
An Ab Initio Investigation of the Hydration of Antimony(III)



Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
3.1. A Survey of Structures
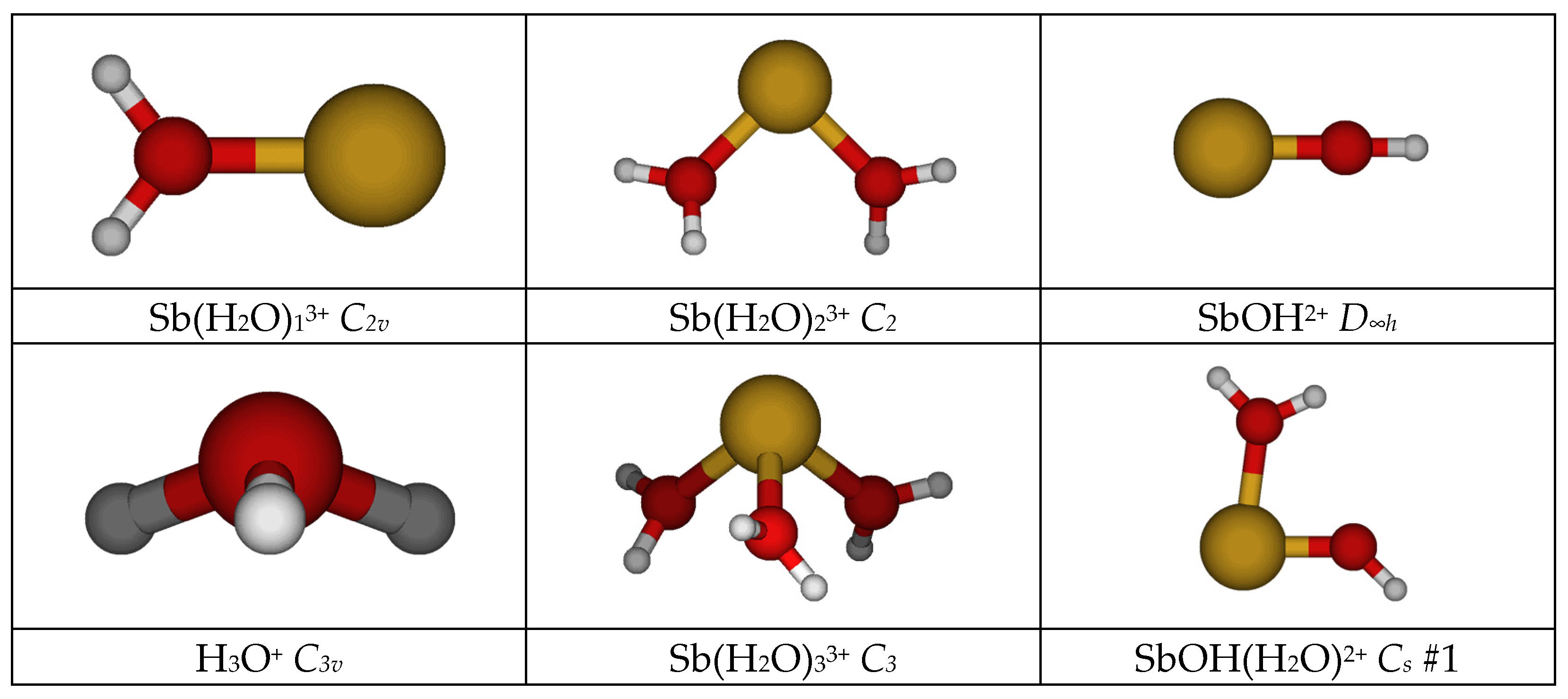
- The monoaquaantimony(III) remains C2v at all levels.
- The most stable diaquaantimony(III) remains the bent C2 at all levels except HF/LANL2MB (C2v planar). The linear holodirected D2d structure is approximately 50 kJ/mol higher in energy, but the unstable bent Cs structure is only slightly higher in energy (<1 kJ/mol for nonminimal basis sets). All attempts to generate a [1+1] structure instead result either in proton transfer to give SbOH2+ + H3O+, which was much lower in energy, or recoordination to give the [2+0] Cs structure.
- The most stable triaquaantimony(III) remains the pyramidal C3 at all levels. The two pyramidal C3v structures are 10–30 kJ/mol higher in energy, whereas the planar holodirected D3h and D3 structures are 125–150 kJ/mol higher in energy still. The unstable [2+1] C2v structure was ~90 kJ/mol higher in energy, and upon desymmetrization, underwent proton transfer to give the much-lower-in-energy SbOH(H2O)2+ + H3O+.
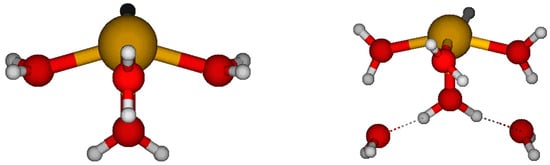
- The most stable tetraaquaantimony(III) is usually the seesaw C2v #3 (all mixed basis sets, HF/SDD), but it can be C2 (all CEP, HF/LANL2DZ) or Cs (HF/LANL2MB). The other C2v structures are higher in energy (10–25 kJ/mol). The holodirected D2d #1, #2, and S4 structures are much higher in energy (75–125 kJ/mol). The unstable Cs #2 [3+1] structures are up to 50 kJ/mol higher in energy, and upon desymmetrization, undergo a proton transfer to give the much-lower-in-energy SbOH(H2O)22+ + H3O+.
- The most stable pentaaquaantimony(III) is the square pyramidal C2v #1, with the other three C2v structures which are 25–50 kJ/mol higher in energy. The usually unstable [4+1] structures are 15–50 kJ/mol higher in energy. To our surprise, the C2v #1 [4+1] is stable at HF/CEP-121G*, HF/LANL2MB, HF/A, HF/B, HF/C, and HF/A+. At the other levels, there is a B1 imaginary mode, along which a proton transfer occurs to give SbOH(H2O)32+ + H3O+. At the MP2 levels, desymmetrization along an A2 mode gives rise to a C2 structure, which is slightly lower in energy.
- The most stable hexaaquaantimony(III) is the distorted octahedral C3. The octahedral Th structure is ~18–50 kJ/mol higher in energy. The two Cs [5+1] structures examined often underwent proton transfer to give SbOH(H2O)42+ + H3O+. However, if the [5+1] structures were stationary points, they were lower in energy than the [6+0] forms by about 25 kJ/mol. In some cases, they were actually minima (HF/CEP-121G*, all HF mixed basis sets). Given how shallow (or in some cases, nonexistent) the barrier to proton transfer is to give the deprotonated forms, it is reasonable to conclude that the aquaantimony(III) species would only exist in an extremely acidic solution. Combined with the results for the pentaaquaantimony(III), this suggests that antimony(III) is actually pentacoordinate square pyramidal.
- Of the 16 different C2v heptaaquaantimony(III) structures tried, none were stable, either possessing imaginary modes or dissociating to a [6+1], [5+2], or [4+3] structure. Structures #1, #2, and #4, nearly always dissociated. The remainder usually remained 7-coordinate, except occasionally dissociating at HF/CEP-4G and HF/LANL2MB. In some cases, the [5+2] structures underwent a double proton transfer and possibly a water elimination to give Sb(OH)2(H2O)2/3+ + 1/0H2O + 2H3O+. Of the 7-coordinate C2v structures, #16 was the lowest in energy, unless #1 was a stationary point, in which case, it was lower in energy. In all cases, the [5+2] structure was 20–80 kJ/mol lower in energy than the 7-coordinate C2v #16 structure. Upon desymmetrization of the remaining 7-coordinate C2v structures to C2, many coalesced to C2 #16, some dissociated to [5+2], but at some levels, C2 #5, #6, #11, and #13 also exist, with at least one imaginary B mode. When these were desymmetrized to C1 #1–5, a stable C1 #3 or #5 was found at some levels, or ligand dissociation to a [6+1] or [5+2] structure took place. Desymmetrization of the C2v structures along the B modes gave one of 27 possible Cs structures. Of these, Cs #1, #5, and either #13 or #20 often coalesce (to Cs #5); Cs #2, #7, #14 and #22 often coalesce (to Cs #7); Cs #12 coalesces to Cs #8; Cs #11, #18, and #26 often coalesce (to Cs #26); Cs #19 and Cs #15 ascend in symmetry to either C2v #10 or #12; Cs #16 and #24 often coalesce to Cs #10 or to each other; Cs #25 coalesces to Cs #21; Cs #27 coalesces to Cs #23; and Cs #17 remains unique. None of these Cs structures are minima, except Cs #13 at HF/A+. The Cs structures desymmetrize to C1 #6–21. Of these possibilities, for the most part, they coalesced to the previously found C1 #3 or #5 structures, or underwent ligand dissociation to give [6+1] or [5+2] structures. This exemplifies the power of the systematic desymmetrization procedure in finding minimum energy structures that would otherwise be difficult to locate. In all cases where a seven-coordinate minimum energy structure exists (HF/CEP-31G*, HF/CEP-121G*, MP2/CEP-31G*, MP2/CEP-121G*, HF/C, HF/A+, HF/B+, HF/C+, MP2/A+), it is less stable than a [5+2] or [6+1] structure (or a proton transferred version thereof). The 7-coordinate structures are unlikely to have any significant population in an aqueous solution under ambient conditions.
- For octaaquaantimony(III), two D4h (square prism) and two D4d (square antiprism) structures were first examined. Multiple imaginary modes were present.
- For the D4d #1 and #2 structures, desymmetrization along the A2 imaginary mode gave the same S8 #1 structure; along the B1 imaginary mode, the same D4 #2 structure; and along the B2 imaginary mode, the C4v #1 and #2 structures. For the D4h #1 and #2 structures, desymmetrization along the A1u imaginary mode gave the D4 #2 structure found before; along the A2g imaginary mode, the same C4h #1 structure; along the A2u imaginary mode, the C4v #3 [4+4] and #4 structures; along the B2g imaginary mode (D4h #1), the D2h #1 structure ascended in symmetry to D4h #2; along the B1u mode, D2d #1 and #3 coalesced; along the B2u mode, D2d #2 and #4, respectively. Another D2d structure (#5) was formed by combining other D2d structures.
- Desymmetrization of the C4h #1 structure along the Au mode usually gives the S8 #1 structure (via C4 #1), and along the Bu mode, D2d #5 (via S4 #1); the S8 #1 structure along the B mode at two levels, C4 #1; the D4 #1 structure along the A2 mode, C4 #1, and along the B2 mode, D2 #1; the D2d #1 structure along the A2 mode, S4 #2, the B1 mode, D2 #1, and the B2 mode, [4+4] C2v #1; the D2d #2 structure along the A2 mode, S4 #2, the B1 mode, D2 #1, and the B2 mode, C2v #2; the D2d #4 structure along the A2 mode, S4 #2, the B1 mode, D2 #1, and the B2 mode, C2v #3; the D2d #5 structure along the B1 mode, D2 #1, and the B2 mode, [6+2] C2v #4. All of these structures had at least one imaginary frequency.
- Desymmetrization of the S4 #2 structure along the B mode, C2 #1 (which was close in structure to the D2 #1 structure); the D2 #1 structure along the B1 mode gave either the [4+4] or [6+2] C2 #1, or more usually the stable S8 #1.
- For enneaaquaantimony(III), four D3h structures were first examined. Desymmetrization along the A1” mode led to the common D3 #1 structure (which was only stable at MP2/CEP-31G*); the A2′ mode led to either the unstable C3h #1 or #2 structures; and the A2” mode led to the unstable C3v #1–4 structures. The C3v #2 and 3 structures were usually of [6+3] coordination. When these structures were desymmetrized, they nearly always resulted in expulsion of three water molecules to the second hydration sphere to give the (usually) stable C3 #1 [6+3]. An unstable [6+3] D3 structure was also found, which desymmetrized to give the even more stable C3 #2 [6+3].
3.2. The Sb-O Distance
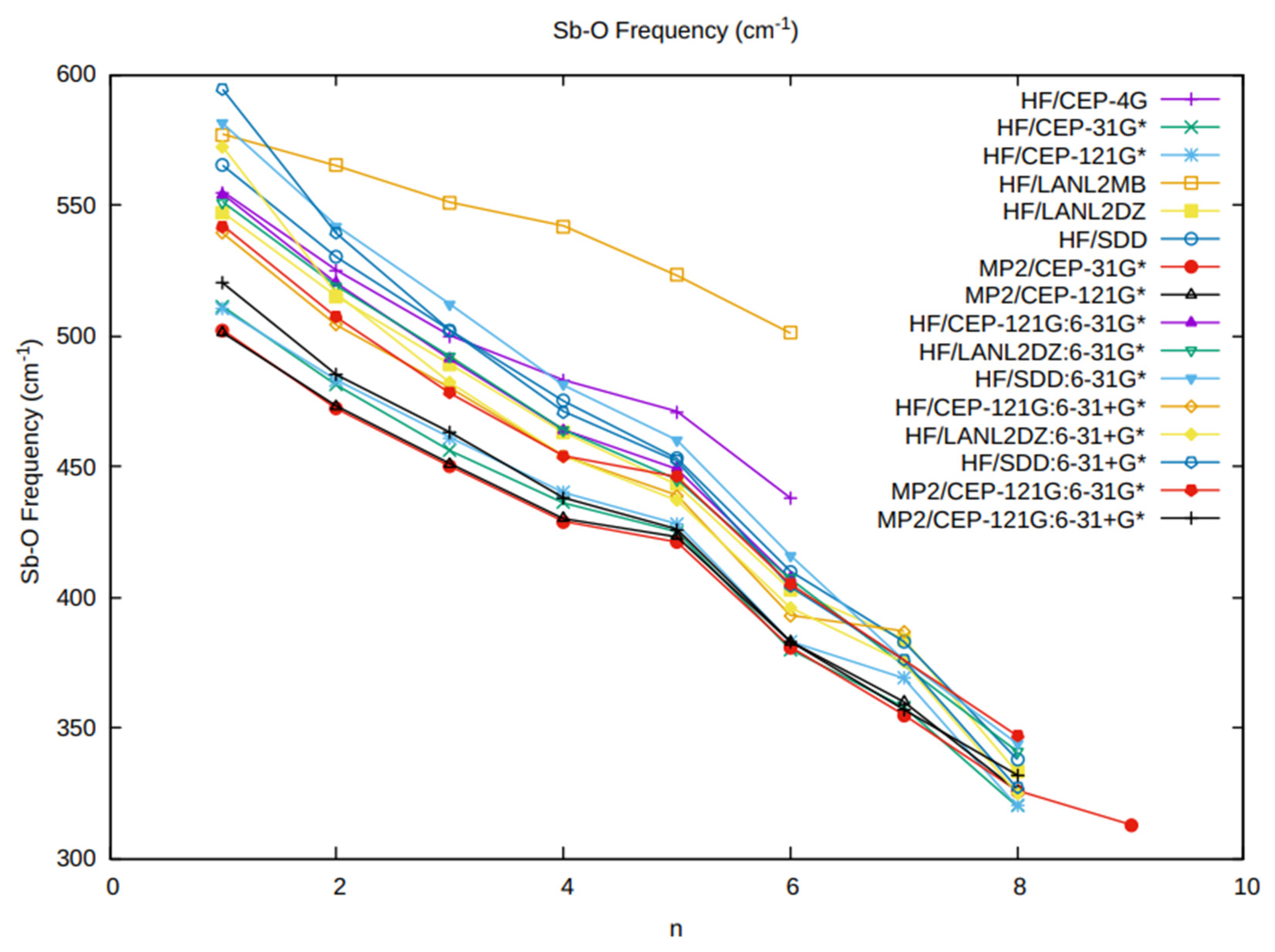
3.3. The Sb-O Vibrational Frequency
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Richens, D.T. The Chemistry of Aqua Ions; Wiley: Chichester, UK, 1997. [Google Scholar]
- Pye, C.C.; Gunasekara, C.M. An Ab Initio Investigation of the Hydration of Thallium(III) and Mercury(II). J. Solut. Chem. 2020, 49, 1419–1429. [Google Scholar] [CrossRef]
- Pye, C.C.; Gunasekara, C.M. An Ab Initio Investigation of the Hydration of Lead(II). Liquids 2022, 2, 39–49. [Google Scholar] [CrossRef]
- Pye, C.C.; Gunasekara, C.M. An Ab Initio Investigation of the Hydration of Tin(II). Liquids 2022, 2, 465–473. [Google Scholar] [CrossRef]
- Gayer, K.H.; Garrett, A.B. The Equilibria of Antimonous Oxide (Rhombic) in Dilute Solutions of Hydrochloric Acid and Sodium Hydroxide at 25°. J. Am. Chem. Soc. 1952, 74, 2353–2354. [Google Scholar] [CrossRef]
- Mishra, S.K.; Gupta, Y.K. Spectrophotometric Study of the Hydrolytic Equilibrium of Sb(III) in Aqueous Perchloric Acid Solution. Ind. J. Chem. 1968, 6, 757–758. [Google Scholar]
- Baes, C.F., Jr.; Mesmer, R.E. The Hydrolysis of Cations; Wiley: New York, NY, USA, 1976. [Google Scholar]
- Ahrland, S.; Bovin, J.-O. The Complex Formation of Antimony(III) in Perchloric Acid and Nitric Acid Solutions. A Solubility Study. Acta Chem. Scand. A 1974, 28, 1089–1100. [Google Scholar] [CrossRef]
- Zakaznova-Herzog, V.P.; Seward, T.M. Antimonous acid protonation/deprotonation equilibria in hydrothermal solutions to 300 °C. Geochim. Cosmochim. Acta 2006, 70, 2298–2310. [Google Scholar] [CrossRef]
- Natta, G.; Baccaredda, M. Composti chimici interstiziali. Struttura del pentossido di antimonio idrato e di alcuni antimoniati. [Interstitial chemical compounds. Structure of hydrated antimony pentoxide and some antimoniates]. Gazz. Chim. Ital. 1936, 66, 308–316. (In Italian) [Google Scholar]
- Dihlstrom, K.; Westgren, A. Uber den Bau des sogenannten Antimontetroxyds und der damit isomorphen Verbindung BiTa2O6F. [About the construction of the so-called antimony tetroxide and the isomorphic compound BiTa2O6F]. Z. Anorg. Allg. Chem. 1937, 235, 153–160. (In German) [Google Scholar] [CrossRef]
- England, W.A.; Cross, M.G.; Hamnett, A.; Wiseman, P.J.; Goodenough, J.B. Fast Proton Conduction in Inorganic Ion-Exchange Compounds. Solid State Ion. 1980, 1, 231–249. [Google Scholar] [CrossRef]
- Watelet, H.; Picard, J.-P.; Baud, G.; Besse, J.-P. Chevalier, R. Un nouveau conducteur protonique, [H(H2O)n]12Sb12O36 (n ≤ 1). [A new protonic conductor, [H(H2O)n]12Sb12O36 (n ≤ 1).]. Mater. Res. Bull. 1981, 16, 1131–1137. (In French) [Google Scholar] [CrossRef]
- Jager, G.; Jones, P.G.; Sheldrick, G.M.; Schwarzmann, F. Darstellung und Kristalstruktur von Antimonen(V)oxidhydroxid HSb3O8. [Representation and Crystal Structure of Antimony(V) Oxide Hydroxide HSb3O8.]. Z. Naturforschung B 1983, 38, 698–701. (In German) [Google Scholar] [CrossRef]
- Riviere, M.; Fourquet, J.L.; Grins, J.; Nygren, M. The cubic pyrochlores H2xSb2xW2-2xO6·nH2O: Structural, thermal, and electrical properties. Mater. Res. Bull. 1988, 23, 965–975. [Google Scholar] [CrossRef]
- Svensson, C. Refinement of the Crystal Structure of Cubic Antimony Trioxide, Sb2O3. Acta Cryst. B 1975, 31, 2016–2018. [Google Scholar] [CrossRef]
- Svensson, C. The Crystal Structure of Orthorhombic Antimony Trioxide, Sb2O3. Acta Cryst. B 1974, 30, 458–461. [Google Scholar] [CrossRef]
- Orosel, D.; Dinnebier, R.E.; Blatov, V.A.; Jansen, M. Structure of a new high-pressure-high-temperature modification of antimony(III) oxide, γ-Sb2O3, from high-resolution synchrotron powder diffraction data. Acta Crystallogr. B 2012, 68, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bovin, J.P. The crystal structure of Sb4O4(OH)2(NO3)2. Acta Chem. Scand. A 1974, 28, 267–274. [Google Scholar] [CrossRef]
- Bovin, J.P. The crystal structure of Sb4O5(OH)ClO4·1/2 H2O. Acta Chem. Scand. A 1974, 28, 723–730. [Google Scholar] [CrossRef][Green Version]
- Bovin, J.P. The Crystal Structure of the Antimony(III) Oxide Sulphate Sb6O7(SO4)2. Acta Cryst. B 1976, 32, 1771–1777. [Google Scholar] [CrossRef]
- Mercier, R.; Douglade, J.; Jones, P.G.; Sheldrick, G.M. Structure of an Oxonium Antimony(III) Sulphate, (H3O)2Sb2(SO4)4. Acta Cryst. C 1983, 39, 145–148. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A., Jr.; Stratmann, R.E.; Burant, J.C.; et al. Gaussian 98, Revision A.9; Gaussian, Inc.: Pittsburgh, PA, USA, 1998. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision D.02; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Pye, C.C.; Gunasekara, C.M.; Rudolph, W.W. An ab initio investigation of bismuth hydration. Can. J. Chem. 2007, 85, 945–950. [Google Scholar] [CrossRef]
- Pye, C.C.; Whynot, D.C.M.; Corbeil, C.R.; Mercer, D.J. Desymmetrization in geometry optimization: Application to an ab initio study of copper(I) hydration. Pure Appl. Chem. 2020, 92, 1643–1654. [Google Scholar] [CrossRef]
- Pye, C.C.; Gilbert, C.R. An ab initio investigation of the second hydration shell of metal cations. Comput. Appl. Chem. 2020, 37, 2020. [Google Scholar] [CrossRef]
- Shimony-Livny, L.; Glusker, J.P.; Bock, C.W. Lone Pair Functionality in Divalent Lead Compounds. Inorg. Chem. 1998, 37, 1853–1867. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pye, C.C.; Gunasekara, C.M. An Ab Initio Investigation of the Hydration of Antimony(III). Liquids 2024, 4, 322-331. https://doi.org/10.3390/liquids4020016
Pye CC, Gunasekara CM. An Ab Initio Investigation of the Hydration of Antimony(III). Liquids. 2024; 4(2):322-331. https://doi.org/10.3390/liquids4020016
Chicago/Turabian StylePye, Cory C., and Champika Mahesh Gunasekara. 2024. "An Ab Initio Investigation of the Hydration of Antimony(III)" Liquids 4, no. 2: 322-331. https://doi.org/10.3390/liquids4020016
APA StylePye, C. C., & Gunasekara, C. M. (2024). An Ab Initio Investigation of the Hydration of Antimony(III). Liquids, 4(2), 322-331. https://doi.org/10.3390/liquids4020016