
Low Selectivity Indices of Ivermectin and Macrocyclic Lactones on SARS-CoV-2 Replication In Vitro

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture
2.2. SARS-CoV-2 Virus
2.3. Virus Titration
2.4. Assessment of Antiviral Activity
2.5. Quantitative Reverse Transcription Polymerase Chain Reaction (RT-qPCR)
2.6. Immunofluorescence and Confocal Microscopy Infected Cell Imaging
3. Results
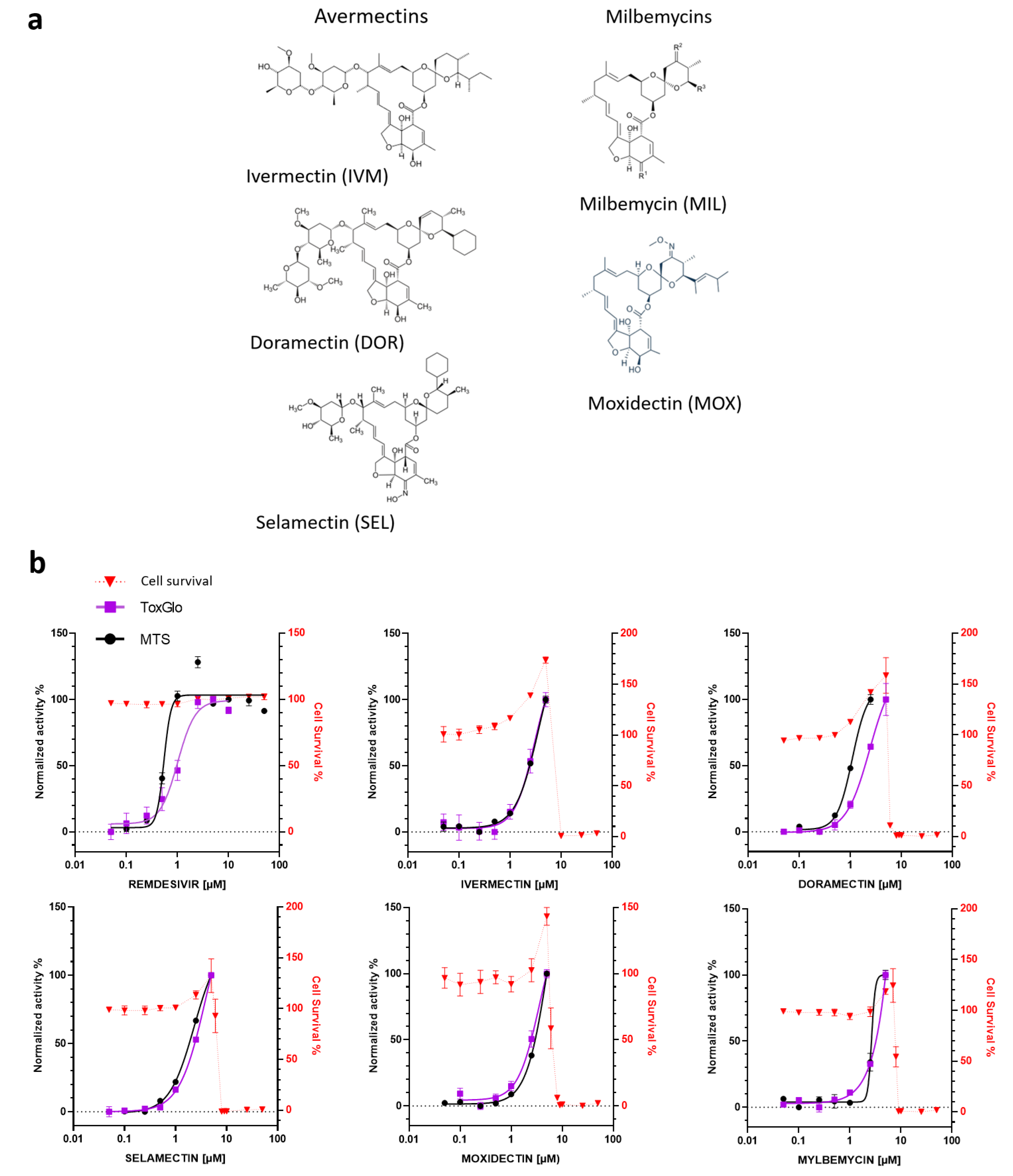
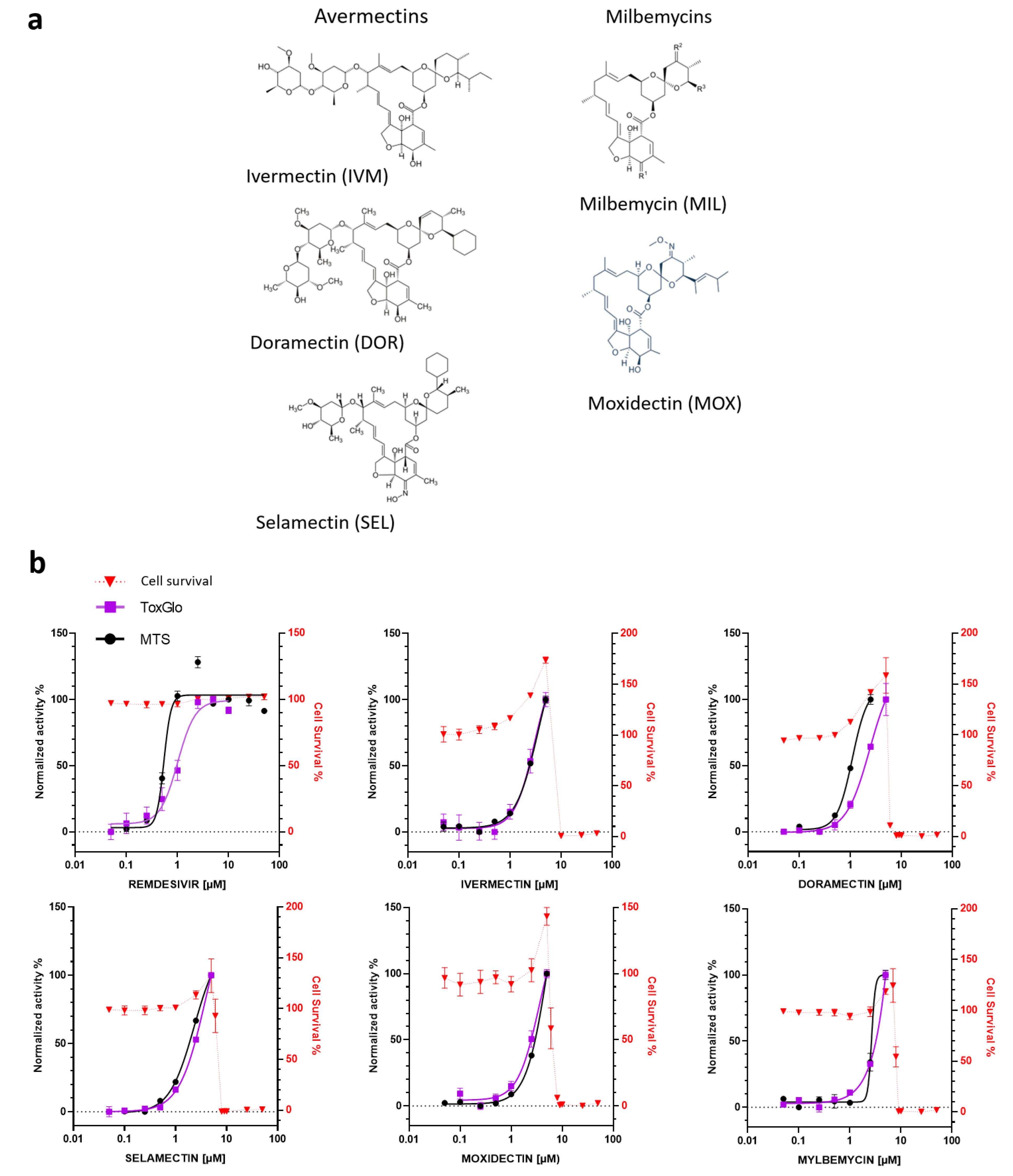
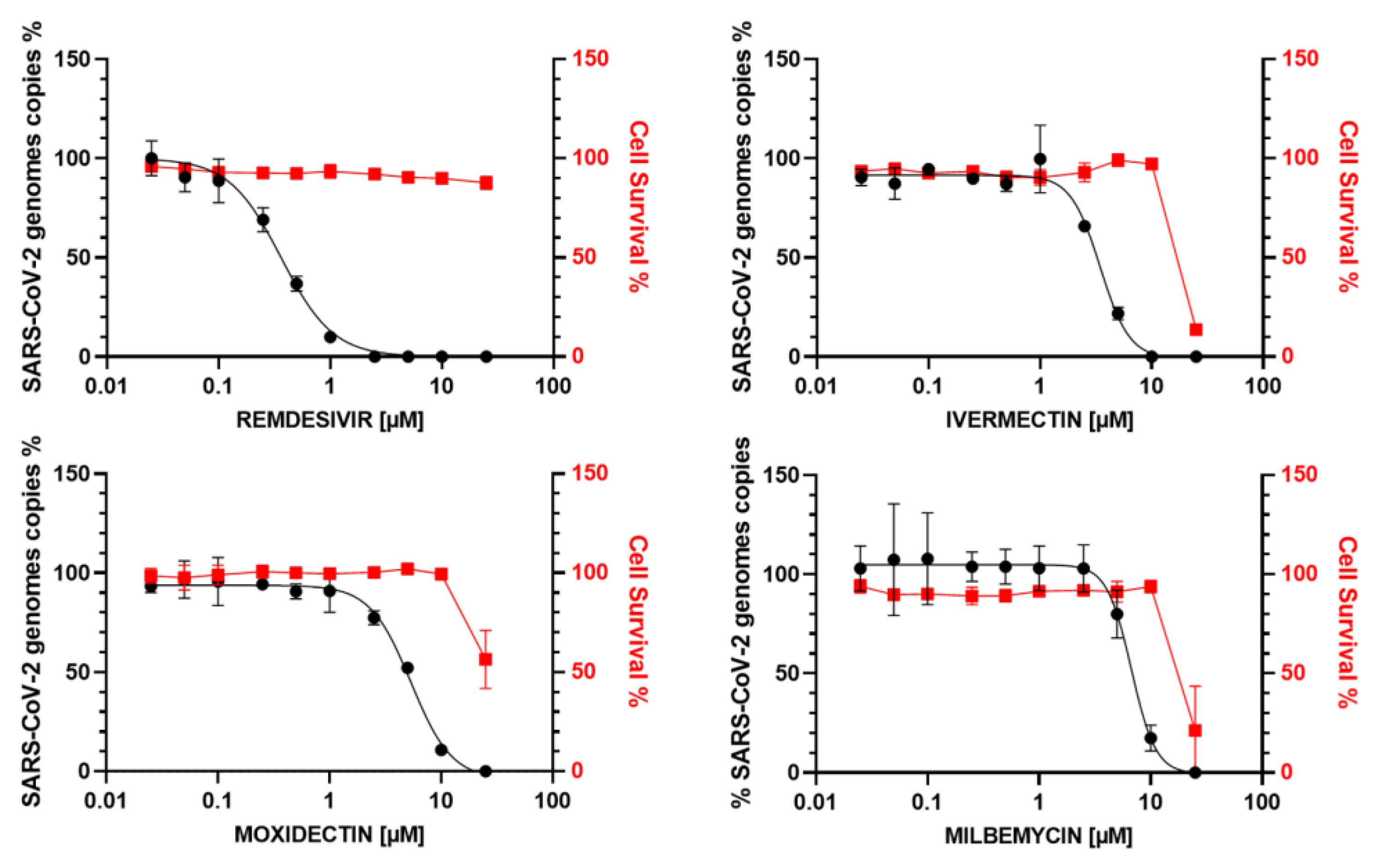
3.1. In Vitro Antiviral Activity of Avermectins or Milbemycins in Simian Vero E6 Cells
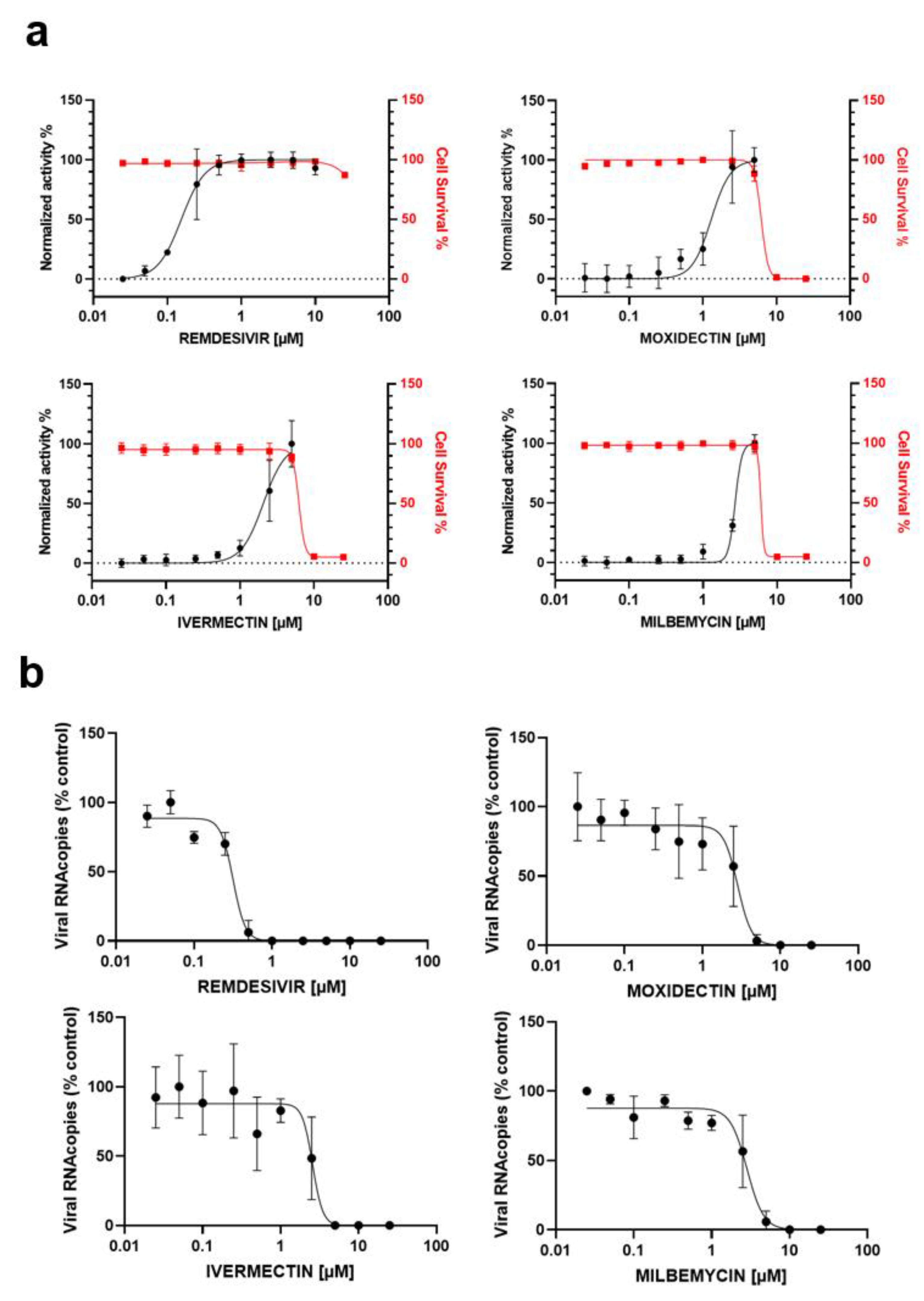
3.2. In Vitro Antiviral Activity of IVM, MOX and MIL in Human Pulmonary Alveolar A549-hACE2 Cells
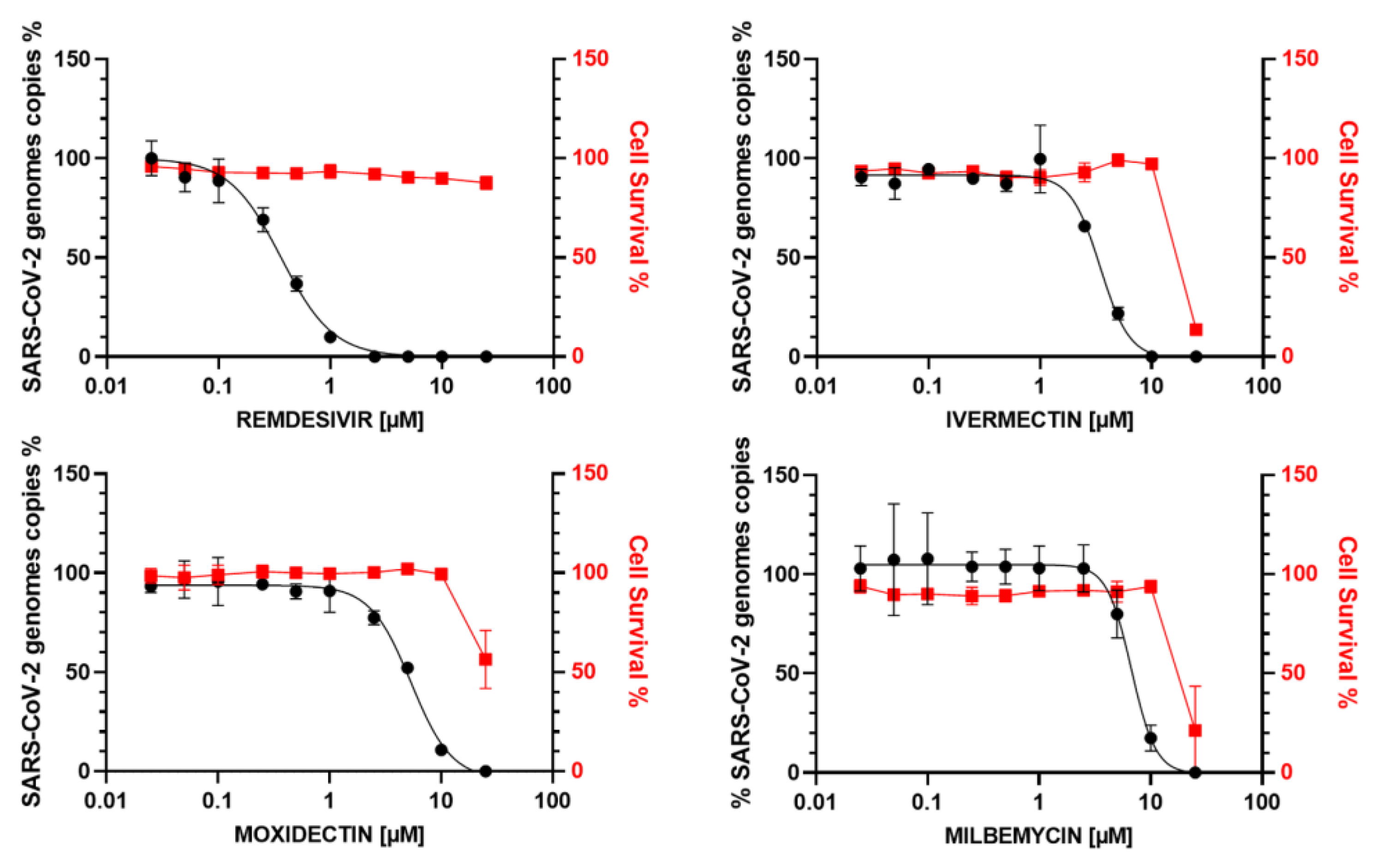
3.3. In Vitro Antiviral Activity of IVM, MOX and MIL in Human Bronchial Calu-3 Cells
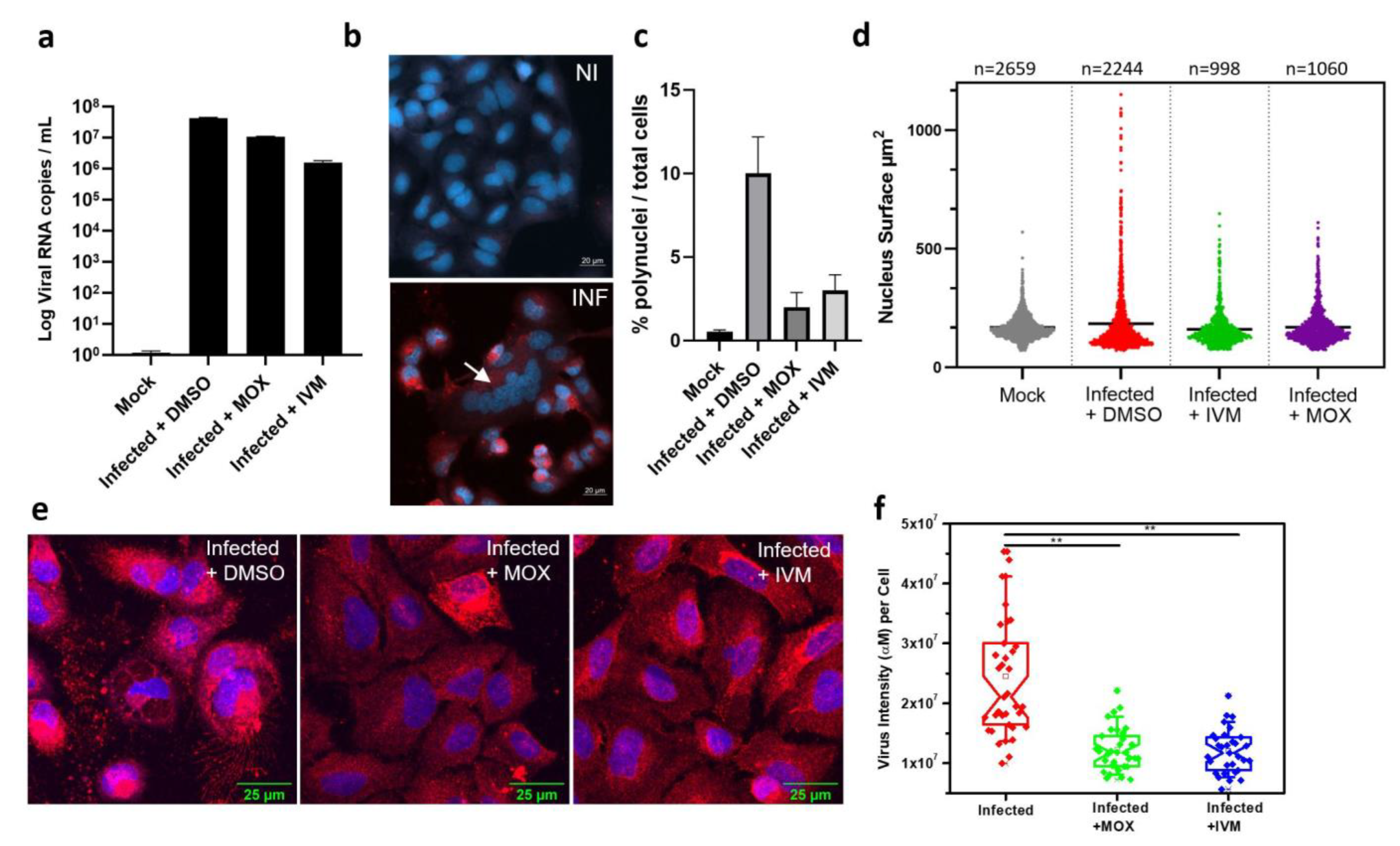
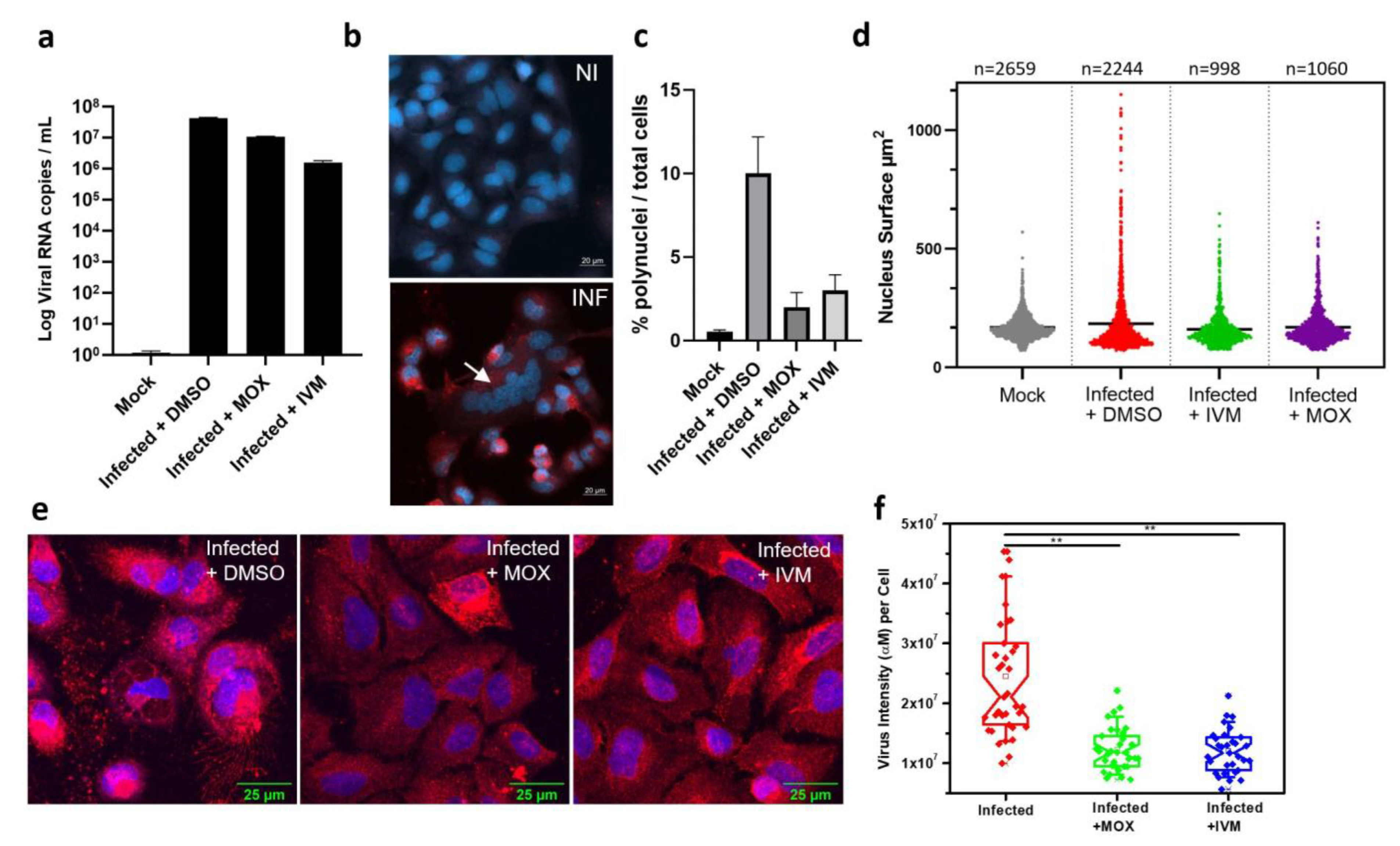
3.4. In Vitro Activity of IVM and MOX on SARS-CoV2 Shows a Reduction in Virus-Induced Cell Syncytia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir–Ritonavir in Adults Hospitalized with Severe COVID-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef]
- The RECOVERY Collaborative Group Effect of Hydroxychloroquine in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2020, 383, 2030–2040. [CrossRef] [PubMed]
- Self, W.H.; Semler, M.W.; Leither, L.M.; Casey, J.D.; Angus, D.C.; Brower, R.G.; Chang, S.Y.; Collins, S.P.; Eppensteiner, J.C.; Filbin, M.R.; et al. Effect of Hydroxychloroquine on Clinical Status at 14 Days in Hospitalized Patients With COVID-19: A Randomized Clinical Trial. JAMA 2020, 324, 2165. [Google Scholar] [CrossRef]
- López-Medina, E.; López, P.; Hurtado, I.C.; Dávalos, D.M.; Ramirez, O.; Martínez, E.; Díazgranados, J.A.; Oñate, J.M.; Chavarriaga, H.; Herrera, S.; et al. Effect of Ivermectin on Time to Resolution of Symptoms Among Adults with Mild COVID-19: A Randomized Clinical Trial. JAMA 2021, 325, 1426. [Google Scholar] [CrossRef]
- The RECOVERY Collaborative Group. Dexamethasone in Hospitalized Patients with COVID-19—Preliminary Report. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef]
- Tsatsakis, A.; Calina, D.; Falzone, L.; Petrakis, D.; Mitrut, R.; Siokas, V.; Pennisi, M.; Lanza, G.; Libra, M.; Doukas, S.G.; et al. SARS-CoV-2 Pathophysiology and Its Clinical Implications: An Integrative Overview of the Pharmacotherapeutic Management of COVID-19. Food Chem. Toxicol. 2020, 146, 111769. [Google Scholar] [CrossRef]
- Heidary, F.; Gharebaghi, R. Ivermectin: A Systematic Review from Antiviral Effects to COVID-19 Complementary Regimen. J. Antibiot. 2020, 73, 593–602. [Google Scholar] [CrossRef]
- Caly, L.; Druce, J.D.; Catton, M.G.; Jans, D.A.; Wagstaff, K.M. The FDA-Approved Drug Ivermectin Inhibits the Replication of SARS-CoV-2 in vitro. Antiviral Res. 2020, 178, 104787. [Google Scholar] [CrossRef]
- Schmith, V.D.; Zhou, J.J.; Lohmer, L.R.L. The Approved Dose of Ivermectin Alone Is not the Ideal Dose for the Treatment of COVID-19. Clin. Pharmacol. Ther. 2020, 108, 762–765. [Google Scholar] [CrossRef]
- Chaccour, C.; Hammann, F.; Ramón-García, S.; Rabinovich, N.R. Ivermectin and COVID-19: Keeping Rigor in Times of Urgency. Am. J. Trop. Med. Hyg. 2020, 102, 1156–1157. [Google Scholar] [CrossRef]
- Bray, M.; Rayner, C.; Noël, F.; Jans, D.; Wagstaff, K. Ivermectin and COVID-19: A Report in Antiviral Research, Widespread Interest, an FDA Warning, Two Letters to the Editor and the Authors’ Responses. Antiviral Res. 2020, 178, 104805. [Google Scholar] [CrossRef]
- Jermain, B.; Hanafin, P.O.; Cao, Y.; Lifschitz, A.; Lanusse, C.; Rao, G.G. Development of a Minimal Physiologically-Based Pharmacokinetic Model to Simulate Lung Exposure in Humans Following Oral Administration of Ivermectin for COVID-19 Drug Repurposing. J. Pharm. Sci. 2020, 109, 3574–3578. [Google Scholar] [CrossRef] [PubMed]
- Campillo, J.T.; Boussinesq, M.; Bertout, S.; Faillie, J.-L.; Chesnais, C.B. Serious Adverse Reactions Associated with Ivermectin: A Systematic Pharmacovigilance Study in Sub-Saharan Africa and in the Rest of the World. PLoS Negl. Trop. Dis. 2021, 15, e0009354. [Google Scholar] [CrossRef]
- Wagstaff, K.M.; Sivakumaran, H.; Heaton, S.M.; Harrich, D.; Jans, D.A. Ivermectin Is a Specific Inhibitor of Importin α/β-Mediated Nuclear Import Able to Inhibit Replication of HIV-1 and Dengue Virus. Biochem. J. 2012, 443, 851–856. [Google Scholar] [CrossRef] [Green Version]
- Hamming, I.; Timens, W.; Bulthuis, M.L.C.; Lely, A.T.; Navis, G.J.; Van Goor, H. Tissue Distribution of ACE2 Protein, the Functional Receptor for SARS Coronavirus. A First Step in Understanding SARS Pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-Converting Enzyme 2 (ACE2) as a SARS-CoV-2 Receptor: Molecular Mechanisms and Potential Therapeutic Target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.N.Y.; Atkinson, S.C.; Wang, C.; Lee, A.; Bogoyevitch, M.A.; Borg, N.A.; Jans, D.A. The Broad Spectrum Antiviral Ivermectin Targets the Host Nuclear Transport Importin α/Β1 Heterodimer. Antiviral Res. 2020, 177, 104760. [Google Scholar] [CrossRef]
- Jans, D.A.; Wagstaff, K.M. Ivermectin as a Broad-Spectrum Host-Directed Antiviral: The Real Deal? Cells 2020, 9, 2100. [Google Scholar] [CrossRef]
- Krause, R.M.; Buisson, B.; Bertrand, S.; Corringer, P.J.; Galzi, J.L.; Changeux, J.P.; Bertrand, D. Ivermectin: A Positive Allosteric Effector of the Alpha7 Neuronal Nicotinic Acetylcholine Receptor. Mol. Pharmacol. 1998, 53, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Changeux, J.-P.; Amoura, Z.; Rey, F.A.; Miyara, M. A Nicotinic Hypothesis for COVID-19 with Preventive and Therapeutic Implications. Comptes Rendus. Biol. 2020, 343, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Awadzi, K.; Opoku, N.O.; Attah, S.K.; Lazdins-Helds, J.; Kuesel, A.C. A Randomized, Single-Ascending-Dose, Ivermectin-Controlled, Double-Blind Study of Moxidectin in Onchocerca Volvulus Infection. PLoS Negl. Trop. Dis. 2014, 8, e2953. [Google Scholar] [CrossRef]
- Kuesel, A.C. Research for New Drugs for Elimination of Onchocerciasis in Africa. Int. J. Parasitol. Drugs Drug Resist. 2016, 6, 272–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opoku, N.O.; Bakajika, D.K.; Kanza, E.M.; Howard, H.; Mambandu, G.L.; Nyathirombo, A.; Nigo, M.M.; Kasonia, K.; Masembe, S.L.; Mumbere, M.; et al. Single Dose Moxidectin versus Ivermectin for Onchocerca Volvulus Infection in Ghana, Liberia, and the Democratic Republic of the Congo: A Randomised, Controlled, Double-Blind Phase 3 Trial. Lancet 2018, 392, 1207–1216. [Google Scholar] [CrossRef] [Green Version]
- Pérez, R.; Cabezas, I.; García, M.; Rubilar, L.; Sutra, J.F.; Galtier, P.; Alvinerie, M. Comparison of the Pharmacokinetics of Moxidectin (Equest) and Ivermectin (Eqvalan) in Horses. J. Vet. Pharmacol. Ther. 1999, 22, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Al-Azzam, S.I.; Fleckenstein, L.; Cheng, K.-J.; Dzimianski, M.T.; McCall, J.W. Comparison of the Pharmacokinetics of Moxidectin and Ivermectin after Oral Administration to Beagle Dogs. Biopharm. Drug Dispos. 2007, 28, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Lyonnais, S.; Hénaut, M.; Neyret, A.; Merida, P.; Cazevieille, C.; Gros, N.; Chable-Bessia, C.; Muriaux, D. Atomic Force Microscopy Analysis of Native Infectious and Inactivated SARS-CoV-2 Virions. Sci. Rep. 2021, 11, 11885. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 Novel Coronavirus (2019-NCoV) by Real-Time RT-PCR. Eurosurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and Chloroquine Effectively Inhibit the Recently Emerged Novel Coronavirus (2019-NCoV) in Vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Choy, K.-T.; Wong, A.Y.-L.; Kaewpreedee, P.; Sia, S.F.; Chen, D.; Hui, K.P.Y.; Chu, D.K.W.; Chan, M.C.W.; Cheung, P.P.-H.; Huang, X.; et al. Remdesivir, Lopinavir, Emetine, and Homoharringtonine Inhibit SARS-CoV-2 Replication in Vitro. Antivir. Res. 2020, 178, 104786. [Google Scholar] [CrossRef]
- Zhang, P.; Ni, H.; Zhang, Y.; Xu, W.; Gao, J.; Cheng, J.; Tao, L. Ivermectin Confers Its Cytotoxic Effects by Inducing AMPK/MTOR-Mediated Autophagy and DNA Damage. Chemosphere 2020, 259, 127448. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, Y.; Liu, K.; Liu, B.; Xu, W.; Gao, J.; Ding, L.; Tao, L. Ivermectin Induces Cell Cycle Arrest and Apoptosis of HeLa Cells via Mitochondrial Pathway. Cell Prolif. 2019, 52, e12543. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell Entry Mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Murgolo, N.; Therien, A.G.; Howell, B.; Klein, D.; Koeplinger, K.; Lieberman, L.A.; Adam, G.C.; Flynn, J.; McKenna, P.; Swaminathan, G.; et al. SARS-CoV-2 Tropism, Entry, Replication, and Propagation: Considerations for Drug Discovery and Development. PLoS Pathog. 2021, 17, e1009225. [Google Scholar] [CrossRef]
- Hoffmann, M.; Mösbauer, K.; Hofmann-Winkler, H.; Kaul, A.; Kleine-Weber, H.; Krüger, N.; Gassen, N.C.; Müller, M.A.; Drosten, C.; Pöhlmann, S. Chloroquine Does Not Inhibit Infection of Human Lung Cells with SARS-CoV-2. Nature 2020, 585, 588–590. [Google Scholar] [CrossRef]
- Buchrieser, J.; Dufloo, J.; Hubert, M.; Monel, B.; Planas, D.; Rajah, M.M.; Planchais, C.; Porrot, F.; Guivel-Benhassine, F.; Van der Werf, S.; et al. Syncytia Formation by SARS-CoV-2-Infected Cells. EMBO J. 2020, 39, e106267. [Google Scholar] [CrossRef]
- Swann, H.; Sharma, A.; Preece, B.; Peterson, A.; Eldridge, C.; Belnap, D.M.; Vershinin, M.; Saffarian, S. Minimal System for Assembly of SARS-CoV-2 Virus like Particles. Sci. Rep. 2020, 10, 21877. [Google Scholar] [CrossRef]
- Xu, R.; Shi, M.; Li, J.; Song, P.; Li, N. Construction of SARS-CoV-2 Virus-Like Particles by Mammalian Expression System. Front. Bioeng. Biotechnol. 2020, 8, 862. [Google Scholar] [CrossRef]
- Plescia, C.B.; David, E.A.; Patra, D.; Sengupta, R.; Amiar, S.; Su, Y.; Stahelin, R.V. SARS-CoV-2 Viral Budding and Entry Can Be Modeled Using BSL-2 Level Virus-like Particles. J. Biol. Chem. 2020, 296, 100103. [Google Scholar] [CrossRef] [PubMed]
- Boson, B.; Legros, V.; Zhou, B.; Siret, E.; Mathieu, C.; Cosset, F.-L.; Lavillette, D.; Denolly, S. The SARS-CoV-2 Envelope and Membrane Proteins Modulate Maturation and Retention of the Spike Protein, Allowing Assembly of Virus-like Particles. J. Biol. Chem. 2020, 296, 100111. [Google Scholar] [CrossRef]
- Zhu, Y.; Chidekel, A.; Shaffer, T.H. Cultured Human Airway Epithelial Cells (Calu-3): A Model of Human Respiratory Function, Structure, and Inflammatory Responses. Crit. Care Res. Pract. 2010, 2010, 394578. [Google Scholar] [CrossRef] [Green Version]
- Arshad, U.; Pertinez, H.; Box, H.; Tatham, L.; Rajoli, R.K.R.; Curley, P.; Neary, M.; Sharp, J.; Liptrott, N.J.; Valentijn, A.; et al. Prioritization of Anti-SARS-Cov-2 Drug Repurposing Opportunities Based on Plasma and Target Site Concentrations Derived from Their Established Human Pharmacokinetics. Clin. Pharmacol. Ther. 2020, 108, 775–790. [Google Scholar] [CrossRef]
- Alvinerie, M.; Escudero, E.; Sutra, J.-F.; Eeckhoutte, C.; Galtier, P. The Pharmacokinetics of Moxidectin after Oral and Subcutaneous Administration to Sheep. Vet. Res. 1998, 29, 113–118. [Google Scholar]
- Lifschitz, A.; Virkel, G.; Imperiale, F.; Sutra, J.F.; Galtier, P.; Lanusse, C.; Alvinerie, M. Moxidectin in Cattle: Correlation between Plasma and Target Tissues Disposition. J. Vet. Pharmacol. Ther. 1999, 22, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Korth-Bradley, J.M.; Parks, V.; Chalon, S.; Gourley, I.; Matschke, K.; Cailleux, K.; Fitoussi, S.; Fleckenstein, L. The Effect of a High-Fat Breakfast on the Pharmacokinetics of Moxidectin in Healthy Male Subjects: A Randomized Phase I Trial. Am. J. Trop. Med. Hyg. 2012, 86, 122–125. [Google Scholar] [CrossRef] [Green Version]
- Cotreau, M.M.; Warren, S.; Ryan, J.L.; Fleckenstein, L.; Vanapalli, S.R.; Brown, K.R.; Rock, D.; Chen, C.-Y.; Schwertschlag, U.S. The Antiparasitic Moxidectin: Safety, Tolerability, and Pharmacokinetics in Humans. J. Clin. Pharmacol. 2003, 43, 1108–1115. [Google Scholar] [CrossRef] [Green Version]
- Korth-Bradley, J.M.; Parks, V.; Chalon, S.; Gourley, I.; Matschke, K.; Gossart, S.; Bryson, P.; Fleckenstein, L. Excretion of Moxidectin into Breast Milk and Pharmacokinetics in Healthy Lactating Women. Antimicrob. Agents Chemother. 2011, 55, 5200–5204. [Google Scholar] [CrossRef] [Green Version]
- Korth-Bradley, J.M.; Parks, V.; Patat, A.; Matschke, K.; Mayer, P.; Fleckenstein, L. Relative Bioavailability of Liquid and Tablet Formulations of the Antiparasitic Moxidectin. Clin. Pharmacol. Drug. Dev. 2012, 1, 32–37. [Google Scholar] [CrossRef]
- Korth-Bradley, J.M.; Parks, V.; Wagner, F.; Chalon, S.; Gourley, I.; Matschke, K.; Gossart, S.; Ripp, S.L.; Fleckenstein, L. Effect of Moxidectin on CYP3A4 Activity as Evaluated by Oral Midazolam Pharmacokinetics in Healthy Subjects. Clin. Pharmacol. Drug Dev. 2014, 3, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Vercruysse, J.; Rew, R.S. Macrocyclic Lactones in Antiparasitic Therapy; CABI Publishing: Oxon, UK; New York, NY, USA, 2002; ISBN 978-0-85199-617-2. [Google Scholar]
- Sarasola, P.; Jernigan, A.D.; Walker, D.K.; Castledine, J.; Smith, D.G.; Rowan, T.G. Pharmacokinetics of Selamectin Following Intravenous, Oral and Topical Administration in Cats and Dogs. J. Vet. Pharmacol. Ther. 2002, 25, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Toutain, P.L.; Upson, D.W.; Terhune, T.N.; McKenzie, M.E. Comparative Pharmacokinetics of Doramectin and Ivermectin in Cattle. Vet. Parasitol. 1997, 72, 3–8. [Google Scholar] [CrossRef]
- Pérez, R.; Cabezas, I.; Godoy, C.; Rubilar, L.; Muñoz, L.; Arboix, M.; Castells, G.; Alvinerie, M. Pharmacokinetics of Doramectin and Ivermectin After Oral Administration in Horses. Vet. J. 2002, 163, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Holmstrom, S.D.; Totten, M.L.; Newhall, K.B.; Qiao, M.; Riggs, K.L. Pharmacokinetics of Spinosad and Milbemycin Oxime Administered in Combination and Separately per Os to Dogs. J. Vet. Pharmacol. Ther. 2012, 35, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Ide, J.; Okasaki, T.; Ono, M. Milbemycin: Discovery and Development. Annu. Rep. Sankyo Res. Lab. 1993, 45, 1–98. [Google Scholar]
- Gardon, J.; Gardon-Wendel, N.; Demanga-Ngangue; Kamgno, J.; Chippaux, J.P.; Boussinesq, M. Serious Reactions after Mass Treatment of Onchocerciasis with Ivermectin in an Area Endemic for Loa Loa Infection. Lancet 1997, 350, 18–22. [Google Scholar] [CrossRef]
- Baudou, E.; Lespine, A.; Durrieu, G.; André, F.; Gandia, P.; Durand, C.; Cunat, S. Serious Ivermectin Toxicity and Human ABCB1 Nonsense Mutations. N. Engl. J. Med. 2020, 383, 787–789. [Google Scholar] [CrossRef] [PubMed]
- Bishop, B.F.; Bruce, C.I.; Evans, N.A.; Goudie, A.C.; Gration, K.A.F.; Gibson, S.P.; Pacey, M.S.; Perry, D.A.; Walshe, N.D.A.; Witty, M.J. Selamectin: A Novel Broad-Spectrum Endectocide for Dogs and Cats. Vet. Parasitol. 2000, 91, 163–176. [Google Scholar] [CrossRef]
- El-Saber Batiha, G.; Alqahtani, A.; Ilesanmi, O.B.; Saati, A.A.; El-Mleeh, A.; Hetta, H.F.; Magdy Beshbishy, A. Avermectin Derivatives, Pharmacokinetics, Therapeutic and Toxic Dosages, Mechanism of Action, and Their Biological Effects. Pharmaceuticals 2020, 13, 196. [Google Scholar] [CrossRef] [PubMed]
- Merola, V.M.; Eubig, P.A. Toxicology of Avermectins and Milbemycins (Macrocylic Lactones) and the Role of P-Glycoprotein in Dogs and Cats. Vet. Clin. N. Am. Small Anim. Pract. 2012, 42, 313–333. [Google Scholar] [CrossRef] [Green Version]
- Prichard, R.; Ménez, C.; Lespine, A. Moxidectin and the Avermectins: Consanguinity but Not Identity. Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 134–153. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, S.; Rheinstein, P.H. Ivermectin Docks to the SARS-CoV-2 Spike Receptor-Binding Domain Attached to ACE2. In Vivo 2020, 34, 3023–3026. [Google Scholar] [CrossRef]
- WHO Solidarity Trial Consortium; Pan, H.; Peto, R.; Henao-Restrepo, A.-M.; Preziosi, M.-P.; Sathiyamoorthy, V.; Abdool Karim, Q.; Alejandria, M.M.; Hernández García, C.; Kieny, M.-P.; et al. Repurposed Antiviral Drugs for COVID-19—Interim WHO Solidarity Trial Results. N. Engl. J. Med. 2020, 384, 497–511. [Google Scholar] [CrossRef]
- Chaccour, C.; Abizanda, G.; Irigoyen-Barrio, Á.; Casellas, A.; Aldaz, A.; Martínez-Galán, F.; Hammann, F.; Gil, A.G. Nebulized Ivermectin for COVID-19 and Other Respiratory Diseases, a Proof of Concept, Dose-Ranging Study in Rats. Sci. Rep. 2020, 10, 17073. [Google Scholar] [CrossRef]
- Dinesh Kumar, N.; Ter Ellen, B.M.; Bouma, E.M.; Troost, B.; Van De Pol, D.P.I.; Van Der Ende-Metselaar, H.H.; Van Gosliga, D.; Apperloo, L.; Carpaij, O.A.; Van Den Berge, M.; et al. Moxidectin and Ivermectin Inhibit Sars-Cov-2 Replication in Vero E6 Cells But Not in Human Primary Airway Epithelium Cells. Antimicrob. Agents Chemother. 2021, AAC0154321. [Google Scholar] [CrossRef]
- De Melo, G.D.; Lazarini, F.; Larrous, F.; Feige, L.; Kornobis, E.; Levallois, S.; Marchio, A.; Kergoat, L.; Hardy, D.; Cokelaer, T.; et al. Attenuation of clinical and immunological outcomes during SARS-CoV-2 infection by ivermectin. EMBO Mol. Med. 2021, 13, e14122. [Google Scholar] [CrossRef]
- Tellez, D.; Dayal, S.; Phan, P.; Mawley, A.; Shah, K.; Consunji, G.; Tellez, C.; Ruiz, K.; Sabnis, R.; Dayal, S.; et al. Analysis of COVID-19 on Diagnosis, Vaccine, Treatment, and Pathogenesis with Clinical Scenarios. Clin. Pract. 2021, 11, 309–321. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| COMPOUND | EC50 (µM) MTS | EC50 (µM) ToxGlo | EC50 (µM) RT-qPCR | CC50 (µM) | SI |
|---|---|---|---|---|---|
| VERO E6 cells | |||||
| REMDESIVIR | 0.55 | 1 | >50 | >49 | |
| MOXIDECTIN | 5.3 | 3.5 | 8.8 | 1.66 | |
| DORAMECTIN | 2.4 | 1.1 | 8.3 | 3.45 | |
| SELAMECTIN | 3.9 | 2.3 | 8.3 | 2.1 | |
| IVERMECTIN | 3.6 | 3 | 8 | 2.2 | |
| MILBEMYCIN | 3.8 | 2.6 | 7.8 | 2.05 | |
| A549 hACE2 cells | |||||
| REMDESIVIR | 0.15 | 0.31 | >10 | >30 | |
| MOXIDECTIN | 1.3 | 1.27 | 6.2 | 4.7 | |
| IVERMECTIN | 2.1 | 2.36 | 6.2 | 2.8 | |
| MILBEMYCIN | 2.7 | 2.8 | 6.1 | 2.2 | |
| Calu-3 cells | |||||
| REMDESIVIR | 0.35 | >25 | >70 | ||
| MOXIDECTIN | 5.55 | 26 | 4.7 | ||
| IVERMECTIN | 3.36 | 22 | 5 | ||
| MILBEMYCIN | 6.78 | 23 | 3.3 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chable-Bessia, C.; Boullé, C.; Neyret, A.; Swain, J.; Hénaut, M.; Merida, P.; Gros, N.; Makinson, A.; Lyonnais, S.; Chesnais, C.; et al. Low Selectivity Indices of Ivermectin and Macrocyclic Lactones on SARS-CoV-2 Replication In Vitro. COVID 2022, 2, 60-75. https://doi.org/10.3390/covid2010005
Chable-Bessia C, Boullé C, Neyret A, Swain J, Hénaut M, Merida P, Gros N, Makinson A, Lyonnais S, Chesnais C, et al. Low Selectivity Indices of Ivermectin and Macrocyclic Lactones on SARS-CoV-2 Replication In Vitro. COVID. 2022; 2(1):60-75. https://doi.org/10.3390/covid2010005
Chicago/Turabian StyleChable-Bessia, Christine, Charlotte Boullé, Aymeric Neyret, Jitendriya Swain, Mathilde Hénaut, Peggy Merida, Nathalie Gros, Alain Makinson, Sébastien Lyonnais, Cédric Chesnais, and et al. 2022. "Low Selectivity Indices of Ivermectin and Macrocyclic Lactones on SARS-CoV-2 Replication In Vitro" COVID 2, no. 1: 60-75. https://doi.org/10.3390/covid2010005
APA StyleChable-Bessia, C., Boullé, C., Neyret, A., Swain, J., Hénaut, M., Merida, P., Gros, N., Makinson, A., Lyonnais, S., Chesnais, C., & Muriaux, D. (2022). Low Selectivity Indices of Ivermectin and Macrocyclic Lactones on SARS-CoV-2 Replication In Vitro. COVID, 2(1), 60-75. https://doi.org/10.3390/covid2010005