
The Role of Surface in the Pathogenesis and Treatment of COVID-19


Abstract
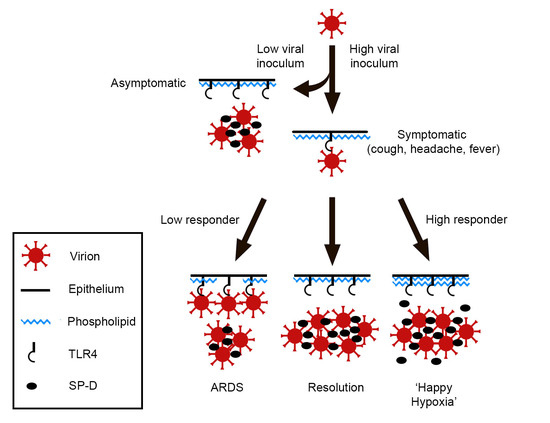
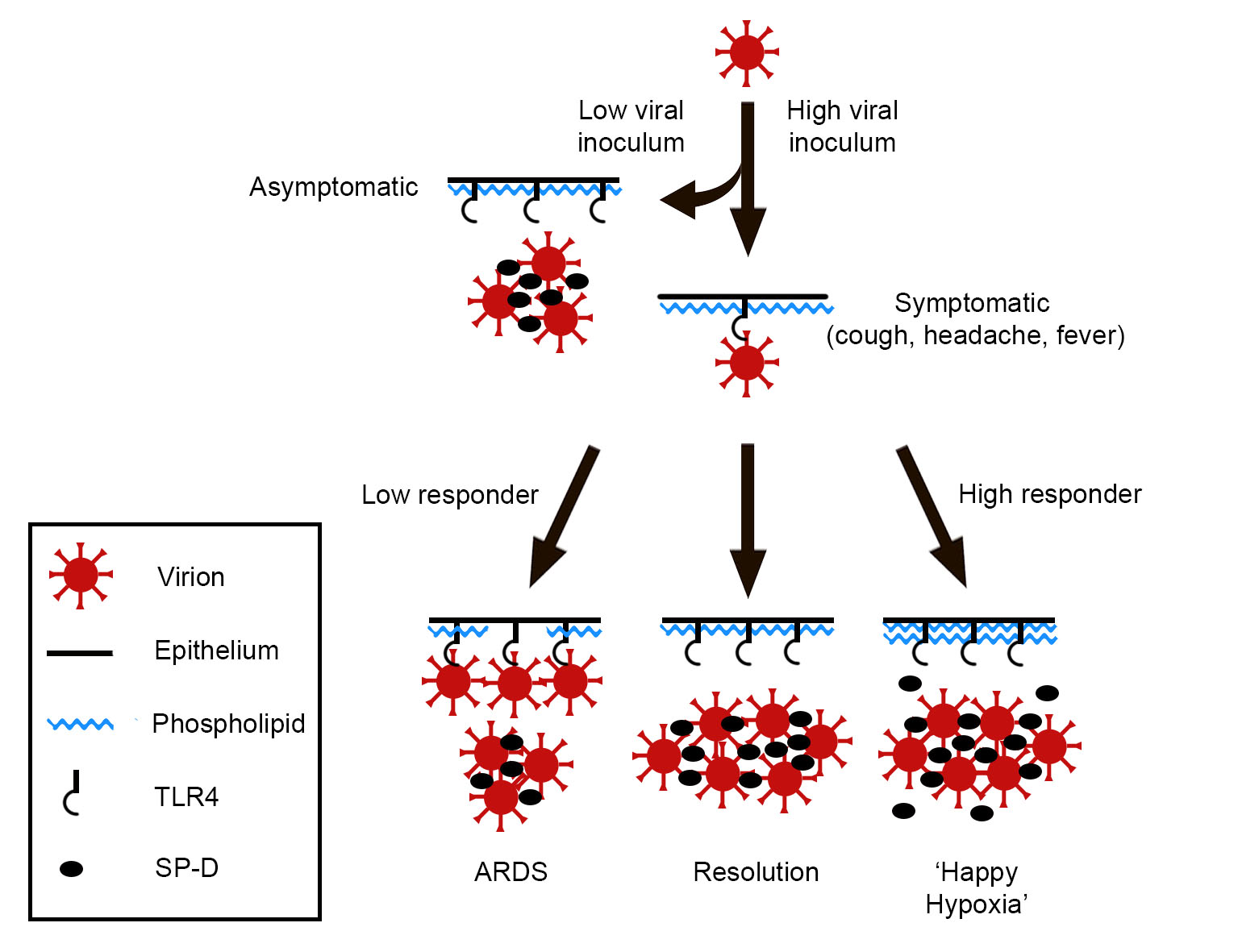{kind=link}
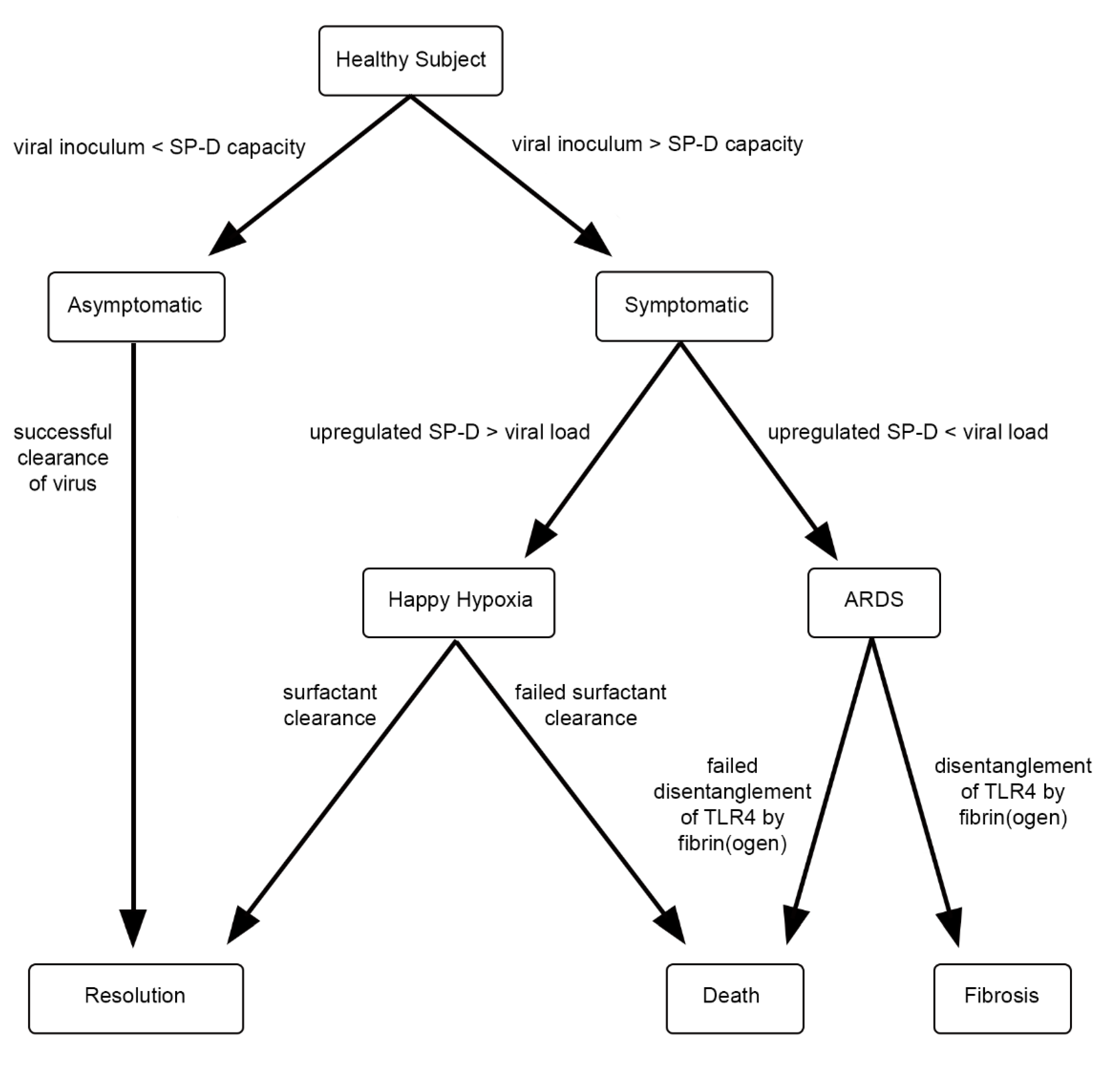{kind=link}
1. Background, Introduction, and Rationale
2. Hypothesis
3. Discussion
4. Closing
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Eythorsson, E.; Helgason, D.; Ingvarsson, R.F.; Bjornsson, H.K.; Olafsdottir, L.B.; Bjarnadottir, V.; Runolfsdottir, H.L.; Bjarnadottir, S.; Agustsson, A.S.; Oskarsdottir, K.; et al. Clinical spectrum of coronavirus disease 2019 in Iceland: Population based cohort study. BMJ 2020, 371, m4529. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.B.; Shah, R.D.; Retzinger, D.G.; Retzinger, A.C.; Retzinger, D.A.; Retzinger, G.S. Competing bioaerosols may influence the seasonality of influenza-like illnesses, including COVID-19. The Chicago experience. Pathogens 2021, 10, 1204. [Google Scholar] [CrossRef] [PubMed]
- Sohn, K.M.; Lee, S.-G.; Kim, H.J.; Cheon, S.; Jeong, H.; Lee, J.; Kim, I.S.; Silwal, P.; Kim, Y.J.; Paik, S.; et al. COVID-19 patients upregulate toll-like receptor 4-mediated inflammatory signaling that mimics bacterial sepsis. J. Korean Med Sci. 2020, 35, e343. [Google Scholar] [CrossRef]
- Zhao, Y.; Kuang, M.; Li, J.; Zhu, L.; Jia, Z.; Guo, X.; Hu, Y.; Kong, J.; Yin, H.; Wang, X.; et al. SARS-CoV-2 spike protein interacts with and activates TLR41 [published correction appears in Cell Res. 27 April 2021]. Cell Res. 2021, 31, 818–820. [Google Scholar] [CrossRef]
- Aboudounya, M.M.; Heads, R.J. COVID-19 and toll-like receptor 4 (TLR4): SARS-CoV-2 may bind and activate TLR4 to increase ACE2 expression, facilitating entry and causing hyperinflammation. Mediat. Inflamm. 2021, 2021, 8874339. [Google Scholar] [CrossRef]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef]
- Marchant, D.; Singhera, G.K.; Utokaparch, S.; Hackett, T.L.; Boyd, J.H.; Luo, Z.; Si, X.; Dorscheid, D.R.; McManus, B.M.; Hegele, R.G. Toll-Like Receptor 4-Mediated Activation of p38 mitogen-activated protein kinase is a determinant of respiratory virus entry and tropism. J. Virol. 2010, 84, 11359–11373. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Armstrong, L.; Medford, A.; Uppington, K.M.; Robertson, J.; Witherden, I.R.; Tetley, T.D.; Millar, A.B. Expression of functional toll-like receptor-2 and -4 on alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 2004, 31, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Juarez, E.; Nuñez, C.; Sada, E.; Ellner, J.J.; Schwander, S.K.; Torres, M. Differential expression of Toll-like receptors on human alveolar macrophages and autologous peripheral monocytes. Respir. Res. 2010, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Min, H.; Cho, W.-H.; Lee, H.; Choi, B.; Kim, Y.-J.; Lee, H.; Joo, Y.; Jung, S.J.; Choi, S.-Y.; Lee, S.; et al. Association of TRPV1 and TLR4 through the TIR domain potentiates TRPV1 activity by blocking activation-induced desensitization. Mol. Pain 2018, 14, 1744806918812636. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H. The emerging role of TRPV1 in airway inflammation. Allergy Asthma Immunol. Res. 2018, 10, 187–188. [Google Scholar] [CrossRef]
- Omar, S.; Clarke, R.; Abdullah, H.; Brady, C.; Corry, J.; Wintrt, H.; Touzelet, O.; Power, U.F.; Lundy, F.; McGarvey, L.P.A.; et al. Respiratory virus infection up-regulates TRPV1, TRPA1 and ASICS3 receptors on airway cells. PLoS ONE 2017, 12, e0171681. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.T.; Nakayama, T.; Wu, C.-T.; Goltsev, Y.; Jiang, S.; Gall, P.A.; Liao, C.-K.; Shih, L.-C.; Schürch, C.M.; McIlwain, D.R.; et al. ACE2 localizes to the respiratory cilia and is not increased by ACE inhibitors or ARBs. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Kuek, L.E.; Lee, R.J. First contact: The role of respiratory cilia in host-pathogen interactions in the airways. Am. J. Physiol. Cell. Mol. Physiol. 2020, 319, L603–L619. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, Y.; Liu, Q.; Yao, Q.; Wang, X.; Zhang, H.; Chen, R.; Ren, L.; Min, J.; Deng, F.; et al. SARS-CoV-2 cell tropism and multiorgan infection. Cell Discov. 2021, 7, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, Y.; Gao, R.; Lu, R.; Han, K.; Wu, G.; Tan, W. Detection of SARS-CoV-2 in different types of clinical specimens. JAMA 2020, 323, 1843–1844. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Serrano, D.A.; Roy-Vallejo, E.; Cruz, N.D.Z.; Ramírez, A.M.; Rodríguez-García, S.C.; Arevalillo-Fernández, N.; Galván-Román, J.M.; García-Rodrigo, L.F.; Vega-Piris, L.; Llano, M.C.; et al. Detection of SARS-CoV-2 RNA in serum is associated with increased mortality risk in hospitalized COVID-19 patients. Sci. Rep. 2021, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Guallar, M.P.; Meiriño, R.; Donat-Vargas, C.; Corral, O.; Jouvé, N.; Soriano, V. Inoculum at the time of SARS-CoV-2 exposure and risk of disease severity. Int. J. Infect. Dis. 2020, 97, 290–292. [Google Scholar] [CrossRef]
- Ryan, K.A.; Bewley, K.R.; Fotheringham, S.A.; Slack, G.S.; Brown, P.; Hall, Y.; Wand, N.I.; Marriott, A.C.; Cavell, B.E.; Tree, J.A.; et al. Dose-dependent response to infection with SARS-CoV-2 in the ferret model and evidence of protective immunity. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Van Damme, W.; Dahake, R.; van de Pas, R.; Vanham, G.; Assefa, Y. COVID-19: Does the infectious inoculum dose-response relationship contribute to understanding heterogeneity in disease severity and transmission dynamics? Med Hypotheses 2020, 146, 110431. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Mallampalli, R.K. The role of surfactant in lung disease and host defense against pulmonary infections. Ann. Am. Thorac. Soc. 2015, 12, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.M.; Whitsett, J.A.; Hartshorn, K.; Crouch, E.C.; Korfhagen, T.R. Surfactant protein D enhances clearance of influenza a virus from the lung in vivo. J. Immunol. 2001, 167, 5868–5873. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.M.; Elliott, J.; Whitsett, J.A.; Srikiatkhachorn, A.; Crouch, E.; DeSilva, N.; Korfhagen, T. Surfactant protein-D enhances phagocytosis and pulmonary clearance of respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol. 2004, 31, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.; Phipps, M.J.; Clark, H.W.; Skylaris, C.-K.; Madsen, J. Surfactant proteins A and D: Trimerized innate immunity proteins with an affinity for viral fusion proteins. J. Innate Immun. 2018, 11, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Leth-Larsen, R.; Zhong, F.; Chow, V.; Holmskov, U.; Lu, J. The SARS coronavirus spike glycoprotein is selectively recognized by lung surfactant protein D and activates macrophages. Immunobiology 2007, 212, 201–211. [Google Scholar] [CrossRef]
- Arroyo, R.; Grant, S.; Kingma, P. Late breaking abstract—The innate immune collectin surfactant protein SP-D binds to SARS-CoV-2 spike-protein. Eur. Resp. J. 2020, 56, 1055. [Google Scholar]
- Wu, Y.P.; Liu, Z.H.; Wei, R.; Pan, S.D.; Mao, N.Y.; Chen, B.; Han, J.J.; Zhang, F.S.; Holmskov, U.; Xia, Z.L.; et al. Elevated Plasma Surfactant Protein D (SP-D) levels and a direct correlation with anti-severe acute respiratory syndrome coronavirus-specific IgG antibody in SARS patients. Scand. J. Immunol. 2009, 69, 508–515. [Google Scholar] [CrossRef]
- Hsieh, M.H.; Beirag, N.; Murugaiah, V.; Chou, Y.C.; Kuo, W.S.; Kao, H.F.; Madan, T.; Kishore, U.; Wang, J.Y. Human Surfactant Protein D Binds Spike Protein and Acts as an Entry Inhibitor of SARS-CoV-2 Pseudotyped Viral Particles. Front Immunol. 2021, 12, 641360. [Google Scholar] [CrossRef]
- Madan, T.; Biswas, B.; Varghese, P.M.; Subedi, R.; Pandit, H.; Idicula-Thomas, S.; Kundu, I.; Rooge, S.; Agarwal, R.; Tripathi, D.M.; et al. A Recombinant Fragment of Human Surfactant Protein D Binds Spike Protein and Inhibits Infectivity and Replication of SARS-CoV-2 in Clinical Samples. Am J Respir Cell Mol Biol. 2021, 65, 41–53. [Google Scholar] [CrossRef]
- Ohya, M.; Nishitani, C.; Sano, H.; Yamada, C.; Mitsuzawa, H.; Shimizu, T.; Saito, T.; Smith, K.; Crouch, E.; Kuroki, Y. Human pulmonary surfactant protein D binds the extracellular domains of toll-like receptors 2 and 4 through the Carbohydrate Recognition domain by a mechanism different from its binding to phosphatidylinositol and lipopolysaccharide. Biochemistry 2006, 45, 8657–8664. [Google Scholar] [CrossRef]
- Yamazoe, M.; Nishitani, C.; Takahashi, M.; Katoh, T.; Ariki, S.; Shimizu, T.; Mitsuzawa, H.; Sawada, K.; Voelker, D.R.; Takahashi, H.; et al. Pulmonary Surfactant Protein D Inhibits Lipopolysaccharide (LPS)-induced Inflammatory cell responses by altering lps binding to its receptors. J. Biol. Chem. 2008, 283, 35878–35888. [Google Scholar] [CrossRef]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020, 92, 2105–2113. [Google Scholar] [CrossRef]
- Saka, R.; Wakimoto, T.; Nishiumi, F.; Sasaki, T.; Nose, S.; Fukuzawa, M.; Oue, T.; Yanagihara, I.; Okuyama, H. Surfactant protein-D attenuates the lipopolysaccharide-induced inflammation in human intestinal cells overexpressing toll-like receptor 4. Pediatr. Surg. Int. 2015, 32, 59–63. [Google Scholar] [CrossRef]
- Korfhagen, T.R.; Sheftelyevich, V.; Burhans, M.S.; Bruno, M.D.; Ross, G.F.; Wert, S.E.; Stahlman, M.T.; Jobe, A.H.; Ikegami, M.; Whitsett, J.A.; et al. Surfactant protein-D regulates surfactant phospholipid homeostasis in vivo. J. Biol. Chem. 1998, 273, 28438–28443. [Google Scholar] [CrossRef]
- Voelker, D.R.; Numata, M. Phospholipid regulation of innate immunity and respiratory viral infection. J. Biol. Chem. 2019, 294, 4282–4289. [Google Scholar] [CrossRef]
- Borczuk, A.C.; Salvatore, S.P.; Seshan, S.V.; Patel, S.S.; Bussel, J.B.; Mostyka, M.; Elsoukkary, S.; He, B.; DEL Vecchio, C.; Fortarezza, F.; et al. COVID-19 pulmonary pathology: A multi-institutional autopsy cohort from Italy and New York City. Mod. Pathol. 2020, 33, 1–13. [Google Scholar] [CrossRef]
- Winkler, C.; Atochina-Vasserman, E.N.; Holz, O.; Beers, M.F.; Erpenbeck, V.J.; Krug, N.; Roepcke, S.; Lauer, G.; Elmlinger, M.; Hohlfeld, J.M. Comprehensive characterisation of pulmonary and serum surfactant protein D in COPD. Respir. Res. 2011, 12, 29. [Google Scholar] [CrossRef] [PubMed]
- Jawed, S.; Mannan, N.; Qureshi, M.A. Association of surfactant protein-D with obesity. J. Ayub. Med. Coll. Abbottabad JAMC 2017, 28, 489–492. [Google Scholar]
- Kale, K.; Vishwekar, P.; Balsarkar, G.; Jassawalla, M.J.; Alkahtani, S.; Kishore, U.; Sawant, G.; Madan, T. Serum levels of collectins are sustained during pregnancy: Surfactant protein D levels are dysregulated prior to missed abortion. Reprod. Sci. 2020, 27, 1894–1908. [Google Scholar] [CrossRef] [PubMed]
- Alberca, R.W.; Lima, J.C.; de Oliveira, E.A.; Gozzi-Silva, S.C.; Ramos, Y.L.; Andrade, M.M.D.S.; Beserra, D.R.; Oliveira, L.D.M.; Branco, A.C.C.C.; Pietrobon, A.J.; et al. COVID-19 Disease course in former smokers, smokers and COPD patients. Front. Physiol. 2021, 11, 637627. [Google Scholar] [CrossRef]
- Ellington, S.; Strid, P.; Tong, V.T.; Woodworth, K.; Galang, R.R.; Zambrano, L.D.; Nahabedian, J.; Anderson, K.; Gilboa, S.M. Characteristics of women of reproductive age with laboratory-confirmed SARS-CoV-2 infection by pregnancy status—United States, January 22–June 7, 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 769–775. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, T.; Wang, Y.; Xia, L. The centrality of obesity in the course of severe COVID-19. Front. Endocrinol. 2021, 12, 620566. [Google Scholar] [CrossRef]
- Notter, R.H. Lung Surfactants: Basic Science and Clinical Applications; Dekker, M., Ed.; CRC Press: New York, NY, USA, 2008; pp. 140–142. [Google Scholar]
- Jiang, N.; Liu, Y.N.; Bao, J.; Li, R.; Ni, W.-T.; Tan, X.-Y.; Xu, Y.; Peng, L.-P.; Wang, X.-R.; Zeng, Y.-M.; et al. Clinical features and risk factors associated with severe COVID-19 patients in China [published online ahead of print, 1 April 2021. Chin. Med. J. 2021, 134, 944. [Google Scholar] [CrossRef]
- Sterne, J.A.C.; Murthy, S.; Diaz, S.; Slutsky, A.S.; Villar, J.; Angus, D.C.; Annane, D.; Azevedo, L.C.P.; Berwanger, O.; Cavalcanti, A.B.; et al. WHO rapid evidence appraisal for COVID-19 therapies (REACT) working group. Association between administration of systemic corticosteroids and mortality among critically Ill patients with COVID-19: A meta-analysis. JAMA 2020, 324, 1330–1341. [Google Scholar] [CrossRef]
- The RECOVERY Collaborative Group dexamethasone in hospitalized patients with Covid-19. New. Engl. J. Med. 2021, 384, 693–704. [CrossRef]
- Sorensen, G.L. Surfactant protein D in respiratory and non-respiratory diseases. Front. Med. 2018, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Huang, I.; Lim, M.A.; Pranata, R. Diabetes mellitus is associated with increased mortality and severity of disease in COVID-19 pneumonia—A systematic review, meta-analysis, and meta-regression. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Whyte, C.S.; Morrow, G.B.; Mitchell, J.L.; Chowdary, P.; Mutch, N.J. Fibrinolytic abnormalities in acute respiratory distress syndrome (ARDS) and versatility of thrombolytic drugs to treat COVID-19. J. Thromb. Haemost. 2020, 18, 1548–1555. [Google Scholar] [CrossRef] [PubMed]
- Smiley, S.T.; King, J.A.; Hancock, W.W. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J. Immunol. 2001, 167, 2887–2894. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.M.; Chu, C.M.; Cheng, V.; Chan, K.; Hung, I.F.N.; Poon, L.; Law, K.; Tang, B.; Hon, T.; Chan, C.; et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: A prospective study. Lancet 2003, 361, 1767–1772. [Google Scholar] [CrossRef]
- Ojo, A.S.; Balogun, S.A.; Williams, O.T.; Ojo, O.S. Pulmonary Fibrosis in COVID-19 Survivors: Predictive factors and risk reduction strategies. Pulm. Med. 2020, 2020, 1–10. [Google Scholar] [CrossRef]
- Salvaterra, E.; Campo, I. Pulmonary alveolar proteinosis: From classification to therapy. Breathe 2020, 16, 200018. [Google Scholar] [CrossRef]
- Gillespie, M.; Flannery, P.; Schumann, J.A.; Dincher, N.; Mills, R.; Can, A. Crazy-Paving: A computed tomographic finding of coronavirus disease 2019. Clin. Pr. Cases Emerg. Med. 2020, 4, 461–463. [Google Scholar] [CrossRef]
- Kawamoto, T.; Ii, M.; Kitazaki, T.; Iizawa, Y.; Kimura, H. TAK-242 selectively suppresses Toll-like receptor 4-signaling mediated by the intracellular domain. Eur. J. Pharmacol. 2008, 584, 40–48. [Google Scholar] [CrossRef]
- Raja, S.G.; Dreyfus, G.D. Eritoran: The evidence of its therapeutic potential in sepsis. Core Évid. 2008, 2, 199–207. [Google Scholar] [CrossRef]
- Neal, M.D.; Jia, H.; Eyer, B.; Good, M.; Guerriero, C.; Sodhi, C.P.; Afrazi, A.; Prindle, T., Jr.; Ma, C.; Branca, M.; et al. Discovery and Validation of a New Class of Small Molecule Toll-Like Receptor 4 (TLR4) Inhibitors. PLoS ONE 2013, 8, e65779. [Google Scholar] [CrossRef]
- Doolittle, R.F.; McNamara, K.; Lin, K. Correlating structure and function during the evolution of fibrinogen-related domains. Protein Sci. 2012, 21, 1808–1823. [Google Scholar] [CrossRef]
- Khan, S.; Wardill, H.R.; Bowen, J.M. Role of toll-like receptor 4 (TLR4)-mediated interleukin-6 (IL-6) production in chemotherapy-induced mucositis. Cancer Chemother. Pharmacol. 2018, 82, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Cruz, A.S.; Mendes-Frias, A.; Oliveira, A.I.; Dias, L.; Matos, A.R.; Carvalho, A.; Capela, C.; Pedrosa, J.; Gil Castro, A.; Silvestre, R. Interleukin-6 is a biomarker for the development of fatal severe acute respiratory syndrome coronavirus 2 pneumonia. Front. Immunol. 2021, 12, 613422. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Retzinger, A.C.; Retzinger, G.S. The Role of Surface in the Pathogenesis and Treatment of COVID-19. COVID 2021, 1, 465-471. https://doi.org/10.3390/covid1020040
Retzinger AC, Retzinger GS. The Role of Surface in the Pathogenesis and Treatment of COVID-19. COVID. 2021; 1(2):465-471. https://doi.org/10.3390/covid1020040
Chicago/Turabian StyleRetzinger, Andrew C., and Gregory S. Retzinger. 2021. "The Role of Surface in the Pathogenesis and Treatment of COVID-19" COVID 1, no. 2: 465-471. https://doi.org/10.3390/covid1020040
APA StyleRetzinger, A. C., & Retzinger, G. S. (2021). The Role of Surface in the Pathogenesis and Treatment of COVID-19. COVID, 1(2), 465-471. https://doi.org/10.3390/covid1020040