


Nanoheterogeneity in Protic and Aprotic Alkylimidazolium Bistriflimide Ionic Liquids



Abstract
1. Introduction
2. Methodology
2.1. Molecular Dynamics Simulations
2.2. Functions Characterizing Nanoheterogeneity
2.3. The Gibbs Free Energy of Cavity Formation
3. Results and Discussion
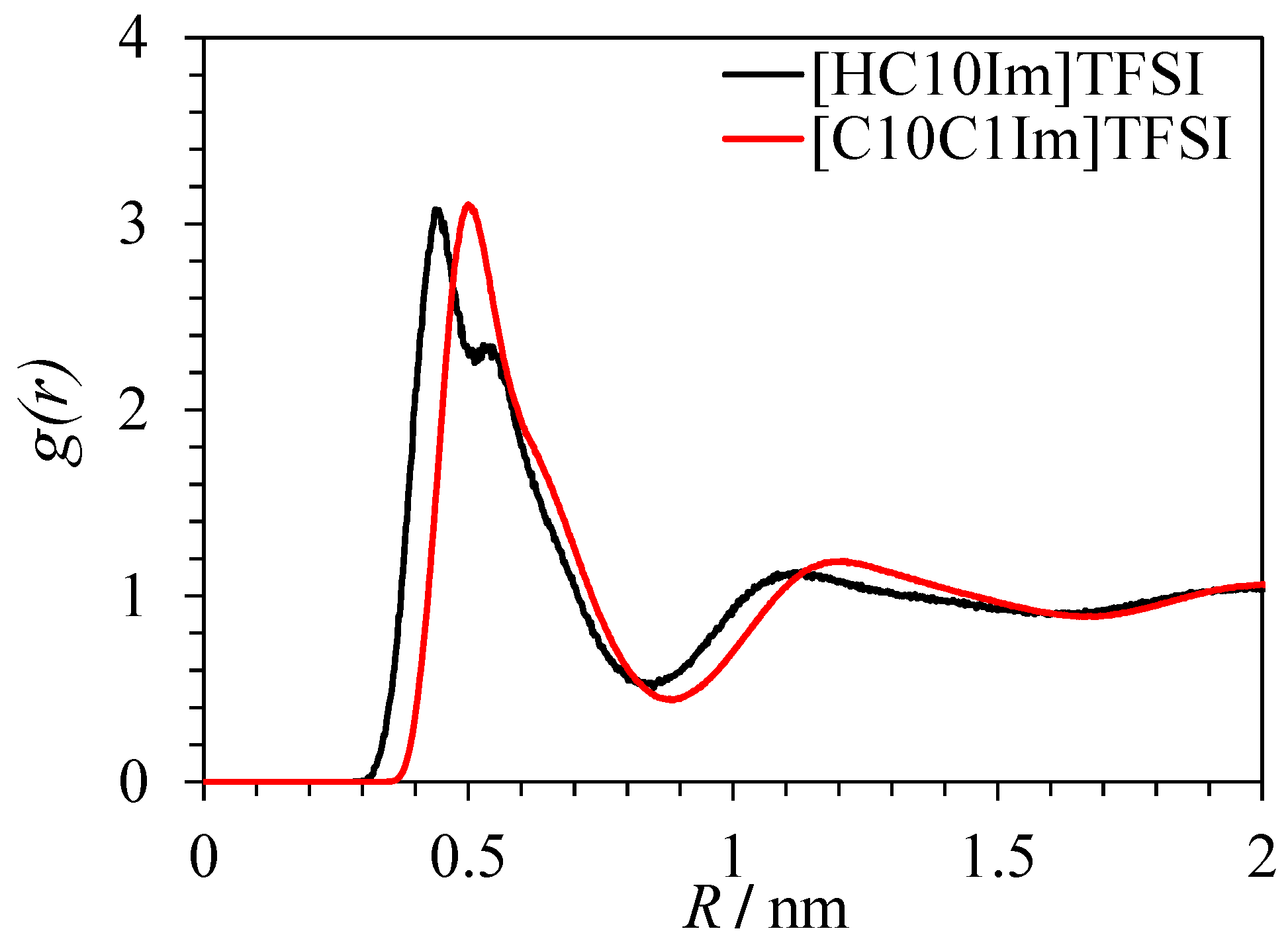
3.1. Scattering Curves Calculated from Simulations
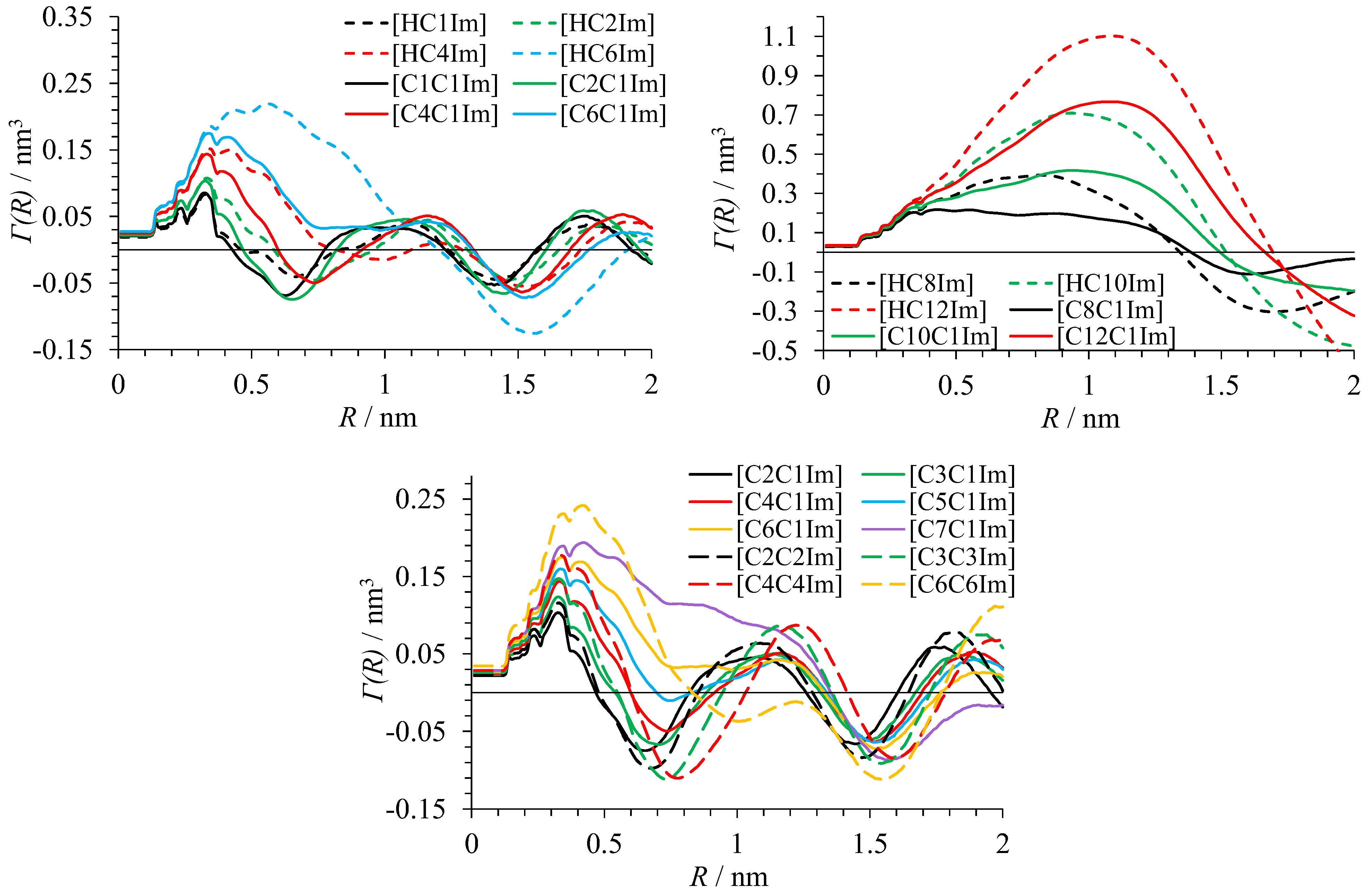
3.2. Heterogeneity of Polar/Apolar Atom Distribution
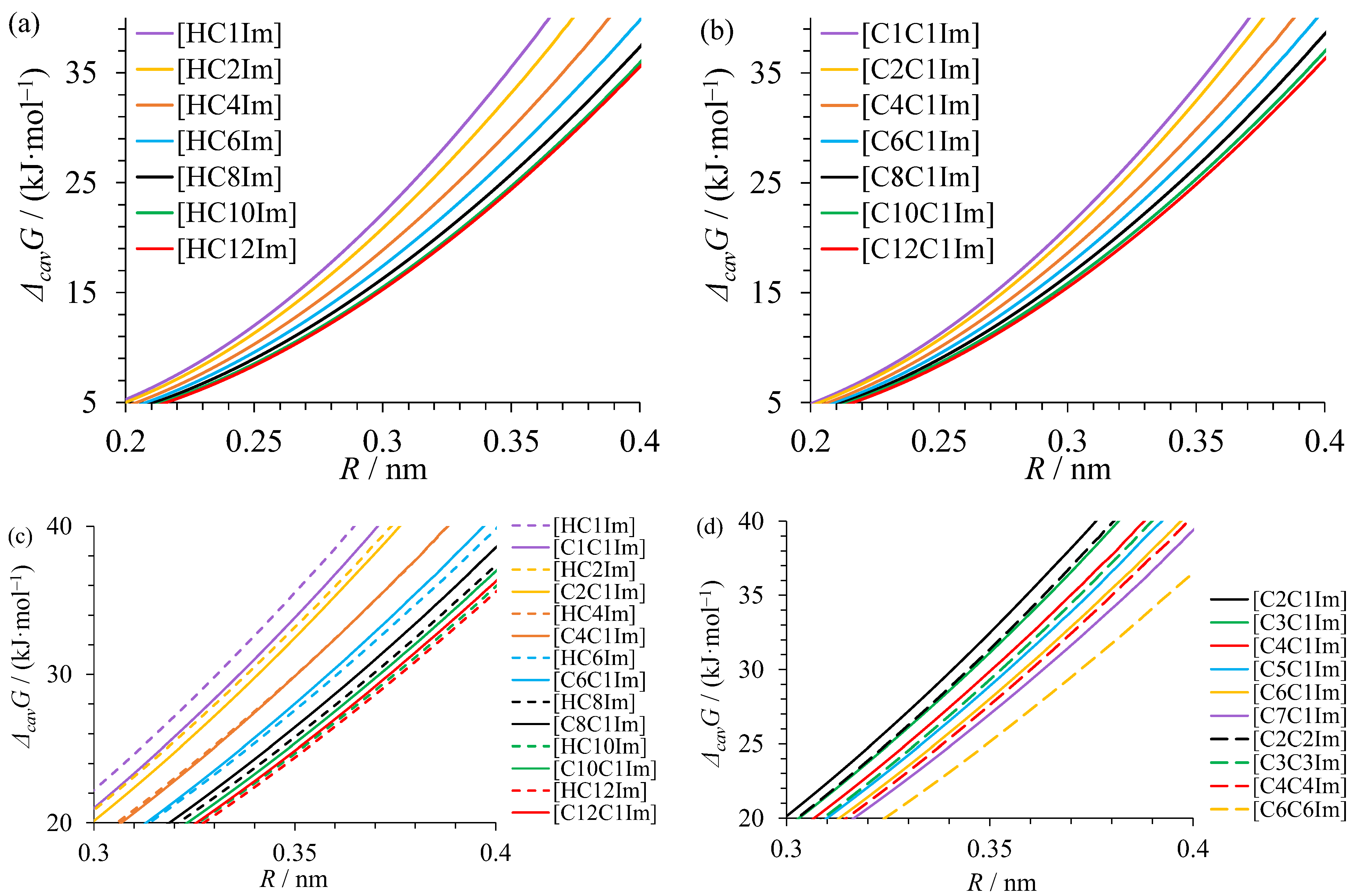
3.3. Gibbs Free Energy of Cavity Formation
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hayes, R.; Warr, G.G.; Atkin, R. Structure and Nanostructure in Ionic Liquids. Chem. Rev. 2015, 115, 6357–6426. [Google Scholar] [CrossRef] [PubMed]
- Greaves, T.L.; Kennedy, D.F.; Mudie, S.T.; Drummond, C.J. Diversity Observed in the Nanostructure of Protic Ionic Liquids. J. Phys. Chem. B 2010, 114, 10022–10031. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-L.; Li, B.; Sarman, S.; Mocci, F.; Lu, Z.-Y.; Yuan, J.; Laaksonen, A.; Fayer, M.D. Microstructural and Dynamical Heterogeneities in Ionic Liquids. Chem. Rev. 2020, 120, 5798–5877. [Google Scholar] [CrossRef] [PubMed]
- Triolo, A.; Russina, O.; Bleif, H.-J.; Di Cola, E. Nanoscale Segregation in Room Temperature Ionic Liquids. J. Phys. Chem. B 2007, 111, 4641–4644. [Google Scholar] [CrossRef] [PubMed]
- Atkin, R.; Warr, G.G. The Smallest Amphiphiles: Nanostructure in Protic Room-Temperature Ionic Liquids with Short Alkyl Groups. J. Phys. Chem. B 2008, 112, 4164–4166. [Google Scholar] [CrossRef]
- Rocha, M.A.A.; Neves, C.M.S.S.; Freire, M.G.; Russina, O.; Triolo, A.; Coutinho, J.A.P.; Santos, L.M.N.B.F. Alkylimidazolium Based Ionic Liquids: Impact of Cation Symmetry on Their Nanoscale Structural Organization. J. Phys. Chem. B 2013, 117, 10889–10897. [Google Scholar] [CrossRef]
- Abdurrokhman, I.; Elamin, K.; Danyliv, O.; Hasani, M.; Swenson, J.; Martinelli, A. Protic Ionic Liquids Based on the Alkyl-Imidazolium Cation: Effect of the Alkyl Chain Length on Structure and Dynamics. J. Phys. Chem. B 2019, 123, 4044–4054. [Google Scholar] [CrossRef]
- Martinelli, A.; Maréchal, M.; Östlund, Å.; Cambedouzou, J. Insights into the Interplay between Molecular Structure and Diffusional Motion in 1-Alkyl-3-Methylimidazolium Ionic Liquids: A Combined PFG NMR and X-Ray Scattering Study. Phys. Chem. Chem. Phys. 2013, 15, 5510. [Google Scholar] [CrossRef]
- Triolo, A.; Russina, O.; Caminiti, R.; Shirota, H.; Lee, H.Y.; Santos, C.S.; Murthy, N.S.; Castner, E.W., Jr. Comparing Intermediate Range Order for Alkyl- vs. Ether-Substituted Cations in Ionic Liquids. Chem. Commun. 2012, 48, 4959. [Google Scholar] [CrossRef] [PubMed]
- Sedov, I.A.; Magsumov, T.I.; Salikov, T.M.; Solomonov, B.N. Solvation of Apolar Compounds in Protic Ionic Liquids: The Non-Synergistic Effect of Electrostatic Interactions and Hydrogen Bonds. Phys. Chem. Chem. Phys. 2017, 19, 25352–25359. [Google Scholar] [CrossRef]
- Sedov, I.A.; Salikov, T.M.; Solomonov, B.N. Contrasting the Solvation Properties of Protic Ionic Liquids with Different Nanoscale Structure. J. Mol. Liq. 2019, 290, 111361. [Google Scholar] [CrossRef]
- Xiao, D.; Hines, L.G.; Li, S.; Bartsch, R.A.; Quitevis, E.L.; Russina, O.; Triolo, A. Effect of Cation Symmetry and Alkyl Chain Length on the Structure and Intermolecular Dynamics of 1,3-Dialkylimidazolium Bis(Trifluoromethanesulfonyl)Amide Ionic Liquids. J. Phys. Chem. B 2009, 113, 6426–6433. [Google Scholar] [CrossRef]
- Garaga, M.N.; Dracopoulos, V.; Werner-Zwanziger, U.; Zwanziger, J.W.; Maréchal, M.; Persson, M.; Nordstierna, L.; Martinelli, A. A Long-Chain Protic Ionic Liquid inside Silica Nanopores: Enhanced Proton Mobility Due to Efficient Self-Assembly and Decoupled Proton Transport. Nanoscale 2018, 10, 12337–12348. [Google Scholar] [CrossRef]
- Ghatee, M.H.; Zolghadr, A.R.; Moosavi, F.; Ansari, Y. Studies of Structural, Dynamical, and Interfacial Properties of 1-Alkyl-3-Methylimidazolium Iodide Ionic Liquids by Molecular Dynamics Simulation. J. Chem. Phys. 2012, 136, 124706. [Google Scholar] [CrossRef] [PubMed]
- Dudariev, D.; Koverga, V.; Kalugin, O.; Miannay, F.-A.; Polok, K.; Takamuku, T.; Jedlovszky, P.; Idrissi, A. Insight to the Local Structure of Mixtures of Imidazolium-Based Ionic Liquids and Molecular Solvents from Molecular Dynamics Simulations and Voronoi Analysis. J. Phys. Chem. B 2023, 127, 2534–2545. [Google Scholar] [CrossRef] [PubMed]
- Sedov, I.A.; Magsumov, T.I. Highlighting the Difference in Nanostructure between Domain-Forming and Domainless Protic Ionic Liquids. Phys. Chem. Chem. Phys. 2022, 24, 21477–21494. [Google Scholar] [CrossRef]
- Kashyap, H.K.; Santos, C.S.; Murthy, N.S.; Hettige, J.J.; Kerr, K.; Ramati, S.; Gwon, J.; Gohdo, M.; Lall-Ramnarine, S.I.; Wishart, J.F.; et al. Structure of 1-Alkyl-1-Methylpyrrolidinium Bis(Trifluoromethylsulfonyl)Amide Ionic Liquids with Linear, Branched, and Cyclic Alkyl Groups. J. Phys. Chem. B 2013, 117, 15328–15337. [Google Scholar] [CrossRef]
- Gontrani, L.; Russina, O.; Lo Celso, F.; Caminiti, R.; Annat, G.; Triolo, A. Liquid Structure of Trihexyltetradecylphosphonium Chloride at Ambient Temperature: An X-Ray Scattering and Simulation Study. J. Phys. Chem. B 2009, 113, 9235–9240. [Google Scholar] [CrossRef]
- Wang, Y.-L.; Li, B.; Sarman, S.; Laaksonen, A. Microstructures and Dynamics of Tetraalkylphosphonium Chloride Ionic Liquids. J. Chem. Phys. 2017, 147, 224502. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, Y.; Zhang, X.; Zhou, G.; Zhang, S. Microstructures and Interaction Analyses of Phosphonium-Based Ionic Liquids: A Simulation Study. J. Phys. Chem. B 2012, 116, 4934–4942. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Padua, A. Fftool v1.0.0. 2015. Available online: https://zenodo.org/records/18618 (accessed on 15 May 2024).
- Canongia Lopes, J.N.; Pádua, A.A.H. Molecular Force Field for Ionic Liquids III: Imidazolium, Pyridinium, and Phosphonium Cations; Chloride, Bromide, and Dicyanamide Anions. J. Phys. Chem. B 2006, 110, 19586–19592. [Google Scholar] [CrossRef] [PubMed]
- Canongia Lopes, J.N.; Deschamps, J.; Pádua, A.A.H. Modeling Ionic Liquids Using a Systematic All-Atom Force Field. J. Phys. Chem. B 2004, 108, 2038–2047. [Google Scholar] [CrossRef]
- Canongia Lopes, J.N.; Pádua, A.A.H. Molecular Force Field for Ionic Liquids Composed of Triflate or Bistriflylimide Anions. J. Phys. Chem. B 2004, 108, 16893–16898. [Google Scholar] [CrossRef]
- Gouveia, A.S.L.; Bernardes, C.E.S.; Tomé, L.C.; Lozinskaya, E.I.; Vygodskii, Y.S.; Shaplov, A.S.; Lopes, J.N.C.; Marrucho, I.M. Ionic Liquids with Anions Based on Fluorosulfonyl Derivatives: From Asymmetrical Substitutions to a Consistent Force Field Model. Phys. Chem. Chem. Phys. 2017, 19, 29617–29624. [Google Scholar] [CrossRef]
- Bhargava, B.L.; Balasubramanian, S. Refined Potential Model for Atomistic Simulations of Ionic Liquid [Bmim][PF6]. J. Chem. Phys. 2007, 127, 114510. [Google Scholar] [CrossRef] [PubMed]
- Chaban, V. Polarizability versus Mobility: Atomistic Force Field for Ionic Liquids. Phys. Chem. Chem. Phys. 2011, 13, 16055. [Google Scholar] [CrossRef]
- Doherty, B.; Zhong, X.; Gathiaka, S.; Li, B.; Acevedo, O. Revisiting OPLS Force Field Parameters for Ionic Liquid Simulations. J. Chem. Theory Comput. 2017, 13, 6131–6145. [Google Scholar] [CrossRef]
- Schröder, C. Comparing Reduced Partial Charge Models with Polarizable Simulations of Ionic Liquids. Phys. Chem. Chem. Phys. 2012, 14, 3089. [Google Scholar] [CrossRef]
- Son, C.Y.; McDaniel, J.G.; Schmidt, J.R.; Cui, Q.; Yethiraj, A. First-Principles United Atom Force Field for the Ionic Liquid BMIM+ BF4–: An Alternative to Charge Scaling. J. Phys. Chem. B 2016, 120, 3560–3568. [Google Scholar] [CrossRef]
- McDaniel, J.G.; Yethiraj, A. Influence of Electronic Polarization on the Structure of Ionic Liquids. J. Phys. Chem. Lett. 2018, 9, 4765–4770. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Doi, H.; Saito, S.; Matsugami, M.; Fujii, K.; Kanzaki, R.; Kameda, Y.; Umebayashi, Y. Hydrogen Bond in Imidazolium Based Protic and Aprotic Ionic Liquids. J. Mol. Liq. 2016, 217, 35–42. [Google Scholar] [CrossRef]
- Rodrigues, A.S.M.C.; Rocha, M.A.A.; Almeida, H.F.D.; Neves, C.M.S.S.; Lopes-da-Silva, J.A.; Freire, M.G.; Coutinho, J.A.P.; Santos, L.M.N.B.F. Effect of the Methylation and N–H Acidic Group on the Physicochemical Properties of Imidazolium-Based Ionic Liquids. J. Phys. Chem. B 2015, 119, 8781–8792. [Google Scholar] [CrossRef]
- Kolbeck, C.; Lehmann, J.; Lovelock, K.R.J.; Cremer, T.; Paape, N.; Wasserscheid, P.; Fröba, A.P.; Maier, F.; Steinrück, H.-P. Density and Surface Tension of Ionic Liquids. J. Phys. Chem. B 2010, 114, 17025–17036. [Google Scholar] [CrossRef]
- Součková, M.; Klomfar, J.; Pátek, J. Measurements and Group Contribution Analysis of 0.1MPa Densities for Still Poorly Studied Ionic Liquids with the [PF6] and [NTf2] Anions. J. Chem. Thermodyn. 2014, 77, 31–39. [Google Scholar] [CrossRef]
- Fröba, A.P.; Kremer, H.; Leipertz, A. Density, Refractive Index, Interfacial Tension, and Viscosity of Ionic Liquids [EMIM][EtSO4], [EMIM][NTf2], [EMIM][N(CN)2], and [OMA][NTf2] in Dependence on Temperature at Atmospheric Pressure. J. Phys. Chem. B 2008, 112, 12420–12430. [Google Scholar] [CrossRef]
- Zorębski, E.; Geppert-Rybczyńska, M.; Zorębski, M. Acoustics as a Tool for Better Characterization of Ionic Liquids: A Comparative Study of 1-Alkyl-3-Methylimidazolium Bis[(Trifluoromethyl)Sulfonyl]Imide Room-Temperature Ionic Liquids. J. Phys. Chem. B 2013, 117, 3867–3876. [Google Scholar] [CrossRef]
- Zorębski, M.; Zorębski, E.; Dzida, M.; Skowronek, J.; Jężak, S.; Goodrich, P.; Jacquemin, J. Ultrasonic Relaxation Study of 1-Alkyl-3-Methylimidazolium-Based Room-Temperature Ionic Liquids: Probing the Role of Alkyl Chain Length in the Cation. J. Phys. Chem. B 2016, 120, 3569–3581. [Google Scholar] [CrossRef] [PubMed]
- Esperança, J.M.S.S.; Visak, Z.P.; Plechkova, N.V.; Seddon, K.R.; Guedes, H.J.R.; Rebelo, L.P.N. Density, Speed of Sound, and Derived Thermodynamic Properties of Ionic Liquids over an Extended Pressure Range. 4. [C3Mim][NTf2] and [C5Mim][NTf2]. J. Chem. Eng. Data 2006, 51, 2009–2015. [Google Scholar] [CrossRef]
- Xue, L.; Gurung, E.; Tamas, G.; Koh, Y.P.; Shadeck, M.; Simon, S.L.; Maroncelli, M.; Quitevis, E.L. Effect of Alkyl Chain Branching on Physicochemical Properties of Imidazolium-Based Ionic Liquids. J. Chem. Eng. Data 2016, 61, 1078–1091. [Google Scholar] [CrossRef]
- Gomes De Azevedo, R.; Esperança, J.M.S.S.; Szydlowski, J.; Visak, Z.P.; Pires, P.F.; Guedes, H.J.R.; Rebelo, L.P.N. Thermophysical and Thermodynamic Properties of Ionic Liquids over an Extended Pressure Range: [Bmim][NTf2] and [Hmim][NTf2]. J. Chem. Thermodyn. 2005, 37, 888–899. [Google Scholar] [CrossRef]
- Zorębski, E.; Zorębski, M.; Dzida, M.; Goodrich, P.; Jacquemin, J. Isobaric and Isochoric Heat Capacities of Imidazolium-Based and Pyrrolidinium-Based Ionic Liquids as a Function of Temperature: Modeling of Isobaric Heat Capacity. Ind. Eng. Chem. Res. 2017, 56, 2592–2606. [Google Scholar] [CrossRef]
- Santos, D.; Santos, M.; Franceschi, E.; Dariva, C.; Barison, A.; Mattedi, S. Experimental Density of Ionic Liquids and Thermodynamic Modeling with Group Contribution Equation of State Based on the Lattice Fluid Theory. J. Chem. Eng. Data 2016, 61, 348–353. [Google Scholar] [CrossRef]
- Khalil, R.; Chaabene, N.; Azar, M.; Malham, I.B.; Turmine, M. Effect of the Chain Lengthening on Transport Properties of Imidazolium-Based Ionic Liquids. Fluid Phase Equilibria 2020, 503, 112316. [Google Scholar] [CrossRef]
- Lago, S.; Rodríguez, H.; Soto, A.; Arce, A. Deterpenation of Citrus Essential Oil by Liquid−Liquid Extraction with 1-Alkyl-3-Methylimidazolium Bis(Trifluoromethylsulfonyl)Amide Ionic Liquids. J. Chem. Eng. Data 2011, 56, 1273–1281. [Google Scholar] [CrossRef]
- Tariq, M.; Forte, P.A.S.; Gomes, M.F.C.; Lopes, J.N.C.; Rebelo, L.P.N. Densities and Refractive Indices of Imidazolium- and Phosphonium-Based Ionic Liquids: Effect of Temperature, Alkyl Chain Length, and Anion. J. Chem. Thermodyn. 2009, 41, 790–798. [Google Scholar] [CrossRef]
- Domańska, U.; Królikowski, M.; Wlazło, M.; Więckowski, M. Phase Equilibrium Investigation on 2-Phenylethanol in Binary and Ternary Systems: Influence of High Pressure on Density and Solid–Liquid Phase Equilibrium. J. Phys. Chem. B 2018, 122, 6188–6197. [Google Scholar] [CrossRef]
- Tomé, L.I.N.; Carvalho, P.J.; Freire, M.G.; Marrucho, I.M.; Fonseca, I.M.A.; Ferreira, A.G.M.; Coutinho, J.A.P.; Gardas, R.L. Measurements and Correlation of High-Pressure Densities of Imidazolium-Based Ionic Liquids. J. Chem. Eng. Data 2008, 53, 1914–1921. [Google Scholar] [CrossRef]
- Hasse, B.; Lehmann, J.; Assenbaum, D.; Wasserscheid, P.; Leipertz, A.; Fröba, A.P. Viscosity, Interfacial Tension, Density, and Refractive Index of Ionic Liquids [EMIM][MeSO3], [EMIM][MeOHPO2], [EMIM][OcSO4], and [BBIM][NTf2] in Dependence on Temperature at Atmospheric Pressure. J. Chem. Eng. Data 2009, 54, 2576–2583. [Google Scholar] [CrossRef]
- Cromer, D.T.; Mann, J.B. X-Ray Scattering Factors Computed from Numerical Hartree–Fock Wave Functions. Acta Crystallogr. A 1968, 24, 321–324. [Google Scholar] [CrossRef]
- Ben-Naim, A. The Kirkwood–Buff Integrals for One-Component Liquids. J. Chem. Phys. 2008, 128, 234501. [Google Scholar] [CrossRef] [PubMed]
- Widom, B. Some Topics in the Theory of Fluids. J. Chem. Phys. 1963, 39, 2808–2812. [Google Scholar] [CrossRef]
- Annapureddy, H.V.R.; Kashyap, H.K.; De Biase, P.M.; Margulis, C.J. What Is the Origin of the Prepeak in the X-Ray Scattering of Imidazolium-Based Room-Temperature Ionic Liquids? J. Phys. Chem. B 2010, 114, 16838–16846. [Google Scholar] [CrossRef]
- Sedov, I.; Magsumov, T. The Gibbs Free Energy of Cavity Formation in a Diverse Set of Solvents. J. Chem. Phys. 2020, 153, 134501. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ionic Liquid | ρ/(g∙mL−1) | |
|---|---|---|
| MD | Experimental | |
| 1-methylimidazolium bis(trifluoromethanesulfonyl)imide ([HC1Im][TFSI]) | 1.607 | 1.614 [33] |
| 1-ethylimidazolium bis(trifluoromethanesulfonyl)imide ([HC2Im][TFSI]) | 1.553 | 1.56913 [7], 1.563 [34] |
| 1-butylimidazolium bis(trifluoromethanesulfonyl)imide ([HC4Im][TFSI]) | 1.467 | 1.47194 [7] |
| 1-hexylimidazolium bis(trifluoromethanesulfonyl)imide ([HC6Im][TFSI]) | 1.399 | 1.40242 [7] |
| 1-octylimidazolium bis(trifluoromethanesulfonyl)imide ([HC8Im][TFSI]) | 1.345 | 1.34633 [7] |
| 1-decylimidazolium bis(trifluoromethanesulfonyl)imide ([HC10Im][TFSI]) | 1.301 | 1.28754 [7] |
| 1-dodecylimidazolium bis(trifluoromethanesulfonyl)imide ([HC12Im][TFSI]) | 1.266 | 1.26068 [7] |
| 1,3-dimethylimidazolium bis(trifluoromethanesulfonyl)imide ([C1C1Im][TFSI]) | 1.525 | 1.5692 [6], 1.56729 [35], 1.557 [33] |
| 1-ethyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide ([C2C1Im][TFSI]) | 1.482 | 1.5195 [36], 1.51845 [37], 1.51859 [38] |
| 1-propyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide ([C3C1Im][TFSI]) | 1.445 | 1.475 [39], 1475.7 [40], 1.4756 [41] |
| 1-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide ([C4C1Im][TFSI]) | 1.412 | 1.43704 [42], 1.43635 [39], 1.4369 [41] |
| 1-pentyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide ([C5C1Im][TFSI]) | 1.382 | 1.40221 [39], 1.4045 [40], 1.4036 [41] |
| 1-hexyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide ([C6C1Im][TFSI]) | 1.355 | 1.3719 [39], 1.3727 [41], 1.37081 [42] |
| 1-heptyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide ([C7C1Im][TFSI]) | 1.33 | 1.3454 [41], 1.3446 [6], 1.34413 [43] |
| 1-octyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide ([C8C1Im][TFSI]) | 1.308 | 1.32032 [38], 1.32054 [44], 1.32081 [45] |
| 1-decyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide ([C10C1Im][TFSI]) | 1.269 | 1.27259 [45], 1.27001 [35], 1.2784 [46] |
| 1-dodecyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide ([C12C1Im][TFSI]) | 1.239 | 1.2447 [47], 1.2423 [48], 1.24355 [35] |
| 1,3-diethylimidazolium bis(trifluoromethanesulfonyl)imide ([C2C2Im][TFSI]) | 1.444 | 1.4632 [49], 1.4749 [6] |
| 1,3-dipropylimidazolium bis(trifluoromethanesulfonyl)imide ([C3C3Im][TFSI]) | 1.381 | 1.399 [6] |
| 1,3-dibutylimidazolium bis(trifluoromethanesulfonyl)imide ([C4C4Im][TFSI]) | 1.329 | 1.3428 [6], 1.340 [50] |
| 1,3-dihexylimidazolium bis(trifluoromethanesulfonyl)imide ([C6C6Im][TFSI]) | 1.249 | 1.2550 [6] |
| Ionic Liquid | Peak I (nm−1) | Peak II (nm−1) | ||
|---|---|---|---|---|
| Calc. | Exp. | Calc. | Exp. | |
| [HC1Im][TFSI] | no peak | – | 8.9 | – |
| [HC2Im][TFSI] | no peak | no peak [7] | 8.7 | 8.7 [7] |
| [HC4Im][TFSI] | 4.9 (weak peak on shoulder) | 5.1 (weak peak) [7] | 8.3 | 8.6 [7] |
| [HC6Im][TFSI] | 3.9 | 4.1 [7] | 8.7 | 8.4 [7] |
| [HC8Im][TFSI] | 3.1 | 3.4 [7] | 8.5 | 8.3 [7] |
| [HC10Im][TFSI] | 2.8 | 2.9 [7] | 8.4 | 8.3 [7] |
| [HC12Im][TFSI] | 2.6 | 2.6 [7] | 8.3 | 8.3 [7] |
| [C1C1Im][TFSI] | no peak | no peak [6] | 9.1 | 9.1 [6] |
| [C2C1Im][TFSI] | no peak | no peak [6,8,12] | 9.0 | 9.0 [8], 9.0 [6], 8.9 [12] |
| [C3C1Im][TFSI] | no peak | no peak [6,12] | 8.7 | 8.7 [12], 8.7 [6] |
| [C4C1Im][TFSI] | no peak | shoulder [6,8,12] | 8.7 | 8.6 [8], 8.5 [6], 8.5 [12] |
| [C5C1Im][TFSI] | 4.4 (weak) | 4.4 (weak) [12], 4.9 (weak) [6] | 8.8 | 8.5 [12], 8.5 [6] |
| [C6C1Im][TFSI] | 3.9 | 4.1 (weak) [8], 4.3 [6] | 8.7 | 8.5 [8], 8.4 [6] |
| [C7C1Im][TFSI] | 3.8 | 3.6 [6] | 8.6 | 8.4 [6] |
| [C8C1Im][TFSI] | 3.0 | 3.5 [8], 3.4 [6] | 8.5 | 8.5 [8], 8.4 [6] |
| [C10C1Im][TFSI] | 3.0 | 2.8 [8], 2.9 [6] | 8.4 | 8.5 [8], 8.5 [6] |
| [C12C1Im][TFSI] | 2.6 | 2.4 [8], 2.5 [6] | 8.3 | 8.5 [8], 8.4 [6] |
| [C2C2Im][TFSI] | no peak | no peak [6,12] | 8.6 | 8.6 [12], 8.7 [6] |
| [C3C3Im][TFSI] | no peak | shoulder [6,12] | 8.1 | 8.1 [12], 8.1 [6] |
| [C4C4Im][TFSI] | shoulder | shoulder [6,12] | 7.9 | 7.9 [12], 8.0 [6] |
| [C6C6Im][TFSI] | 4.5 | 4.6 [6] | 8.0 | 7.8 [6] |
| Ionic Liquid | |||
|---|---|---|---|
| R0/nm | R0/nm | 2Rmax/nm | |
| [HC4Im][TFSI] | 0.8 | 0.82 | 0.80 |
| [HC6Im][TFSI] | 1.23 | 1.21 | 1.06 |
| [HC8Im][TFSI] | 1.34 | 1.35 | 1.40 |
| [HC10Im][TFSI] | 1.54 | 1.52 | 1.60 |
| [HC12Im][TFSI] | 1.71 | 1.70 | 1.80 |
| [C4C1Im][TFSI] | – | 0.60 | – |
| [C5C1Im][TFSI] | 0.78 | 0.70 | – |
| [C6C1Im][TFSI] | 0.87 | 1.33 | 0.76 |
| [C7C1Im][TFSI] | 1.32 | 1.36 | 0.96 |
| [C8C1Im][TFSI] | 1.37 | 1.39 | 1.30 |
| [C10C1Im][TFSI] | 1.52 | 1.50 | 1.56 |
| [C12C1Im][TFSI] | 1.68 | 1.67 | 1.76 |
| [C4C4Im][TFSI] | 0.69 | 0.61 | – |
| [C6C6Im][TFSI] | 0.83 | 0.84 | 0.80 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magsumov, T.I.; Sedov, I.A. Nanoheterogeneity in Protic and Aprotic Alkylimidazolium Bistriflimide Ionic Liquids. Liquids 2024, 4, 632-646. https://doi.org/10.3390/liquids4030035
Magsumov TI, Sedov IA. Nanoheterogeneity in Protic and Aprotic Alkylimidazolium Bistriflimide Ionic Liquids. Liquids. 2024; 4(3):632-646. https://doi.org/10.3390/liquids4030035
Chicago/Turabian StyleMagsumov, Timur I., and Igor A. Sedov. 2024. "Nanoheterogeneity in Protic and Aprotic Alkylimidazolium Bistriflimide Ionic Liquids" Liquids 4, no. 3: 632-646. https://doi.org/10.3390/liquids4030035
APA StyleMagsumov, T. I., & Sedov, I. A. (2024). Nanoheterogeneity in Protic and Aprotic Alkylimidazolium Bistriflimide Ionic Liquids. Liquids, 4(3), 632-646. https://doi.org/10.3390/liquids4030035