
An X-ray and Neutron Scattering Study of Aqueous MgCl2 Solution in the Gigapascal Pressure Range


 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Solutions
2.2. Experimental PT Condition and Density Estimation
2.3. X-ray Scattering Measurements
2.4. Neutron Scattering Measurements
2.5. EPSR Modeling
3. Results
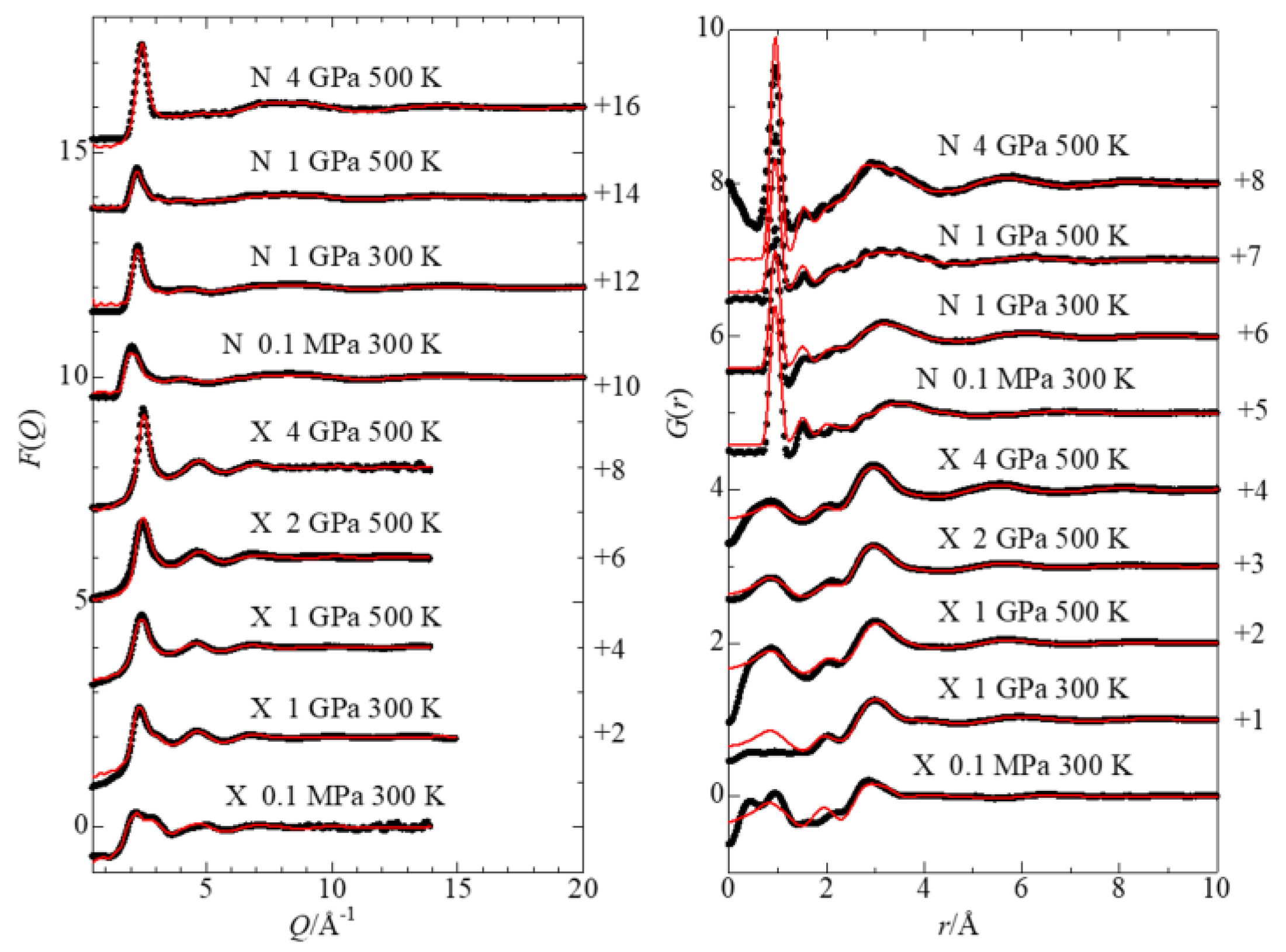
3.1. Interference Functions and Radial Distribution Functions
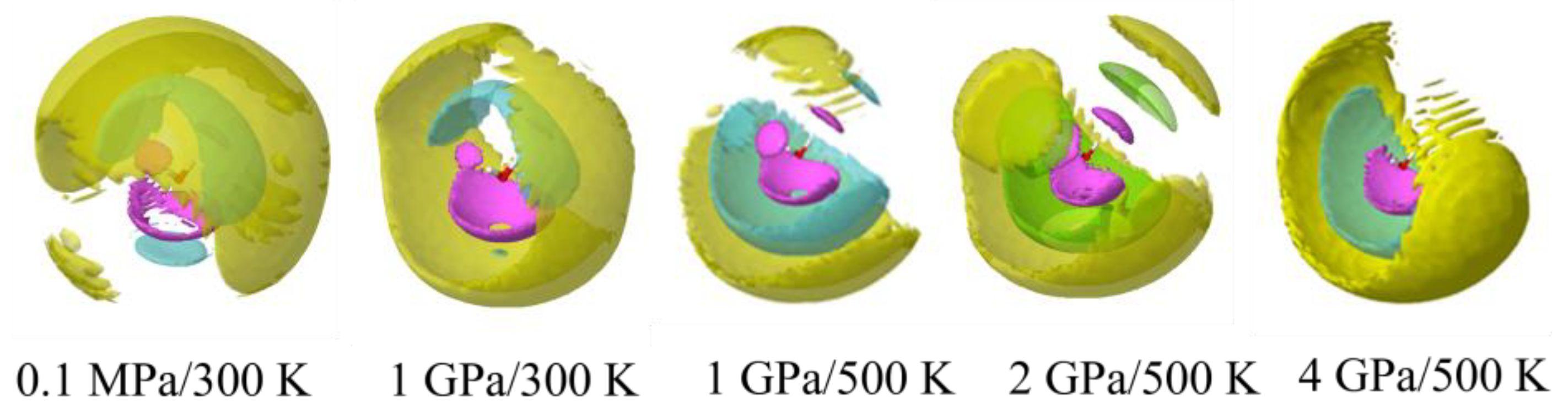
3.2. Mg2+ Solvation
3.3. Cl− Solvation
3.4. Solvent Water
3.5. Ion Association
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saris, N.-E.L.; Mervaala, E.; Karppanen, H.; Khawaja, J.A.; Lewenstam, A. Magnesium, An update on physiological, clinical and analytical aspects. Clin. Chim. Acta 2000, 294, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.-J.; Zhang, Y.-H.; Wei, Z.-F.; Cheng, H.; Li, X.-H. Magnesium sulfate aerosols studied by FTIR spectroscopy: Hygroscopic properties, supersaturated structures, and implications for seawater aerosols. J. Phys. Chem. A 2006, 110, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Hirschmann, M.; Kohlstedt, D. Water in Earth’s Mantle. Phys. Today 2012, 65, 40–45. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Fujimura, K.; Uchi, K.; Yoshida, K.; Katayama, Y. Structure of water from water to 4 GPa revealed by energy-dispersive X-ray diffraction combined with empirical potential structure refinement modeling. J. Mol. Liq. 2012, 176, 44–51. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Fukuyama, N.; Yoshida, K.; Katayama, Y. Ion solvation and water structure in an aqueous sodium chloride solution in the gigapascal pressure range. J. Phys. Chem. Lett. 2021, 12, 250–256. [Google Scholar] [CrossRef]
- Polidori, A.; Rowlands, R.F.; Zeidler, A.; Salanne, M.; Fischer, H.E.; Annighöfer, B.; Klotz, S.; Salmon, P.S. Structure and dynamics of aqueous NaCl solutions at high temperatures and pressures. J. Chem. Phys. 2021, 155, 194506. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Yoshida, K.; Machida, S.; Hattori, T. Neutron scattering on an aqueous sodium chloride solution in the gigapascal pressure range. J. Mol. Liq. 2022, 365, 120181. [Google Scholar] [CrossRef]
- Zhang, W.-Q.; Yamaguchi, T.; Fang, C.-H.; Yoshida, K.; Zhou, Y.-Q.; Zhu, F.-Y.; Machida, S.; Hattori, T.; Li, W. Structure of an aqueous RbCl solution in the gigapascal pressure range by neutron diffraction combined with empirical potential structure refinement modeling. J. Mol. Liq. 2022, 348, 118080. [Google Scholar] [CrossRef]
- Takamuku, T.; Yagafarov, O.F.; Katayama, Y.; Umeki, T. Structure of aqueous calcium chloride solutions by energy-dispersive x-ray diffraction under high temperatures and high pressures. Bunseki Kagaku 2015, 64, 203. (In Japanese) [Google Scholar] [CrossRef]
- Yamaguchi, T.; Nishino, M.; Yoshida, K.; Takumi, M.; Nagata, K.; Hattori, T. Ion hydration and association in an aqueous calcium chloride solution in the GPa range. Eur. J. Inorg. Chem. 2019, 2019, 1170–1177. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Fukuyama, N.; Yoshida, K.; Katayama, Y. Ion solvation and association and water structure in an aqueous cerium(III) chloride solution in the gigapascal pressure range. Anal. Sci. 2022, 38, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Caminiti, R.; Licheri, G.; Piccaluga, G.; Pinna, G. X-ray diffraction study of MgCl2 aqueous solution. J. Appl. Crystallogr. 1979, 12, 34–38. [Google Scholar] [CrossRef]
- Persson, I. Structures of hydrated metal ions in the solid state and aqueous solution. Liquids 2022, 2, 210–242. [Google Scholar] [CrossRef]
- Bruni, F.; Imberti, S.; Mancinelli, R.; Ricci, M.A. Aqueous solutions of divalent chlorides; ions hydration shell and water structure. J. Chem. Phys. 2012, 136, 064520. [Google Scholar] [CrossRef]
- Duboué-Dijon, E.; Mason, P.E.; Fischer, H.E.; Jungwirth, P. Hydration and ion pairing in aqueous Mg2+ and Zn2+ solutions; force-field description aided by neutron scattering experiments and ab initio molecular dynamics simulations. J. Phys. Chem. B 2018, 122, 3296–3306. [Google Scholar] [CrossRef]
- Szász, G.I.; Dietz, W.; Heinzinger, K.; Pálinkás, G.; Tadnai, T. On the orientation of water molecules in the hydration shell of the ions in an MgCl2 solution. Chem. Phys. Lett. 1982, 92, 388–392. [Google Scholar] [CrossRef]
- Jiao, D.; King, C.; Grossfield, A.; Darden, T.A.; Ren, P. Simulation of Ca2+ and Mg2+ solvation using polarizable atomic multipole potential. J. Phys. Chem. B 2006, 110, 18553–18559. [Google Scholar] [CrossRef]
- Callahan, K.M.; Casillas-Ituarte, N.N.; Roeselová, M.; Allen, H.C.; Tobias, D.J. Solvation of magnesium dication; molecular dynamics simulation and vibrational spectroscopic study of magnesium chloride in aqueous solution. J. Phys. Chem. A 2010, 114, 5141–5148. [Google Scholar] [CrossRef]
- Mamatkulov, S.; Fyta, M.; Netz, R.R. Force fields for divalent cations based on single-ion and ion-pair properties. J. Chem. Phys. 2013, 138, 024505. [Google Scholar] [CrossRef]
- Mamatkulov, S.; Scwierz, N. Force fields for monovalent and divalent metal cations in TIP3P water based on thermodynamic and kinetic properties. J. Chem. Phys. 2018, 148, 074504. [Google Scholar] [CrossRef]
- Zeron, I.M.; Abascal, J.L.F.; Vega, C. A force field of Li+, Na+, K+, Mg2+, Ca2+, Cl-, and SO42- in aqueous solution based on the TIP4P/2005 water model and scaled charges for the ions. J. Chem. Phys. 2019, 151, 134504. [Google Scholar] [CrossRef]
- Ikeda, T.; Boero, M.; Terakura, K. Hydration properties of magnesium and calcium ions from constrained first-principles molecular dynamics. J. Chem. Phys. 2007, 127, 074503. [Google Scholar] [CrossRef] [PubMed]
- Tommaso, D.D.; de Leeuw, N.H. Structure and dynamics of the hydrated magnesium ion and of the solvated magnesium carbonates; insights from first principles simulations. Phys. Chem. Chem. Phys. 2010, 12, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Delgado, A.A.A.; Sethio, D.; Kraka, E. Assessing the intrinsic strengths of ion-solvent and solvent0-solvent interactions for hydrated Mg2+ clusters. Inorganics 2021, 9, 31. [Google Scholar] [CrossRef]
- Wagner, W.; Pruβ, A. The IAPWS Formulation 1995 for the Thermodynamic Properties of Ordinary Water Substance for General and Scientific Use. J. Phys. Chem. Ref. Data 2002, 31, 387. [Google Scholar] [CrossRef]
- Eisenberg, D.; Kauzmann, W. The Structure and Properties of Water; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Tsuji, K.; Yaoita, K.; Imai, M.; Shimomura, O.; Kikegawa, T. Measurements of X-ray Diffraction for Liquid Metals under High Pressure. Rev. Sci. Instrum. 1989, 60, 2425. [Google Scholar] [CrossRef]
- Decker, D.L. High-Pressure Equation of State for NaCl, KCl, and CsCl. J. Appl. Phys. 1971, 42, 3239. [Google Scholar] [CrossRef]
- Datchi, F.; Loubeyre, P.; LeToullec, R. Extended and Accurate Determination of the Melting Curves of Argon, Helium, Ice (H2O), and Hydrogen (H2). Phys. Rev. B 2000, 61, 6535. [Google Scholar] [CrossRef]
- Utsumi, W.; Funakoshi, K.; Katayama, Y.; Yamanaka, M.; Okada, T.; Shimomura, O. High-Pressure Science with a Multi-Anvil Apparatus at SPring-8. J. Phys. Condens. Matter 2002, 14, 10497–10504. [Google Scholar] [CrossRef]
- Kaplow, R.; Strong, S.L.; Averbach, B.L. Radial Density Functions for Liquid Mercury and Lead. Phys. Rev. 1965, 138, A1336. [Google Scholar] [CrossRef]
- Sano-Furukawa, A.; Hattori, T.; Arima, H.; Yamada, A.; Tabata, S.; Kondo, M.; Nakamura, A.; Kagi, H.; Yagi, T. Six-axis multi-anvil press for high-pressure, high-temperature neutron diffraction experiments. Rev. Sci. Instrum. 2014, 85, 113905. [Google Scholar] [CrossRef]
- Hattori, T.; Sano-Furukawa, A.; Arima, H.; Komatsu, K.; Yamada, A.; Inamura, Y.; Nakatani, T.; Seto, Y.; Nagai, T.; Tsumi, W.; et al. Design and performance of high-pressure PLANET beamline at pulsed neutron source at J-PARC. Nucl. Instrum. Methods Phys. Res. 2015, A780, 55–67. [Google Scholar] [CrossRef]
- Paalman, H.H.; Pings, C.J. Numerical evaluation of X-ray absorption factors for cylindrical samples and annular sample cells. J. Appl. Phys. 1962, 33, 2635. [Google Scholar] [CrossRef]
- Blech, I.A.; Averbach, B.L. Multiple scattering of neutrons in vanadium and copper. Phys. Rev. 1965, 137, A1113. [Google Scholar] [CrossRef]
- NOVA Group, Institute of Materials Structure Science, Tsukuba, Ibaraki 305-080, Japan. Program nvaSq Manual. Available online: https://research.kek.jp/group/hydrogen/reduction/index.html (accessed on 26 June 2023). (In Japanese).
- Sears, V.F. Neutron scattering lengths and cross sections. Neutron News 1992, 3, 26. [Google Scholar] [CrossRef]
- Granada, J.R.; Gillette, V.H.; Mayer, R.E. Neutron cross sections and thermalization parameters using a synthetic scattering function. II: Applications to H2O, D2O, and C6H6. Phys. Rev. A 1987, 36, 5594. [Google Scholar] [CrossRef] [PubMed]
- Kameda, Y.; Sasaki, M.; Usuki, T.; Othomo, T.; Itoh, K.; Suzuya, K.; Fukunaga, T. Inelasticity effects on neutron scattering intensities of the null-H2O. J. Neutron Res. 2003, 11, 153–163. [Google Scholar] [CrossRef]
- OriginPro. The Ultimate Software for Graphing and Analysis; OriginLab Corp.: Northampton, MA, USA, 2022. [Google Scholar]
- Druchok, M.; Holovko, M. Molecular dynamics study of ion hydration under pressure. J. Mol. Liq. 2011, 159, 24–30. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The Missing Term in Effective Pair Potentials. J. Phys. Chem. 1987, 91, 6269. [Google Scholar] [CrossRef]
- Soper, A.K. Computer simulation as a tool for the interpretation of total scattering data from glasses and liquids. Mol. Simul. 2012, 38, 1171–1185. [Google Scholar] [CrossRef]
- Soper, A.K. Empirical potential Monte Carlo simulation of fluid structure. Chem. Phys. 1996, 202, 295–306. [Google Scholar] [CrossRef]
- Soper, A.K. Partial structure factors from disordered materials diffraction data: An approach using empirical potential structure refinement. Phys. Rev. B 2005, 72, 104204. [Google Scholar] [CrossRef]
- Amann-Winkel, K.; Bellissent-Funel, M.-C.; Bove, L.E.; Loerting, T.; Nilsson, A.; Paciaroni, A.; Schlesinger, D.; Skinner, L. X-ray and Neutron Scattering of Water. Chem. Rev. 2016, 116, 7570–7589. [Google Scholar] [CrossRef] [PubMed]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halide and chalcogenides. Acta Crystallogr. 1976, A32, 751–767. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | c/mol dm–3 | dX/g cm–3 | dN/g cm–3 | ρS/atoms Å–3 | PX/GPa | PN/GPa | TX/K | TN/K | Method |
|---|---|---|---|---|---|---|---|---|---|
| 0.1 MPa 300 K | 2.024 | 1.113 | 1.204 | 0.09577 | 1.0 × 10−4 | 1.0 × 10−4 | 300 | 298 | X, N |
| 1 GPa 300 K | 2.397 | 1.377 | 1.426 | 0.1134 | 1.0 | 0.65 | 300 | 298 | X, N |
| 1 GPa 500 K | 2.125 | 1.269 | 1.264 | 0.1005 | 1.0 | 0.63 | 500 | 523 | X, N |
| 2 GPa 500 K | 2.020 | 1.396 | ⋯ | 0.1386 | 2.0 | ⋯ | 500 | ⋯ | X |
| 4 GPa 500 K | 2.439 | 1.459 | 1.451 | 0.1388 | 3.8 | 4.5 | 500 | 523 | X, N |
| Atoms | ε/kJ mol–1 | Atomic Mass | Coulomb Charge/e | Refs. | |
|---|---|---|---|---|---|
| Mg2+ | 0.0040 | 2.63 | 24.31 | 2 | [20] |
| Cl− | 0.4187 | 4.40 | 35 | −1 | [41] |
| Ow | 0.65 | 3.16 | 16.00 | −0.8476 | [42] |
| Hw | 0.00 | 0.00 | 2.00 | 0.4238 | [42] |
| P, T | Parameter | Mg–Ow(I) | Mg–Hw(I) | Mg–Ow(II) | Cl–Ow(I) | Cl–Hw(I) | Cl–Ow(II) | Mg–Cl(I) | Mg–Cl(II) |
|---|---|---|---|---|---|---|---|---|---|
| 0.1 MPa/ 300 K | r/Å | 1.92 | 2.58 | 3.78 | 3.18 | 2.32 | 5.76 | 2.37 | 4.32 |
| C.N. | 6.0 ± 0.2 | 12.0 ± 0.4 | 8.2 ± 1.2 | 6.7 ± 1.1 | 0.01 ± 0.12 | ||||
| rmax/Å | 2.40 | 3.15 | 4.05 | 3.00 | 3.21 | ||||
| 1 GPa/ 300 K | r/Å | 1.92 | 2.58 | 4.53 | 3.14 | 2.24 | 4.41, 5.75 | 2.33 | 4.57 |
| C.N. | 5.0 ± 1.3 | 10.2 ± 2.6 | 8.2 ± 1.8 | 6.2 ± 2.0 | 0.78 ± 0.90 | ||||
| rmax/Å | 2.37 | 3.09 | 3.87 | 2.91 | 3.15 | ||||
| 1 GPa/ 500 K | r/Å | 1.92 | 2.61 | 4.14 | 3.21 | 2.31 | 5.86 | 2.19 | 4.50 |
| C.N. | 4.2 ± 2.0 | 8.6 ± 4.0 | 8.4 ± 2.2 | 4.0 ± 2.0 | 1.12 ± 1.27 | ||||
| rmax/Å | 2.58 | 3.18 | 4.17 | 2.88 | 2.85 | ||||
| 2 GPa/ | r/Å | 1.92 | ⋯ | 4.03 | 3.12 | ⋯ | 5.70 | 2.35 | 4.34 |
| 500 K | C.N. | 5.3 ± 0.9 | ⋯ | 14.2 ± 1.5 | ⋯ | 0.65 ± 0.75 | |||
| rmax/Å | 2.32 | ⋯ | 4.19 | ⋯ | 3.00 | ||||
| 4 GPa/500 K | r/Å | 1.93 | 2.61 | 3.99 | 3.14 | 2.18 | 5.73 | 2.36 | 4.34 |
| C.N. | 5.6 ± 0.7 | 11.4 ± 1.2 | 14.0 ± 1.4 | 6.2 ± 1.6 | 0.33 ± 0.54 | ||||
| rmax/Å | 2.31 | 3.07 | 4.15 | 2.72 | 3.30 |
| P, T | Parameters | Ow–Ow(I) | Ow–Hw(I) | Ow–Ow(II) | Ow–Ow(III) |
|---|---|---|---|---|---|
| 0.1 MPa/ 300 K | r/Å | 2.76 | 1.80 | 3.91–4.80 | 6.63 |
| C.N. | 4.6 ± 1.7 | 1.1 ± 0.90 | |||
| rmax/Å | 3.39 | 2.04 | |||
| 1 GPa/ 300 K | r/Å | 2.79 | 2.07 | 3.84 | 6.09 |
| C.N. | 6.3 ± 1.8 | 1.3 ± 1.0 | |||
| rmax/Å | 1.8 | 2.13 | |||
| 1 GPa/ 500 K | r/Å | 2.85 | 1.98 | 5.61 | 8.22 |
| C.N. | 7.6 ± 1.7 | 1.5 ± 1.1 | |||
| rmax/Å | 3.78 | 2.22 | |||
| 2 GPa/ | r/Å | 2.71 | 5.31 | 7.99 | |
| 500 K | C.N. | 9.5 ± 1.5 | |||
| rmax/Å | 3.63 | ||||
| 4 GPa/ 500 K | r/Å | 2.74 | 1.74 | 5.38 | 7.83 |
| C.N. | 8.4 ± 1.6 | 0.88 ± 0.84 | |||
| rmax/Å | 3.69 | 1.98 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamaguchi, T.; Fukuyama, N.; Yoshida, K.; Katayama, Y.; Machida, S.; Hattori, T. An X-ray and Neutron Scattering Study of Aqueous MgCl2 Solution in the Gigapascal Pressure Range. Liquids 2023, 3, 288-302. https://doi.org/10.3390/liquids3030019
Yamaguchi T, Fukuyama N, Yoshida K, Katayama Y, Machida S, Hattori T. An X-ray and Neutron Scattering Study of Aqueous MgCl2 Solution in the Gigapascal Pressure Range. Liquids. 2023; 3(3):288-302. https://doi.org/10.3390/liquids3030019
Chicago/Turabian StyleYamaguchi, Toshio, Nami Fukuyama, Koji Yoshida, Yoshinori Katayama, Shinichi Machida, and Takanori Hattori. 2023. "An X-ray and Neutron Scattering Study of Aqueous MgCl2 Solution in the Gigapascal Pressure Range" Liquids 3, no. 3: 288-302. https://doi.org/10.3390/liquids3030019
APA StyleYamaguchi, T., Fukuyama, N., Yoshida, K., Katayama, Y., Machida, S., & Hattori, T. (2023). An X-ray and Neutron Scattering Study of Aqueous MgCl2 Solution in the Gigapascal Pressure Range. Liquids, 3(3), 288-302. https://doi.org/10.3390/liquids3030019