
Solvation Structure and Ion–Solvent Hydrogen Bonding of Hydrated Fluoride, Chloride and Bromide—A Comparative QM/MM MD Simulation Study

Abstract
1. Introduction
2. Methodology
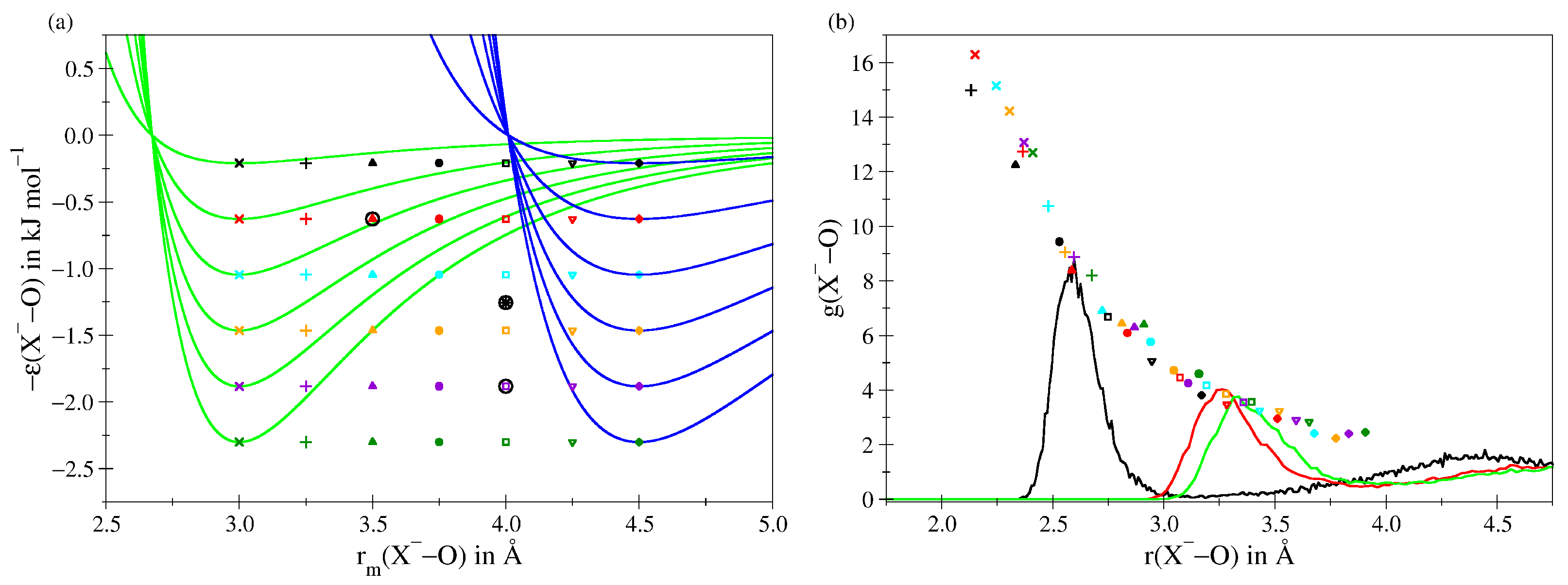
2.1. Ion–Water Interaction Potentials
2.2. QM/MM MD Simulation Protocol
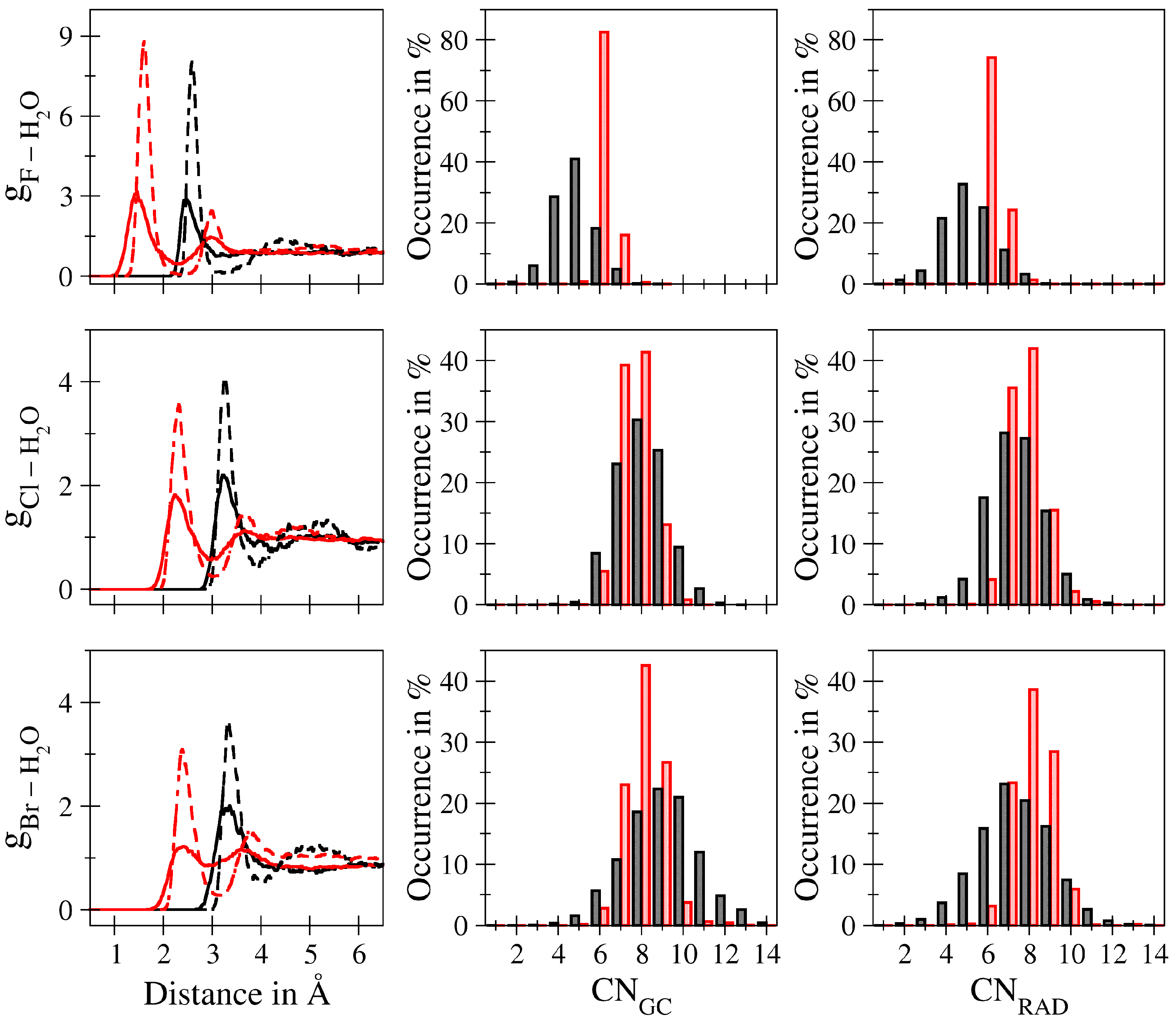
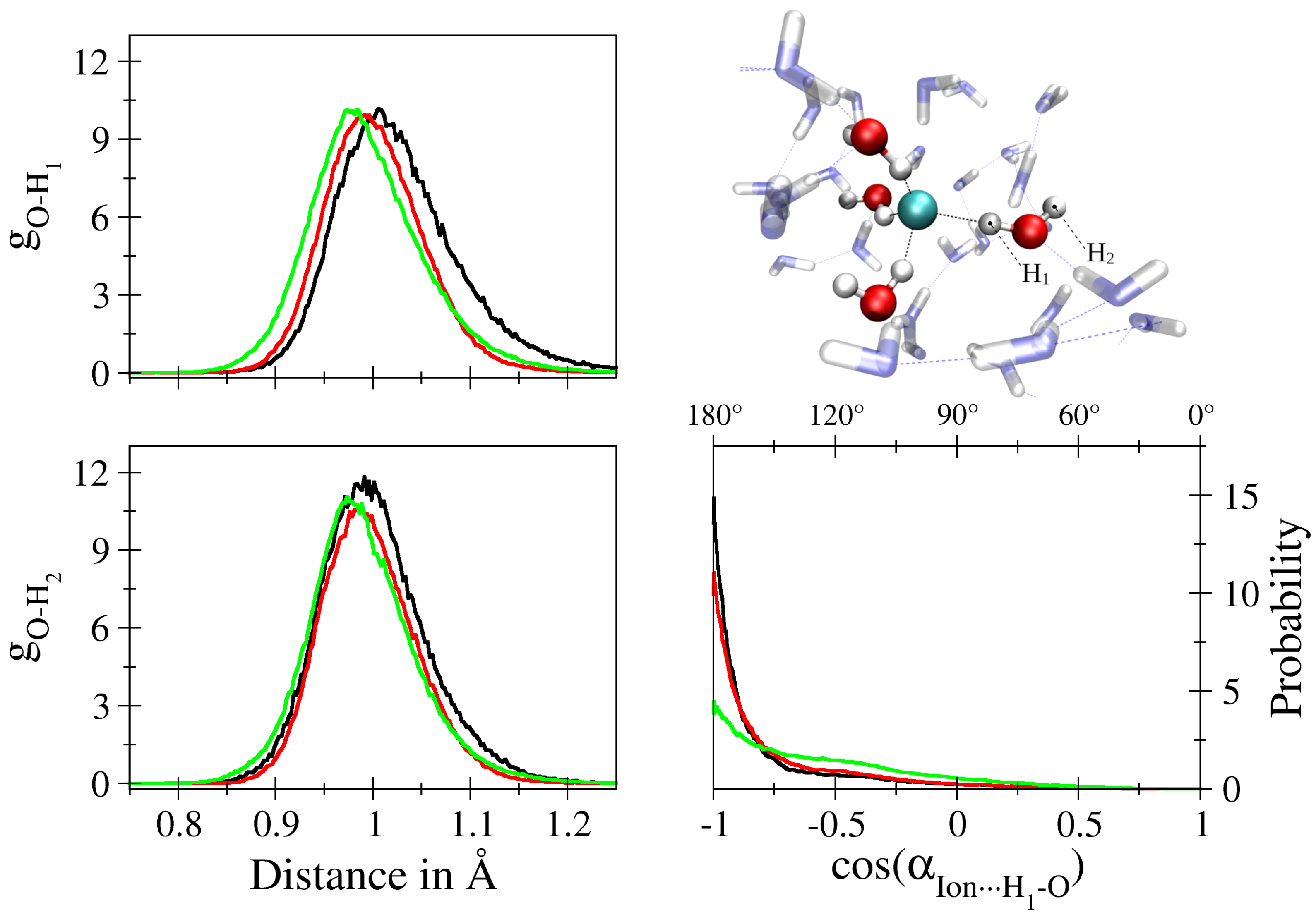
2.3. Analysis
3. Results
4. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hertz, H.G. Water: A Comprehensive Treatise; Plenum Press: New York, NY, USA, 1973. [Google Scholar]
- Stumm, W.; Morgan, J.J. Aqueous Chemistry, 3rd ed.; John Wiley & Sons: New York, NY, USA, 1996. [Google Scholar]
- Reichardt, C. Solvents and Solvent Effects in Organic Chemistry, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Buncel, E.; Stairs, R.A.; Wilson, H. The Role of the Solvent in Chemical Reactions; Oxford University Press: Cambridge, UK, 2003. [Google Scholar]
- Buncel, E.; Stairs, R.A. Solvent Effects in Chemistry, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2016. [Google Scholar]
- Shimizu, S.; Boon, C.L. The Kirkwood-Buff theory and the effect of cosolvents on biochemical reactions. J. Chem. Phys. 2004, 121, 9147–9155. [Google Scholar] [CrossRef] [PubMed]
- Wypych, G. (Ed.) Handbook of Solvents, 2nd ed.; ChemTec Publishing: Oxford, UK, 2014. [Google Scholar]
- Barthel, J.M.G.; Krienke, H.; Kunz, W. (Eds.) Physical Chemistry of Electrolyte Solutions; Steinkopff: Darmstadt, Germany, 1998. [Google Scholar]
- Wright, M.R. (Ed.) An Introduction to Aqueous Electrolyte Solutions; John Wiley & Sons: New York, NY, USA, 1998. [Google Scholar]
- Marcus, Y. Ions in Solution and Their Solvation; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Kastenholz, M.A.; Hünenberger, P.H. Computation of methodology-independent ionic solvation free energies from molecular simulations. I. The electrostatic potential in molecular liquids. J. Chem. Phys. 2006, 124, 124106. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.L. (Ed.) Molecular Thermodynamics of Electrolyte Solutions; World Scientific: Singapore, 2008. [Google Scholar]
- Hünenberger, P.; Reif, M. Single Ion Solvation; RSC Publishing: Cambridge, UK, 2011. [Google Scholar]
- Feldberg, S.W. On the dilemma of the use of the electroneutrality constraint in electrochemical calculations. Electrochem. Commun. 2000, 2, 453–456. [Google Scholar] [CrossRef]
- Jackowska, K.; Krysiński, P. Applied Electrochemistry; De Gruyter: Berlin, Germany, 2020. [Google Scholar]
- Tapia, O.; Bertrán, J. (Eds.) Solvent Effects and Chemical Reactivity; Springer: Darmstadt, The Netherlands, 2003. [Google Scholar]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Oxford Science Publications: Oxford, UK, 1990. [Google Scholar]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation; Academic Press: San Diego, CA, USA; London, UK, 2002. [Google Scholar]
- Tuckerman, M.E. Statistical Mechanics: Theory and Molecular Simulation; Oxford University Press: New York, NY, USA, 2009. [Google Scholar]
- Laurendeau, N.M. Statistical Thermodynamics: Fundamentals and Applications; Cambridge University Press: New York, NY, USA, 2005. [Google Scholar]
- Schwabl, F. Statistical Mechanics, 2nd ed.; Springer: Berlin, Germnay, 2010. [Google Scholar]
- Kastenholz, M.A.; Hünenberger, P.H. Computation of methodology-independent ionic solvation free energies from molecular simulations. II. The hydration free energy of the sodium cation. J. Chem. Phys. 2006, 124, 224501. [Google Scholar] [CrossRef]
- Reif, M.M.; Hünenberger, P.H. Computation of methodology-independent single-ion solvation properties from molecular simulations. III. Correction terms for the solvation free energies, enthalpies, entropies, heat capacities, volumes, compressibilities and expansivities of solvated ions. J. Chem. Phys. 2011, 134, 144103. [Google Scholar] [CrossRef]
- Szabo, A.; Ostlund, N. Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory; Dover Pubn. Inc.: New York, NY, USA, 1996. [Google Scholar]
- Helgaker, T.; Jørgensen, P.; Olsen, J. Molecular Electronic-Structure Theory; Wiley: Hoboken, NJ, USA, 2000. [Google Scholar]
- Koch, W.; Holthausen, M.C. A Chemist’s Guide to Density Functional Theory, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Sholl, D.S.; Steckel, J.A. Density Functional Theory—A Practical Introduction; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Hofer, T.S.; Rode, B.M.; Pribil, A.B.; Randolf, B.R. Simulations of Liquids and Solutions Based on Quantum Mechanical Forces. Adv. Inorg. Chem. 2010, 62, 143–175. [Google Scholar]
- Weiss, A.K.H.; Hofer, T.S. Exploiting the Capabilities of Quantum Chemical Simulations to Characterise the Hydration of Molecular Compounds. RSC Adv. 2014, 3, 1606–1635. [Google Scholar] [CrossRef]
- Hassanali, A.A.; Cuny, J.; Verdolino, V.; Parrinello, M. Aqueous solutions: State of the art in ab initio molecular dynamics. Phil. Trans. R. Soc. A 2014, 372, 20120482. [Google Scholar] [CrossRef]
- Gregory, K.P.; Elliott, G.R.; Wanless, E.J.; Webber, G.B.; Page, A.J. A quantum chemical molecular dynamics repository of solvated ions. Sci. Data 2022, 9, 430. [Google Scholar] [CrossRef]
- Pratt, L.R.; Rempe, S.B. Quasi-chemical theory and implicit solvent models for simulations. AIP Conf. Proc. 1999, 492, 172–201. [Google Scholar]
- Pliego, J.R.; Riveros, J.M. On the Calculation of the Absolute Solvation Free Energy of Ionic Species: Application of the Extrapolation Method to the Hydroxide Ion in Aqueous Solution. J. Phys. Chem. B 2000, 104, 5155–5160. [Google Scholar] [CrossRef]
- Lamoureux, G.; Roux, B. Absolute hydration free energy scale for alkali and halide ions established from simulations with a polarizable force field. J. Phys. Chem. B 2006, 110, 3308–3322. [Google Scholar] [CrossRef]
- Lev, B.; Roux, B.; Noskov, S.Y. Relative Free Energies for Hydration of Monovalent Ions from QM and QM/MM Simulations. J. Chem. Theory Comput. 2013, 9, 4165–4175. [Google Scholar] [CrossRef]
- Vlcek, L.; Chialvo, A.A. Single-ion hydration thermodynamics from clusters to bulk solutions: Recent insights from molecular modeling. Fluid Phase Equilib. 2016, 407, 58–75. [Google Scholar] [CrossRef]
- Duignan, T.T.; Baer, M.D.; Schenter, G.K.; Mundy, C.J. Real single ion solvation free energies with quantum mechanical simulation. Chem. Sci. 2017, 372, 6131–6140. [Google Scholar] [CrossRef] [PubMed]
- Hofer, T.S.; Hünenberger, P.H. Absolute proton hydration free energy, surface potential of water, and redox potential of the hydrogen electrode from first principles: QM/MM MD free-energy simulations of sodium and potassium hydration. J. Chem. Phys. 2018, 148, 222814. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Beck, T.L. Absolute ion hydration free energy scale and the surface potential of water via quantum simulation. Proc. Natl. Acad. Sci. USA 2001, 117, 30151–30158. [Google Scholar] [CrossRef]
- Prasetyo, N.; Hofer, T.S.; Hünenberger, P.H. Single-Ion Thermodynamics from First Principles: Calculation of the Absolute Hydration Free Energy and Single-Electrode Potential of Aqueous Li+ Using ab Initio Quantum Mechanical/Molecular Mechanical Molecular Dynamics Simulations. J. Chem. Theory Comput. 2018, 14, 6443–6459. [Google Scholar] [CrossRef]
- Schwenk, C.F.; Rode, B.M. Extended ab initio quantum mechanical/molecular mechanical molecular dynamics simulations of hydrated Cu2+. J. Chem. Phys. 2003, 119, 9523–9530. [Google Scholar] [CrossRef]
- Schwenk, C.F.; Hofer, T.S.; Rode, B.M. The Structure Breaking Effect of hydrated Cs(I). J. Phys. Chem. A 2004, 108, 1509–1514. [Google Scholar] [CrossRef]
- Schwenk, C.F.; Löffler, H.H.; Rode, B.M. Molecular dynamics simulations of Ca2+ in water: Comparison of a classical simulation including three-body corrections and Born-Oppenheimer ab initio and density functional theory quantum mechanical/molecular mechanics simulations. J. Chem. Phys. 2001, 115, 10808–10813. [Google Scholar] [CrossRef]
- Yoo, S.; Zeng, X.C.; Xantheas, S. On the phase diagram of water with density functional theory potentials: The melting temperature of ice Ih with the Perdew-Ernzerhof and Becke-Lee-Yang-Parr functionals. J. Chem. Phys. 2009, 130, 221102. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.; VandeVondele, J.; Kuo, I.; Sebastiani, D.; Siepmann, J.; Hutter, J.; Mundy, C.J. Isobaric-isothermal molecular dynamics simulations utilising density functional theory: An assessment of the structure and density of water at near-ambient conditions. J. Phys. Chem. B 2009, 113, 11959–11964. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion contribution. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, X.; Goddard, W.A., III. Doubly hybrid density functional for accurate descriptions of nonbond interactions, thermochemistry, and thermochemical kinetics. Proc. Natl. Acad. Sci. USA 2009, 106, 4963–4968. [Google Scholar] [CrossRef]
- Barone, V.; Biczyskoa, M.; Pavone, M. The role of dispersion correction to DFT for modelling weakly bound molecular complexes in the ground and excited electronic states. J. Comput. Chem. 2008, 346, 247–256. [Google Scholar] [CrossRef]
- Yoo, S.; Xantheas, S.S. The effect of dispersion corrections on the melting temperature of liquid water. J. Chem. Phys. 2011, 134, 121105. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Häser, M.; Weigand, F. RI-MP2: First derivatives and global consistency. Theor. Chem. Acc. 1997, 97, 331–340. [Google Scholar]
- Weigend, F.; Häser, M.; Patzelt, H.; Ahlrichs, R. RI-MP2: Optimized auxiliary basis sets and demonstration of efficiency. Chem. Phys. Lett. 1998, 294, 143–152. [Google Scholar] [CrossRef]
- Hättig, A.H.C.; Köhn, A. Distributed memory parallel implementation of energies and gradients for second-order Møller-Plesset perturbation theory with the resolution-of-the-identity approximation. Phys. Chem. Chem. Phys. 2006, 8, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- The Nobel Prize in Chemistry 2013, NobelPrize.org. Available online: https://www.nobelprize.org/prizes/chemistry/2013/summary/ (accessed on 11 November 2022).
- Warshel, A.; Levitt, M. Theoretical Studies of Enzymic Reactions: Dielectric, Electrostatic and Steric Stabilization of the Carbenium Ion in the Reaction of Lysozyme. J. Mol. Biol. 1976, 103, 227–249. [Google Scholar] [CrossRef] [PubMed]
- Åqvist, J.; Warshel, A. Simulation of enzyme reactions using valence bond force fields and other hybrid quantum/classical approaches. Chem. Rev. 1993, 93, 2523–2544. [Google Scholar] [CrossRef]
- Field, M.J.; Bash, P.A.; Karplus, M. A Combined Quantum Mechanical and Molecular Mechanical Potential for Molecular Dynamics Simulations. J. Comput. Chem. 1990, 11, 700–733. [Google Scholar] [CrossRef]
- Lyne, P.D.; Hodoscek, M.; Karplus, M. A Hybrid QM-MM Potential Employing Hartree-Fock or Density Functional Methods in the Quantum Region. J. Phys. Chem. A 1999, 103, 3462–3471. [Google Scholar] [CrossRef]
- Warshel, A. Molecular Dynamics Simulations of Biological Reactions. Acc. Chem. Res. 2002, 35, 385–395. [Google Scholar] [CrossRef]
- Senn, H.M.; Thiel, W. QM/MM studies of enzymes. Curr. Opin. Chem. Biol. 2007, 11, 182–187. [Google Scholar] [CrossRef]
- Lin, H.; Truhlar, D.G. QM/MM: What have we learned, where are we, and where do we go from here? Theor. Chem. Acc. 2007, 117, 185–199. [Google Scholar] [CrossRef]
- Bakowies, D.; Thiel, W. Hybrid Models for Combined Quantum Mechanical and Molecular Mechanical Approaches. J. Phys. Chem. 1996, 100, 10580–10594. [Google Scholar] [CrossRef]
- Rode, B.M.; Schwenk, C.F.; Tongraar, A. Structure and dynamics of hydrated ions—New insights through quantum mechanical simulations. J. Mol. Liq. 2003, 110, 105–122. [Google Scholar] [CrossRef]
- Rode, B.M.; Schwenk, C.F.; Hofer, T.S.; Randolf, B.R. Coordination and ligand exchange dynamics of solvated metal ions. Coord. Chem. Rev. 2005, 249, 2993–3006. [Google Scholar] [CrossRef]
- Guillot, B. A reappraisal of what we have learnt during three decades of computer simulations on water. J. Mol. Liq. 2002, 101, 219–260. [Google Scholar] [CrossRef]
- Cisneros, A.G.; Wikfeldt, K.T.; Ojamäe, L.; Lu, J.; Xu, Y.; Torabifard, H.; Bartók, A.P.; Csányi, G.; Molinero, V.; Paesani, F. Modeling Molecular Interactions in Water: From Pairwise to Many-Body Potential Energy Functions. Chem. Rev. 2016, 116, 7501–7528. [Google Scholar] [CrossRef]
- Demerdash, O.; Wang, L.-P.; Head-Gordon, T. Advanced models for water simulations. WIREs Comput. Mol. Sci. 2018, 8, e1355. [Google Scholar] [CrossRef]
- Kräutler, V.; van Gunsteren, W.; Hünenberger, P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Hofer, T.S. Balancing the structural, vibrational and dielectric properties of an advanced flexible water model. Chem. Phys. Lett. 2021, 762, 138172. [Google Scholar] [CrossRef]
- Rode, B.M.; Hofer, T.S.; Randolf, B.R.; Schwenk, C.F.; Xenides, D.; Vchirawongkwin, V. Ab initio Quantum Mechanical Charge Field Molecular Dynamics—A QM/MM MD Procedure for Accurate Simulations of Ions and Complexes. Theor. Chem. Acc. 2006, 115, 77–85. [Google Scholar] [CrossRef]
- Hofer, T.S.; Pribil, A.B.; Randolf, B.R.; Rode, B.M. Ab Initio Quantum Mechanical Charge Field Molecular Dynamics: A Nonparametrized First-Principle Approach to Liquids and Solutions. Adv. Quant. Chem. 2010, 59, 213–246. [Google Scholar]
- Tongraar, A.; Rode, B.M. The hydration structures of F- and Cl- investigated by ab initio QM/MM molecular dynamics simulations. Phys. Chem. Chem. Phys. 2003, 5, 357–362. [Google Scholar] [CrossRef]
- Wiedemair, M.J.; Weiss, A.K.H.; Rode, B.M. Ab initio quantum mechanical simulations confirm the formation of all postulated species in ionic dissociation. Phys. Chem. Chem. Phys. 2014, 16, 7368–7376. [Google Scholar] [CrossRef] [PubMed]
- Heuft, J.M.; Meijer, E.J. Density functional theory based molecular-dynamics study of aqueous fluoride solvation. J. Chem. Phys. 2005, 122, 094501. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, P.; Migliorati, V.; Guidoni, L. Hydration Properties of the Bromide Aqua Ion: The Interplay of First Principle and Classical Molecular Dynamics, and X-ray Absorption Spectroscopy. Inorg. Chem. 2010, 49, 4224–4231. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, J.R.; Yadav, V.K.; Krmakar, A.; Mallik, B.S.; Chandra, A. A first-principles theoretical study of hydrogen-bond dynamics and vibrational spectral diffusion in aqueous ionic solution: Water in the hydration shell of a fluoride ion. Pure Appl. Chem. 2013, 85, 27–40. [Google Scholar] [CrossRef]
- Bankura, A.; Santra, B.; DiStasio, R.A., Jr.; Swartz, C.W.; Klein, M.L.; Wu, X. A systematic study of chloride ion solvation in water using van der Waals inclusive hybrid density functional theory. Mol. Phys. 2015, 113, 2842–2854. [Google Scholar] [CrossRef]
- DelloStritto, M.; Xu, J.; Wu, X.; Klein, M.L. Aqueous solvation of the chloride ion revisited with density functional theory: Impact of correlation and exchange approximations. Phys. Chem. Chem. Phys. 2020, 22, 10666–10675. [Google Scholar] [CrossRef]
- Merkling, P.J.; Ayala, R.; Martínez, J.M.; Pappalardo, R.; Marcos, E.S. Interplay of computer simulations and X-ray absorption spectra in the study of the bromide hydration structure. J. Chem. Phys. 2003, 119, 6647–6654. [Google Scholar] [CrossRef]
- Soper, A.K.; Weckström, K. Ion solvation and water structure in potassium halide aqueous solutions. Biophys. Chem. 2006, 124, 180–191. [Google Scholar] [CrossRef]
- Antalek, M.; Pace, E.; Hedman, B.; Hodgson, K.; Chillemi, G.; Benfatto, M.; Sarangi, R.; Frank, P. Solvation structure of the halides from X-ray absorption spectroscopy. J. Chem. Phys. 2016, 145, 044318. [Google Scholar] [CrossRef]
- Wallen, S.L.; Palmer, B.J.; Pfund, D.M.; Fulton, J.L.; Newville, M.; Ma, Y.; Stern, E.A. Hydration of Bromide Ion in Supercritical Water: An X-ray Absorption Fine Structure and Molecular Dynamics Study. J. Phys. Chem. A 1997, 101, 9632–9640. [Google Scholar] [CrossRef]
- Ohtaki, H.; Radnai, T. Structure and Dynamics of Hydrated Ions. Chem. Rev. 1993, 93, 1157–1204. [Google Scholar] [CrossRef]
- Ohtaki, H. Ionic Solvation in Aqueous and Nonaqueous Solutions. Monatsh. Chem. 2001, 132, 1237–1268. [Google Scholar] [CrossRef]
- Gaspar, A.M.; Alves Marques, M.; Cabaço, M.I.; de Barros Marques, M.I.; Buslaps, T.; Honkimaki, V. X-ray diffraction investigations of concentrated aqueous solutions of calcium halides. J. Mol. Liq. 2004, 110, 15–22. [Google Scholar] [CrossRef]
- Mile, V.; Pusztai, L.; Dominguez, H.; Pizio, O. Understanding the Structure of Aqueous Cesium Chloride Solutions by Combining Diffraction Experiments, Molecular Dynamics Simulations, and Reverse Monte Carlo Modeling. J. Phys. Chem. B 2009, 113, 10760–10769. [Google Scholar] [CrossRef]
- Mile, V.; Gereben, O.; Kohara, S.; Pusztai, L. On the structure of aqueous cesium bromide solutions: Diffraction experiments, molecular dynamics simulations and Reverse Monte Carlo modeling. J. Mol. Liq. 2010, 157, 36–42. [Google Scholar] [CrossRef]
- Mile, V.; Gereben, O.; Kohara, S.; Pusztai, L. On the Structure of Aqueous Cesium Fluoride and Cesium Iodide Solutions: Diffraction Experiments, Molecular Dynamics Simulations, and Reverse Monte Carlo Modeling. J. Phys. Chem. B 2012, 116, 9758–9767. [Google Scholar] [CrossRef]
- Pethes, I.; Bakó, I.; Pusztai, L. Chloride ions as integral parts of hydrogen bonded networks in aqueous salt solutions: The appearance of solvent separated anion pairs. Phys. Chem. Chem. Phys. 2020, 22, 11038–11044. [Google Scholar] [CrossRef]
- Jensen, F. Introduction to Computational Chemistry; Wiley: Hoboken, NJ, USA, 1999. [Google Scholar]
- Leach, A.R. Molecular Modelling: Priciples and Applications; Prentice Hall: Harlow, UK, 2001. [Google Scholar]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The Missing Term in Effective Pair Potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Barker, J.; Watts, R. Monte Carlo studies of the dielectric properties of water-like models. Mol. Phys. 1973, 26, 789–792. [Google Scholar] [CrossRef]
- Swope, W.C.; Andersen, H.C.; Berens, P.H.; Wilson, K.R. A computer simulation method for the calculation of equilibrium constants for the formation of physical clusters of molecules: Application to small water clusters. J. Chem. Phys. 1982, 76, 637–649. [Google Scholar] [CrossRef]
- Horn, H.; Swope, W.; Pitera, J.; Madura, J.; Dick, T.; Hura, G.; Head-Gordon, T. Development of an improved four-site water model for biomolecular simulations: TIP4P-EW. J. Chem. Phys. 2004, 120, 9665–9678. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Phys. Chem. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Laio, A.; VandeVondele, J.; Rothlisberger, U. A Hamiltonian electrostatic coupling scheme for hybrid Car-Parrinello molecular dynamics simulations. J. Chem. Phys. 2002, 116, 6941–6947. [Google Scholar] [CrossRef]
- Voloshina, E.; Gaston, N.; Paulus, B. Embedding procedure for ab initio correlation calculations in group II metals. J. Chem. Phys. 2007, 126, 134115. [Google Scholar] [CrossRef]
- Hofer, T.S.; Randolf, B.R.; Rode, B.M. Solvation Effects on Molecules and Biomolecules. In Challenges and Advances in Computational Chemistry and Physics; Canuto, S., Ed.; Chapter Molecular Dynamics Simulation Methods Including Quantum Effects; Springer: Heidelberg, Germany, 2008; Volume 6, pp. 247–278. [Google Scholar]
- Hofer, T.S. Perspectives for hybrid ab initio/molecular mechanical simulations of solutions: From complex chemistry to proton-transfer reactions and interfaces. Pure Appl. Chem. 2014, 86, 105–117. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic population analysis on LCAO-MO molecular wave functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic population analysis on LCAO-MO molecular wave functions. II. J. Chem. Phys. 1955, 23, 1841–1846. [Google Scholar] [CrossRef]
- Liew, C.C.; Inomata, H.; Arai, K. Flexible molecular models for molecular dynamics study of near and supercritical water. Fluid Phase Equilibr. 1998, 144, 287–298. [Google Scholar] [CrossRef]
- TURBOMOLE V7.2 2017, a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, Since 2007. Available online: http://www.turbomole.com (accessed on 11 November 2022).
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Hofer, T.S.; Tran, H.T.; Schwenk, C.F.; Rode, B.M. Characterisation of Dynamics and Reactivities of Solvated Ions by ab initio Simulations. J. Comput. Chem. 2004, 25, 211–214. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Hofer, T.; Rode, B. Local density corrected three-body distribution functions for probing local structure reorganization in liquids. Phys. Chem. Chem. Phys. 2008, 10, 6653–6657. [Google Scholar] [CrossRef]
- Higham, J.; Henchman, R.H. Locally adaptive method to define coordination shell. J. Chem. Phys. 2016, 145, 084108. [Google Scholar] [CrossRef]
- Ali, H.S.; Higham, J.; Henchman, R.H. Entropy of simulated liquids using multiscale cell correlation. Entropy 2019, 21, 750. [Google Scholar] [CrossRef] [PubMed]
- Tkatchenko, A.; Scheffler, M. Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef]
- Leontyev, I.V.; Stuchebrukhov, A.A. Electronic continuum model for molecular dynamics simulations. J. Chem. Phys. 2009, 130, 085102. [Google Scholar] [CrossRef]
- Leontyev, I.V.; Stuchebrukhov, A.A. Electronic polarizability and the effective pair potentials of water. J. Chem. Theory Comput. 2010, 6, 3153–3161. [Google Scholar] [CrossRef]
- Leontyev, I.V.; Stuchebrukhov, A.A. Accounting for electronic polarization in non-polarizable force fields. Phys. Chem. Chem. Phys. 2011, 13, 2613–2626. [Google Scholar] [CrossRef] [PubMed]
- Kann, Z.; Skinner, J.L. A scaled-ionic-charge simulation model that reproduces enhanced and suppressed water diffusion in aqueous salt solutions. J. Chem. Phys. 2014, 141, 104507. [Google Scholar] [CrossRef] [PubMed]
- Hofer, T.S.; Randolf, B.R.; Rode, B.M. tructure-Breaking Effects of Solvated Rb(I) in Dilute Aqueous Solution—An Ab Initio QM/MM MD Approach. J. Comput. Chem. 2005, 26, 949–956. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| – | – | – | |
|---|---|---|---|
| F | 0.6276 | 3.1181 | 3.5 |
| Cl | 1.2552 | 3.5636 | 4.0 |
| Br | 1.8828 | 3.5636 | 4.0 |
| CN | CN | R | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| F | MM MD | 2.59 | 2.63 | 3.25 | 6.2 | 6.3 | 15.4 | 10 | 5.4 | this work |
| RIMP2/MM MD | 2.46 | 2.68 | 3.36 | 4.9 | 5.2 | 1.1 | 115 | 5.2 | this work | |
| MM MD | 2.53 | 5.8 ± 0.1 | Ref. [73] | |||||||
| HF/MM MD | 2.68 | 4.6 ± 0.2 | Ref. [73] | |||||||
| BLYP CPMD | 2.66 | 5.1 | Ref. [75] | |||||||
| BLYP CPMD | 2.7 | 3.4 | Ref. [77] | |||||||
| NDIS KF/D0 1.2:100 | 2.54 | 3.27 | 6.9 | Ref. [81] | ||||||
| Cl | MM MD | 3.26 | 3.35 | 3.93 | 7.6 | 7.8 | 4.2 | 46 | 6.7 | this work |
| RIMP2/MM MD | 3.23 | 3.48 | 4.16 | 8.1 | 7.5 | 1.6 | 178 | 4.5 | this work | |
| MM MD | 3.15 | 5.9 ± 0.1 | Ref. [73] | |||||||
| HF/MM MD | 3.24 | 5.9 ± 0.1 | Ref. [73] | |||||||
| HF/MM MD | 3.25 | 3.9 | 6.8 | 2.0 | 4.6 | Ref. [74] | ||||
| PBE-D3 CPMD | 3.14 | 3.78 | 6.0 | Ref. [79] | ||||||
| PBE0-D3 CPMD | 3.17 | 3.85 | 6.1 | Ref. [79] | ||||||
| SCAN CPMD | 3.17 | 3.85 | 6.7 | Ref. [79] | ||||||
| PBE CPMD | 3.11 | 3.64 | 5.5 ± 0.2 | Ref. [78] | ||||||
| PBE+TS-vdW CPMD | 3.14 | 3.78 | 6.3 ± 0.9 | Ref. [78] | ||||||
| PBE0 CPMD | 3.14 | 3.72 | 5.8 ± 0.7 | Ref. [78] | ||||||
| PBE0+TS-vdW CPMD | 3.16 | 3.73 | 6.3 ± 0.8 | Ref. [78] | ||||||
| EXAFS NaCl 40 mM | 2.91/3.11 | 4+3 | Ref. [82] | |||||||
| NDIS KCl/DO 1.2:100 | 3.14 | 3.78 | 7.1 | Ref. [81] | ||||||
| Br | MM MD | 3.33 | 3.45 | 4.05 | 8.1 | 8.1 | 3.0 | 313 | 4.7 | this work |
| RIMP2/MM MD | 3.31 | 3.68 | 4.30 | 9.1 | 7.4 | 0.9 | 390 | 2.9 | this work | |
| MM MD | 3.27 | 3.9 | 7.6 ± 0.5 | 2.6 | Ref. [76] | |||||
| BLYP CPMD | 3.33 | 3.9 | 6.5 ± 0.3 | 5.7 | Ref. [76] | |||||
| XAFS/MM MC YBr 0.3M | 3.44 ± 0.07 | 6 ± 0.5 | Ref. [80] | |||||||
| XAFS RbBr 0.2M | 3.35 | 7.1 ± 1.5 | Ref. [83] | |||||||
| XAFS RbBr 1.5M | 3.36 | 7.2 ± 0.4 | Ref. [83] | |||||||
| XAFS RbCl 0.5 mM | 3.26 | 10 | Ref. [82] | |||||||
| NDIS KBr/DO 1.2/100 | 3.32 | 3.90 | 6.7 | Ref. [81] | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hofer, T.S. Solvation Structure and Ion–Solvent Hydrogen Bonding of Hydrated Fluoride, Chloride and Bromide—A Comparative QM/MM MD Simulation Study. Liquids 2022, 2, 445-464. https://doi.org/10.3390/liquids2040026
Hofer TS. Solvation Structure and Ion–Solvent Hydrogen Bonding of Hydrated Fluoride, Chloride and Bromide—A Comparative QM/MM MD Simulation Study. Liquids. 2022; 2(4):445-464. https://doi.org/10.3390/liquids2040026
Chicago/Turabian StyleHofer, Thomas S. 2022. "Solvation Structure and Ion–Solvent Hydrogen Bonding of Hydrated Fluoride, Chloride and Bromide—A Comparative QM/MM MD Simulation Study" Liquids 2, no. 4: 445-464. https://doi.org/10.3390/liquids2040026
APA StyleHofer, T. S. (2022). Solvation Structure and Ion–Solvent Hydrogen Bonding of Hydrated Fluoride, Chloride and Bromide—A Comparative QM/MM MD Simulation Study. Liquids, 2(4), 445-464. https://doi.org/10.3390/liquids2040026