
Comprehensive Evaluation of End-Point Free Energy Techniques in Carboxylated-Pillar[6]arene Host-Guest Binding: IV. The QM Treatment, GB Models and the Multi-Trajectory Extension


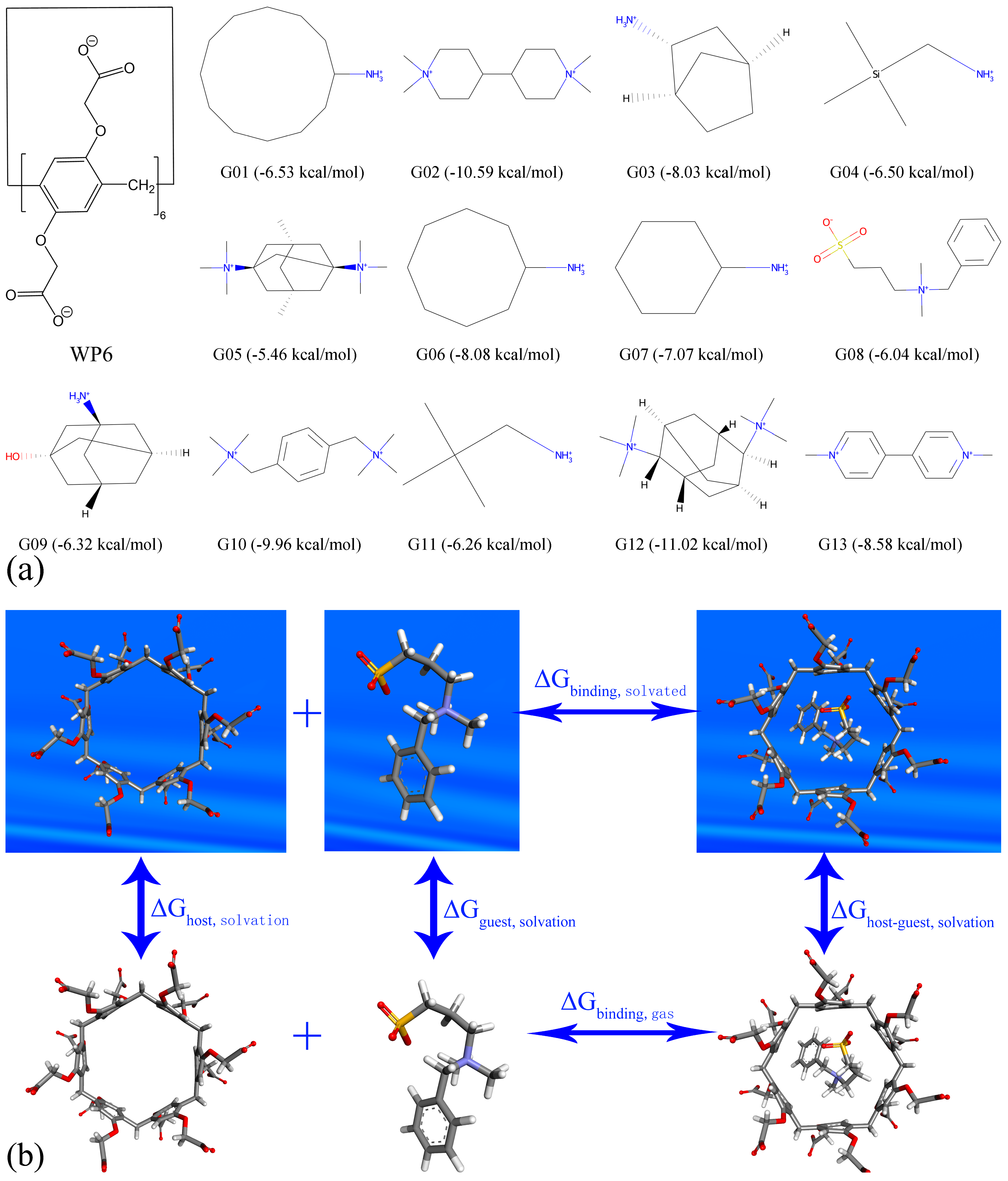{kind=link}
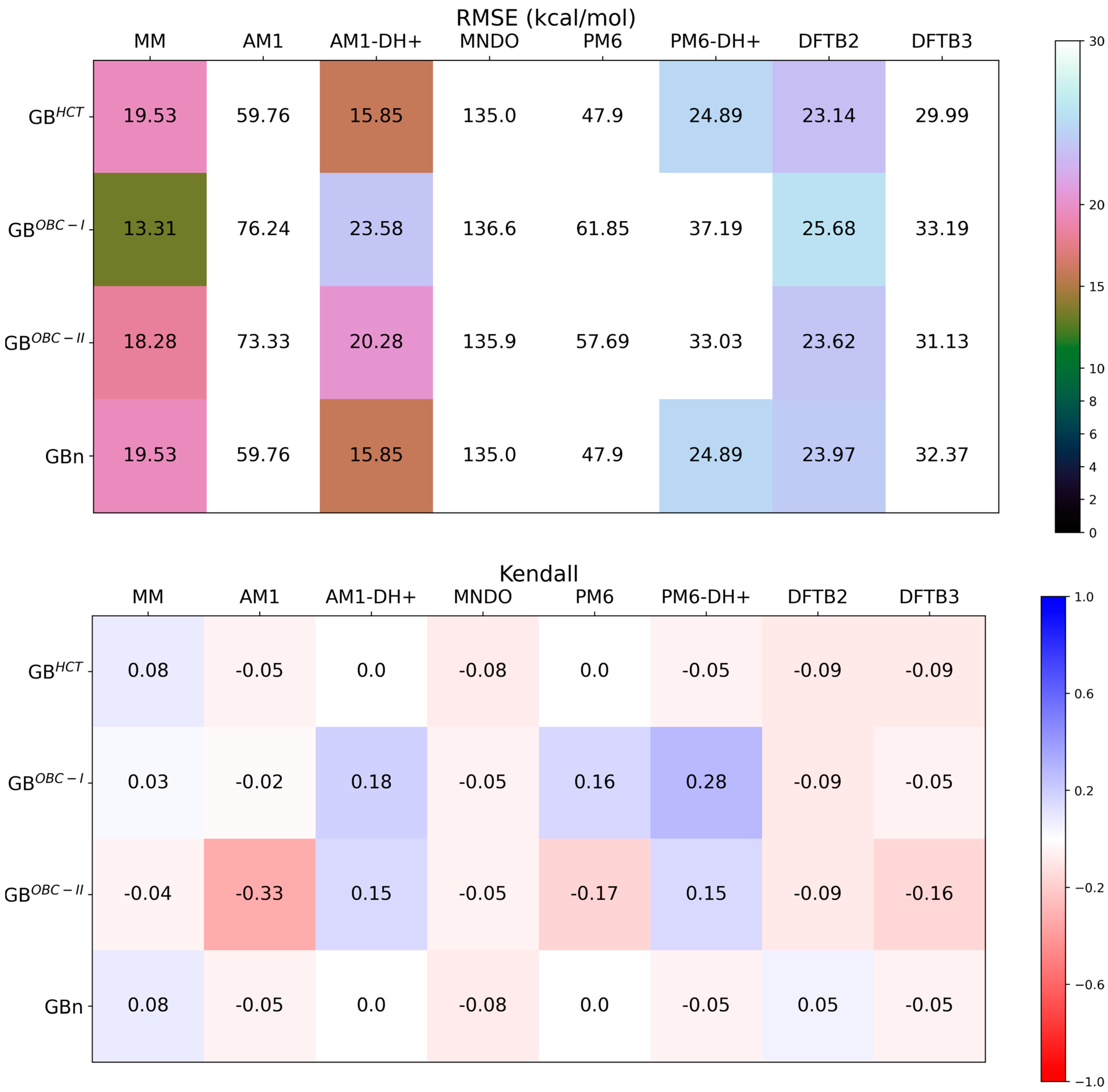{kind=link}
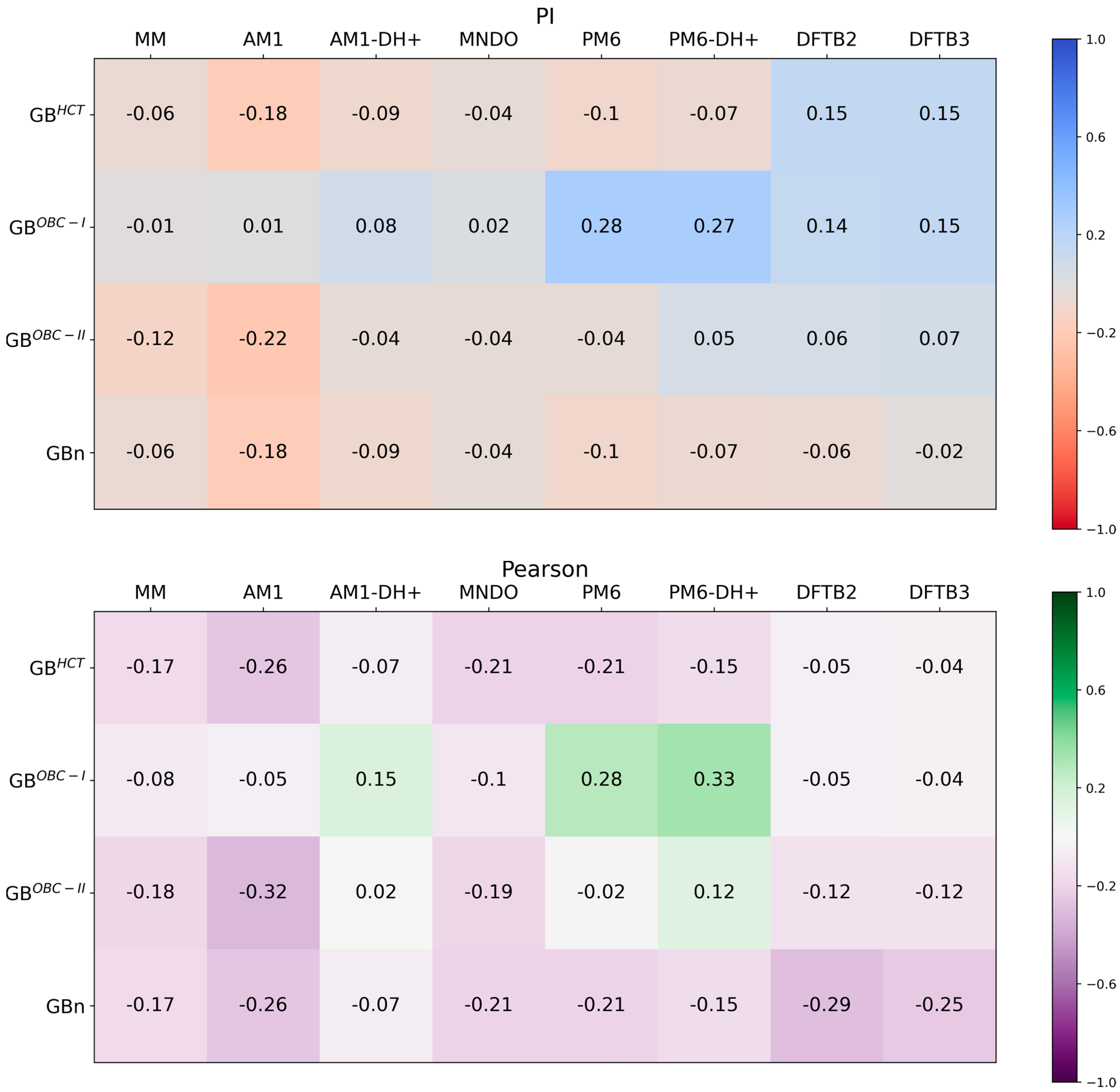{kind=link}
{kind=link}
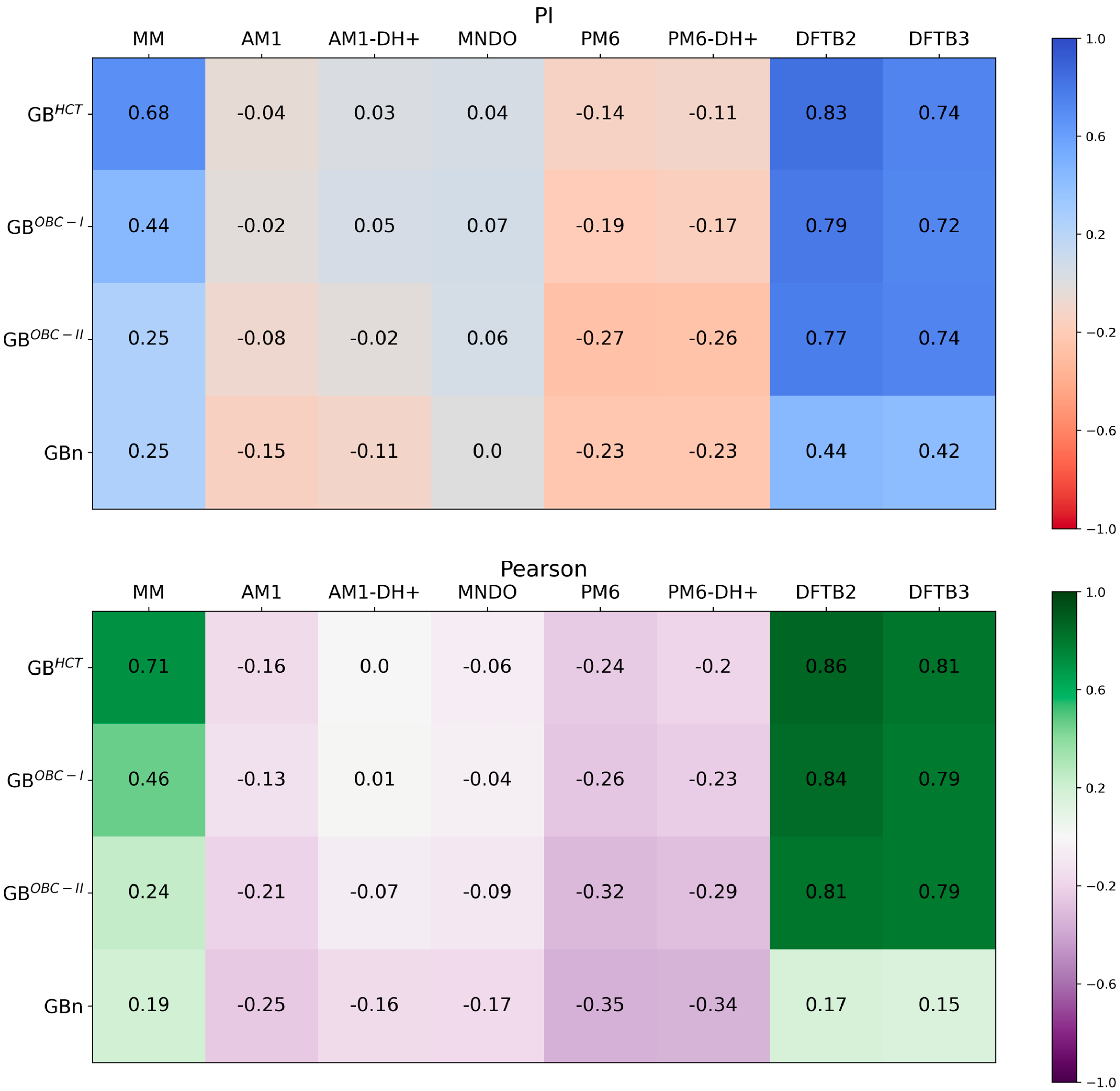{kind=link}
Abstract
:1. Introduction
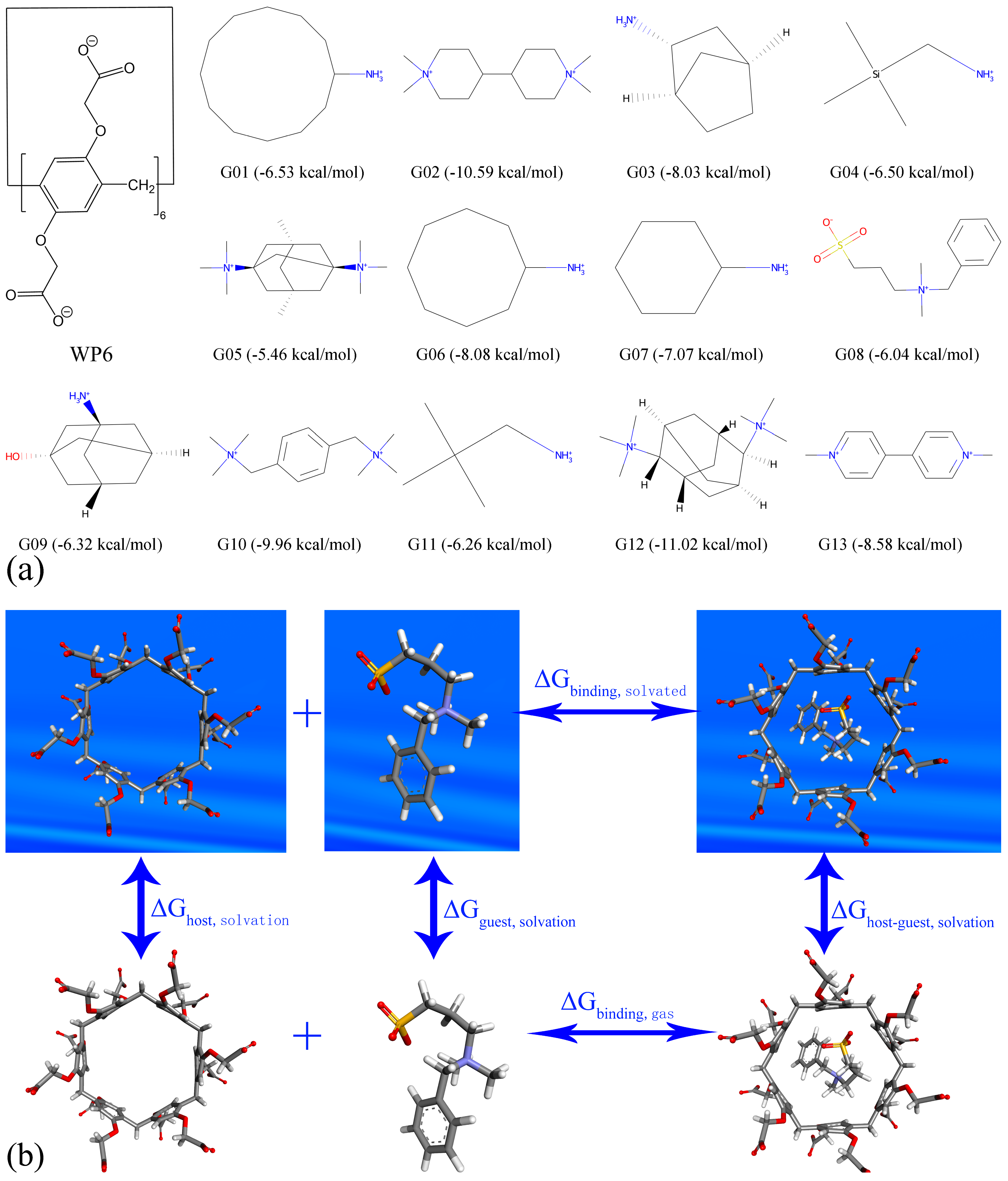
2. Model Construction and Configurational Sampling
3. Results and Discussion
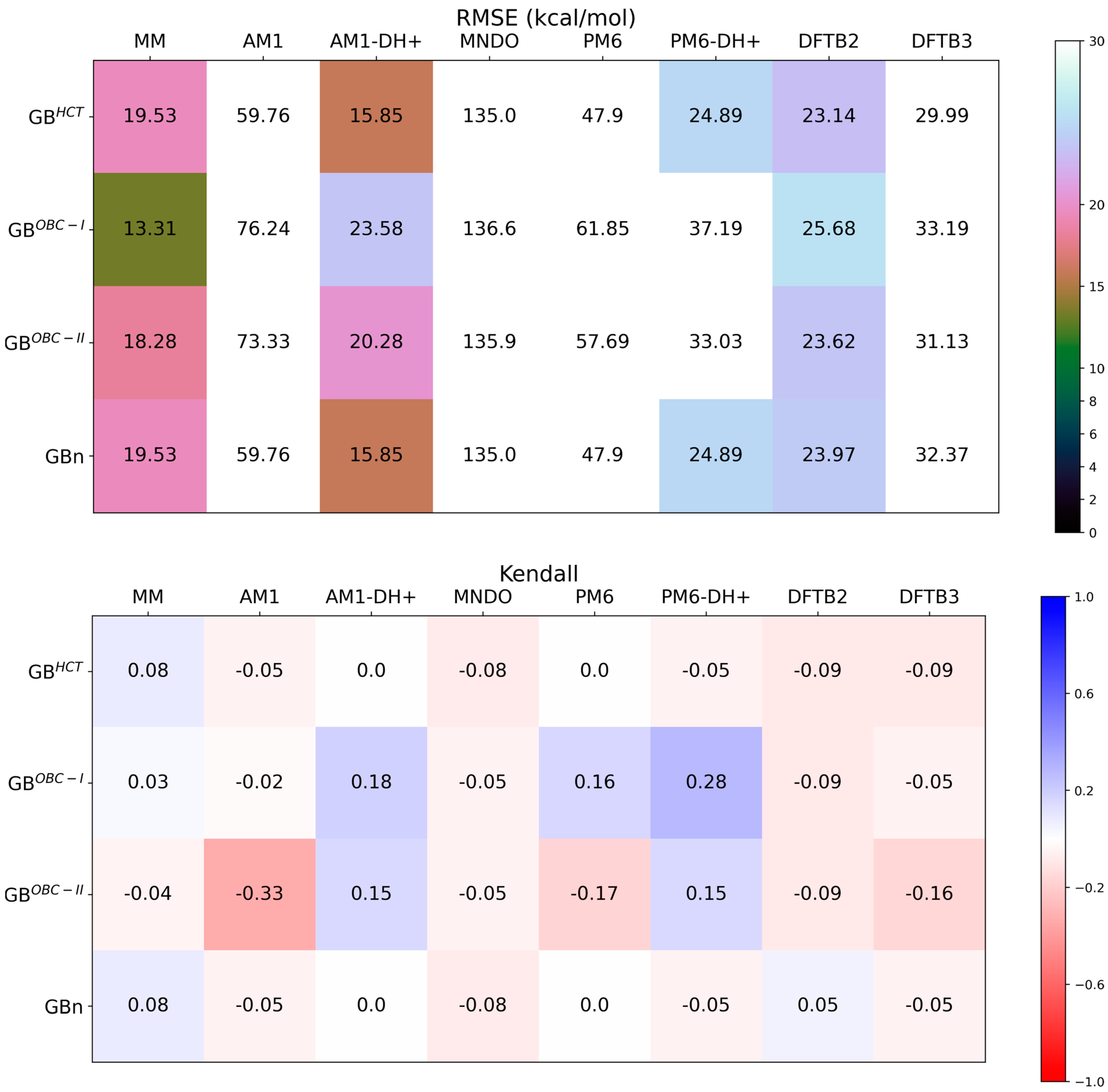
3.1. The QM Treatment
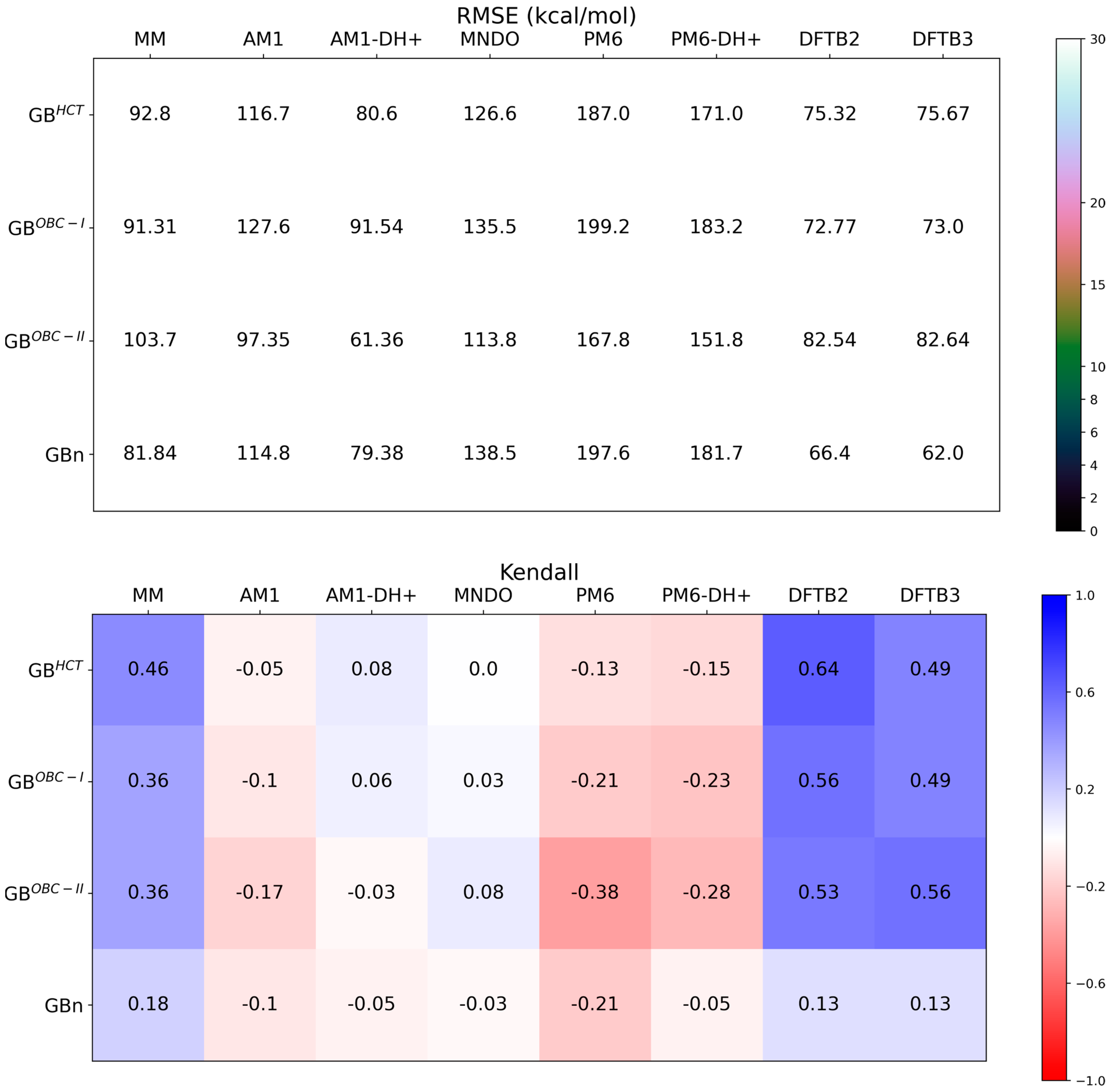
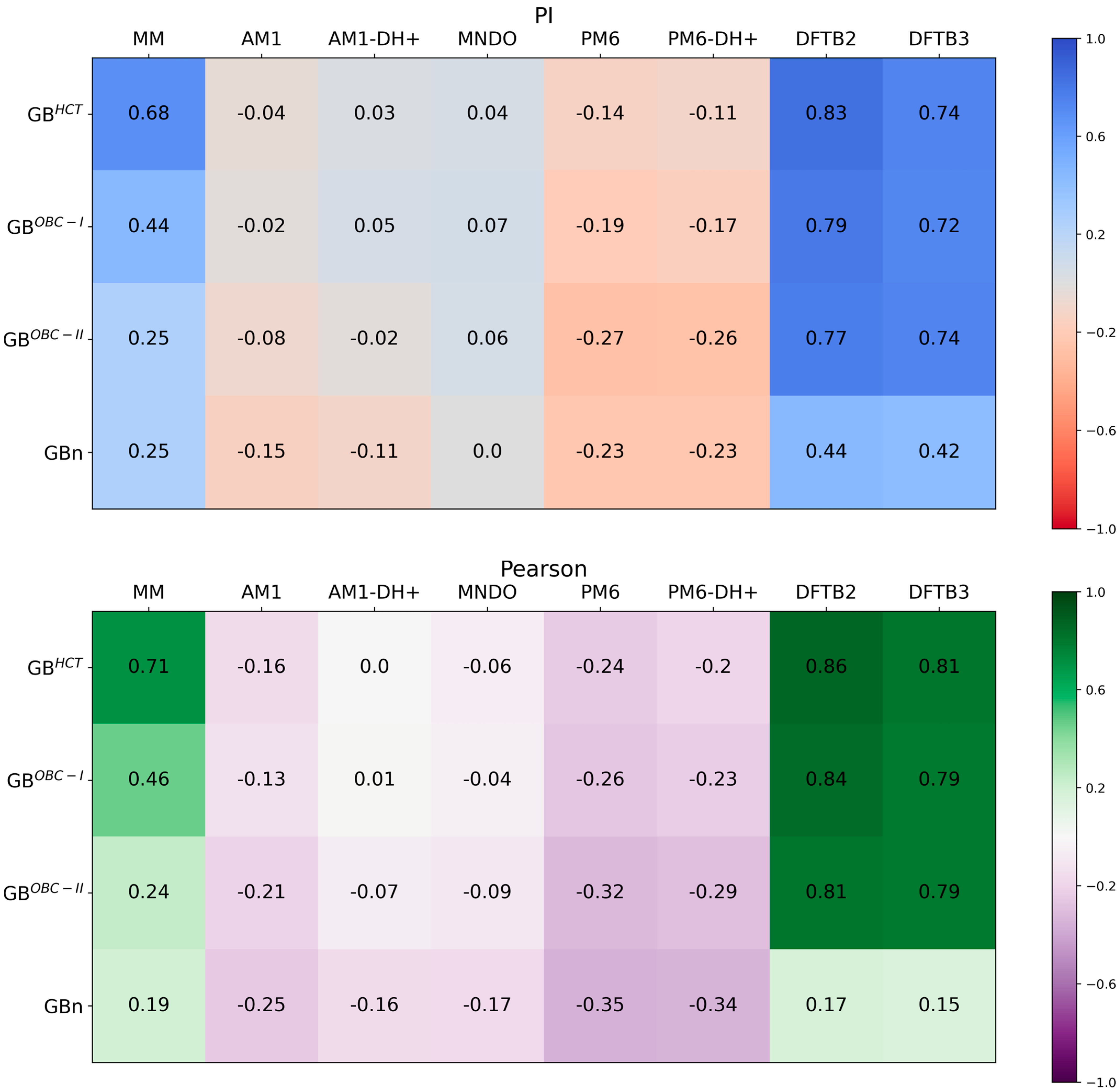
3.2. Three-Trajectory QM/GBSA
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, X.; Huai, Z.; Sun, Z. Host Dynamics under General-Purpose Force Fields. Molecules 2023, 28, 5940. [Google Scholar] [CrossRef] [PubMed]
- Alexov, E.; Mehler, E.L.; Baker, N.; Baptista, A.M.; Huang, Y.; Milletti, F.; Erik Nielsen, J.; Farrell, D.; Carstensen, T.; Olsson, M.H.M.; et al. Progress in the prediction of pKa values in proteins. Proteins Struct. Funct. Bioinform. 2011, 79, 3260–3275. [Google Scholar] [CrossRef]
- Li, C.; Zhang, A.; Wang, L.; Zuo, J.; Zhu, C.; Xu, J.; Wang, M.; Zhang, J.Z.H. Development of a polynomial scoring function P3-Score for improved scoring and ranking powers. Chem. Phys. Lett. 2023, 824, 140547. [Google Scholar] [CrossRef]
- Su, P.C.; Tsai, C.C.; Mehboob, S.; Hevener, K.E.; Johnson, M.E. Comparison of radii sets, entropy, QM methods, and sampling on MM-PBSA, MM-GBSA, and QM/MM-GBSA ligand binding energies of F. tularensis enoyl-ACP reductase (F abI). J. Comput. Chem. 2015, 36, 1859–1873. [Google Scholar] [CrossRef]
- Sangpheak, W.; Khuntawee, W.; Wolschann, P.; Pongsawasdi, P.; Rungrotmongkol, T. Enhanced stability of a naringenin/2, 6-dimethyl β-cyclodextrin inclusion complex: Molecular dynamics and free energy calculations based on MM-and QM-PBSA/GBSA. J. Mol. Graph. Model. 2014, 50, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Tsitsanou, K.E.; Hayes, J.M.; Keramioti, M.; Mamais, M.; Oikonomakos, N.G.; Kato, A.; Leonidas, D.D.; Zographos, S.E. Sourcing the affinity of flavonoids for the glycogen phosphorylase inhibitor site via crystallography, kinetics and QM/MM-PBSA binding studies: Comparison of chrysin and flavopiridol. Food Chem. Toxicol. 2013, 61, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Gundelach, L.; Fox, T.; Tautermann, C.S.; Skylaris, C.-K. BRD4: Quantum mechanical protein–ligand binding free energies using the full-protein DFT-based QM-PBSA method. Phys. Chem. Chem. Phys. 2022, 24, 25240–25249. [Google Scholar] [CrossRef]
- Nutho, B.; Pengthaisong, S.; Tankrathok, A.; Lee, V.S.; Ketudat Cairns, J.R.; Rungrotmongkol, T.; Hannongbua, S. Structural basis of specific glucoimidazole and mannoimidazole binding by Os3BGlu7. Biomolecules 2020, 10, 907. [Google Scholar] [CrossRef]
- Li, J.; Huang, Y.; Wu, M.; Wu, C.; Li, X.; Bao, J. Structure and energy based quantitative missense variant effect analysis provides insights into drug resistance mechanisms of anaplastic lymphoma kinase mutations. Sci. Rep. 2018, 8, 10664. [Google Scholar] [CrossRef]
- Chetter, B.A.; Kyriakis, E.; Barr, D.; Karra, A.G.; Katsidou, E.; Koulas, S.M.; Skamnaki, V.T.; Snape, T.J.; Psarra, A.-M.G.; Leonidas, D.D.; et al. Synthetic flavonoid derivatives targeting the glycogen phosphorylase inhibitor site: QM/MM-PBSA motivated synthesis of substituted 5,7-dihydroxyflavones, crystallography, in vitro kinetics and ex-vivo cellular experiments reveal novel potent inhibitors. Bioorg. Chem. 2020, 102, 104003. [Google Scholar] [CrossRef]
- Dutkiewicz, Z. Computational methods for calculation of protein-ligand binding affinities in structure-based drug design. In Theoretical and Computational Chemistry; Iwona, G., Bartosz, T., Eds.; De Gruyter: Berlin, Germany; Boston, MA, USA, 2021; pp. 1–40. [Google Scholar]
- Maier, S.; Thapa, B.; Erickson, J.; Raghavachari, K. Comparative assessment of QM-based and MM-based models for prediction of protein–ligand binding affinity trends. Phys. Chem. Chem. Phys. 2022, 24, 14525–14537. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, J.; Li, J.; He, X. Fragment-based quantum mechanical calculation of protein–protein binding affinities. J. Comput. Chem. 2018, 39, 1617–1628. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, X.; Zhang, J.Z.H.; He, X. Calculation of protein–ligand binding affinities based on a fragment quantum mechanical method. RSC Adv. 2015, 5, 107020–107030. [Google Scholar] [CrossRef]
- Lukac, I.; Abdelhakim, H.; Ward, R.A.; St-Gallay, S.A.; Madden, J.C.; Leach, A.G. Predicting protein–ligand binding affinity and correcting crystal structures with quantum mechanical calculations: Lactate dehydrogenase A. Chem. Sci. 2019, 10, 2218–2227. [Google Scholar] [CrossRef]
- Thapa, B.; Beckett, D.; Erickson, J.; Raghavachari, K. Theoretical Study of Protein–Ligand Interactions Using the Molecules-in-Molecules Fragmentation-Based Method. J. Chem. Theory Comput. 2018, 14, 5143–5155. [Google Scholar] [CrossRef]
- Okimoto, N.; Otsuka, T.; Hirano, Y.; Taiji, M. Use of the Multilayer Fragment Molecular Orbital Method to Predict the Rank Order of Protein–Ligand Binding Affinities: A Case Study Using Tankyrase 2 Inhibitors. ACS Omega 2018, 3, 4475–4485. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.K.; Koča, J. Assessing the Performance of MM/PBSA, MM/GBSA, and QM–MM/GBSA Approaches on Protein/Carbohydrate Complexes: Effect of Implicit Solvent Models, QM Methods, and Entropic Contributions. J. Phys. Chem. B 2018, 122, 8113–8121. [Google Scholar] [CrossRef]
- Nutho, B.; Khuntawee, W.; Rungnim, C.; Pongsawasdi, P.; Wolschann, P.; Karpfen, A.; Kungwan, N.; Rungrotmongkol, T. Binding mode and free energy prediction of fisetin/β-cyclodextrin inclusion complexes. Beilstein J. Org. Chem. 2014, 10, 2789–2799. [Google Scholar] [CrossRef] [PubMed]
- Rungrotmongkol, T.; Chakcharoensap, T.; Pongsawasdi, P.; Kungwan, N.; Wolschann, P. The inclusion complexation of daidzein with β-cyclodextrin and 2,6-dimethyl-β-cyclodextrin: A theoretical and experimental study. Monatshefte Chem.-Chem. Mon. 2018, 149, 1739–1747. [Google Scholar] [CrossRef]
- Ciancetta, A.; Genheden, S.; Ryde, U. A QM/MM study of the binding of RAPTA ligands to cathepsin B. J. Comput.-Aided Mol. Des. 2011, 25, 729–742. [Google Scholar] [CrossRef]
- Sun, Z.; He, Q.; Gong, Z.; Kalhor, P.; Huai, Z.; Liu, Z. A General Picture of Cucurbit[8]uril Host-Guest Binding: Recalibrating Bonded Interactions. Molecules 2023, 28, 3124. [Google Scholar] [CrossRef]
- Sun, Z.; Zheng, L.; Kai, W.; Huai, Z.; Liu, Z. Primary vs. Secondary: Directionalized Guest Coordination in β-Cyclodextrin Derivatives. Carbohydr. Polym. 2022, 297, 120050. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Huai, Z.; He, Q.; Liu, Z. A General Picture of Cucurbit[8]uril Host–Guest Binding. J. Chem. Inf. Model. 2021, 61, 6107–6134. [Google Scholar] [CrossRef]
- Sun, Z. SAMPL7 TrimerTrip Host-Guest Binding Poses and Binding Affinities from Spherical-Coordinates-Biased Simulations. J. Comput.-Aided Mol. Des. 2021, 35, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.; Kang, Y.; Xu, J.-F.; Zhang, X. pH/ROS Dual-Responsive Supramolecular Vesicles Fabricated by Carboxylated Pillar[6]arene-Based Host–Guest Recognition and Phenylboronic Acid Pinacol Ester Derivative. Langmuir 2020, 36, 4080–4087. [Google Scholar] [CrossRef]
- Zhang, X.; Cheng, Q.; Li, L.; Shangguan, L.; Li, C.; Li, S.; Huang, F.; Zhang, J.; Wang, R. Supramolecular therapeutics to treat the side effects induced by a depolarizing neuromuscular blocking agent. Theranostics 2019, 9, 3107. [Google Scholar] [CrossRef] [PubMed]
- Hua, B.; Shao, L.; Zhang, Z.; Sun, J.; Yang, J. Pillar [6] arene/acridine orange host–guest complexes as colorimetric and fluorescence sensors for choline compounds and further application in monitoring enzymatic reactions. Sens. Actuators B Chem. 2018, 255, 1430–1435. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, X.; Ding, J.; Yu, L.; Ma, D.; Ding, J. Improved solubility and bioactivity of camptothecin family antitumor drugs with supramolecular encapsulation by water-soluble pillar [6] arene. ACS Omega 2017, 2, 5283–5288. [Google Scholar] [CrossRef]
- Liu, X.; Zheng, L.; Qin, C.; Yalong, C.; Zhang, J.Z.; Sun, Z. Comprehensive Evaluation of End-Point Free Energy Techniques in Carboxylated-Pillar[6]arene Host-Guest Binding: III. Force-Field Comparison, Three-Trajectory Realization and Further Dielectric Augmentation. Molecules 2023, 28, 2767. [Google Scholar] [CrossRef]
- Liu, X.; Zheng, L.; Qin, C.; Zhang, J.Z.H.; Sun, Z. Comprehensive Evaluation of End-Point Free Energy Techniques in Carboxylated-Pillar[6]arene Host-guest Binding: I. Standard Procedure. J. Comput.-Aided Mol. Des. 2022, 36, 735–752. [Google Scholar] [CrossRef]
- Liu, X.; Zheng, L.; Cong, Y.; Gong, Z.; Yin, Z.; Zhang, J.Z.; Liu, Z.; Sun, Z. Comprehensive Evaluation of End-Point Free Energy Techniques in Carboxylated-Pillar[6]arene Host-guest Binding: II. Regression and Dielectric Constant. J. Comput.-Aided Mol. Des. 2022, 36, 879–894. [Google Scholar] [CrossRef] [PubMed]
- Procacci, P.; Guarnieri, G. SAMPL9 blind predictions using nonequilibrium alchemical approaches. J. Chem. Phys. 2022, 156, 164104. [Google Scholar] [CrossRef] [PubMed]
- Macchiagodena, M.; Pagliai, M.; Procacci, P. NE-RDFE: A protocol and toolkit for computing relative dissociation free energies with GROMACS between dissimilar molecules using bidirectional nonequilibrium dual topology schemes. J. Comput. Chem. 2023, 44, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1992, 97, 10269–10280. [Google Scholar] [CrossRef]
- Mcweeny, R.; Diercksen, G. Self-Consistent Perturbation Theory. II. Extension to Open Shells. J. Chem. Phys. 1968, 49, 4852–4856. [Google Scholar] [CrossRef]
- Pople, J.A.; Nesbet, R.K. Self-Consistent Orbitals for Radicals. J. Chem. Phys. 1954, 22, 571–572. [Google Scholar] [CrossRef]
- Roothaan, C.C.J. New Developments in Molecular Orbital Theory. Rev. Mod. Phys. 1951, 23, 69–89. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Price, D.J.; Brooks, C.L., III. A Modified TIP3P Water Potential for Simulation with Ewald Summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E., III. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [PubMed]
- Joung, I.S.; Cheatham, T.E. Molecular Dynamics Simulations of the Dynamic and Energetic Properties of Alkali and Halide Ions Using Water-Model-Specific Ion Parameters. J. Phys. Chem. B 2009, 113, 13279–13290. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Tom, D.; Holger, G.; Luo, R.; Merz, K.M.; Alexey, O.; Carlos, S.; Bing, W.; Woods, R.J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of The Cartesian Equations of Motion of A System with Constraints: Molecular Dynamics of n -alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An Analytical Version of The SHAKE and RATTLE Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Pastor, R.W.; Brooks, B.R.; Szabo, A. An analysis of the accuracy of Langevin and molecular dynamics algorithms. Mol. Phys. 1988, 65, 1409–1419. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J.P. Development and use of quantum mechanical molecular models. 76. AM1: A new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Thiel, W. Ground states of molecules. 38. The MNDO method. Approximations and parameters. J. Am. Chem. Soc. 1977, 99, 4899–4907. [Google Scholar] [CrossRef]
- Stewart, J.J. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef]
- Elstner, M.; Porezag, D.; Jungnickel, G.; Elsner, J.; Haugk, M.; Frauenheim, T.; Suhai, S.; Seifert, G. Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Phys. Rev. B 1998, 58, 7260. [Google Scholar] [CrossRef]
- Niehaus, T.A.; Elstner, M.; Frauenheim, T.; Suhai, S. Application of an approximate density-functional method to sulfur containing compounds. J. Mol. Struct. THEOCHEM 2001, 541, 185–194. [Google Scholar] [CrossRef]
- Gaus, M.; Goez, A.; Elstner, M. Parametrization and benchmark of DFTB3 for organic molecules. J. Chem. Theory Comput. 2013, 9, 338–354. [Google Scholar] [CrossRef]
- Gaus, M.; Cui, Q.; Elstner, M. DFTB3: Extension of the self-consistent-charge density-functional tight-binding method (SCC-DFTB). J. Chem. Theory Comput. 2011, 7, 931–948. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, G.D.; Cramer, C.J.; Truhlar, D.G. Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J. Phys. Chem. 1996, 100, 19824–19839. [Google Scholar] [CrossRef]
- Hawkins, G.D.; Cramer, C.J.; Truhlar, D.G. Pairwise solute descreening of solute charges from a dielectric medium. Chem. Phys. Lett. 1995, 246, 122–129. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins Struct. Funct. Bioinform. 2004, 55, 383–394. [Google Scholar] [CrossRef]
- Feig, M.; Onufriev, A.; Lee, M.S.; Im, W.; Case, D.A. Performance comparison of generalized born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. J. Comput. Chem. 2004, 25, 265–284. [Google Scholar] [CrossRef]
- Hai, N.; Pérez, A.; Bermeo, S.; Simmerling, C. Refinement of Generalized Born Implicit Solvation Parameters for Nucleic Acids and their Complexes with Proteins. J. Chem. Theory Comput. 2015, 11, 3714. [Google Scholar]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. Comput. Chem. 1999, 20, 217–230. [Google Scholar] [CrossRef]
- Kendall, M.G. A New Measure of Rank Correlation. Biometrika 1938, 30, 81–93. [Google Scholar] [CrossRef]
- Pearlman, D.A.; Charifson, P.S. Are Free Energy Calculations Useful in Practice? A Comparison with Rapid Scoring Functions for the p38 MAP Kinase Protein System. J. Med. Chem. 2001, 44, 3417–3423. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A. Normal mode analysis of protein dynamics. Curr. Opin. Struct. Biol. 2010, 4, 285–290. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Wang, M.; Sun, Z. Comprehensive Evaluation of End-Point Free Energy Techniques in Carboxylated-Pillar[6]arene Host-Guest Binding: IV. The QM Treatment, GB Models and the Multi-Trajectory Extension. Liquids 2023, 3, 426-439. https://doi.org/10.3390/liquids3040027
Wang X, Wang M, Sun Z. Comprehensive Evaluation of End-Point Free Energy Techniques in Carboxylated-Pillar[6]arene Host-Guest Binding: IV. The QM Treatment, GB Models and the Multi-Trajectory Extension. Liquids. 2023; 3(4):426-439. https://doi.org/10.3390/liquids3040027
Chicago/Turabian StyleWang, Xiaohui, Mao Wang, and Zhaoxi Sun. 2023. "Comprehensive Evaluation of End-Point Free Energy Techniques in Carboxylated-Pillar[6]arene Host-Guest Binding: IV. The QM Treatment, GB Models and the Multi-Trajectory Extension" Liquids 3, no. 4: 426-439. https://doi.org/10.3390/liquids3040027
APA StyleWang, X., Wang, M., & Sun, Z. (2023). Comprehensive Evaluation of End-Point Free Energy Techniques in Carboxylated-Pillar[6]arene Host-Guest Binding: IV. The QM Treatment, GB Models and the Multi-Trajectory Extension. Liquids, 3(4), 426-439. https://doi.org/10.3390/liquids3040027