of Novel Imidazole Derivatives as Inhibitors of SARS-CoV-2 Main Protease: An Integrated Approach Combining Molecular Dynamics and Binding Affinity Analysis" COVID 4, no. 6: 672-695. https://doi.org/10.3390/covid4060046
APA StyleBabalola, B. A., & Adegboyega, A. E. (2024). Computational Discovery of Novel Imidazole Derivatives as Inhibitors of SARS-CoV-2 Main Protease: An Integrated Approach Combining Molecular Dynamics and Binding Affinity Analysis. COVID, 4(6), 672-695. https://doi.org/10.3390/covid4060046