
Structures of Hydrated Metal Ions in Solid State and Aqueous Solution


Abstract

1. Introduction
2. Methods to Investigate the Coordination Chemistry in Solution
3. Structures of Hydrated Metal Ions in Solid State and Aqueous Solution
3.1. Alkali Metal Ions
3.1.1. Lithium
3.1.2. Sodium
3.1.3. Potassium
3.1.4. Rubidium
3.1.5. Cesium
3.2. Alkaline Earth Metal Ions
3.2.1. Beryllium
3.2.2. Magnesium
3.2.3. Calcium
3.2.4. Strontium
3.2.5. Barium
3.2.6. Radium
3.3. Transition Metal Ions
3.3.1. Group 3 Elements
Scandium
Yttrium
3.3.2. Lanthanoids
3.3.3. Actinoids
Actinoid(III) and Actinoid(IV) Ions
Oxoactinoid(V) and Dioxoactinoid(VI)/Actinyl(V) and Actinyl(VI)
3.3.4. Group 4 Elements
Titanium(III)
Titanium(IV)/Titanyl(IV)
Zirconium(IV) and Hafnium(IV)
3.3.5. Group 5 Elements
Vanadium(II) and Vanadium(III)
Oxovanadium(IV) and Dioxovanadium(V)/Vanadyl(IV) and Vanadyl(V)
3.3.6. Group 6 Elements
Chromium(II)
Chromium(III)
Molybenum(II) and Molybdenum(III)
3.3.7. Group 7 Elements
Manganese(II)
Manganese(III)
3.3.8. Group 8 Elements
Iron(II)
Iron(III)
Ruthenium(II) and Ruthenium(III)
3.3.9. Group 9 Elements
Cobalt(II)
Cobalt(III)
Rhodium(III)
Iridium(III)
3.3.10. Group 10 Elements
Nickel
Palladium(II) and Platinum(II)
3.3.11. Group 11 Elements
Copper(I)
Copper(II)
Silver
Gold
3.3.12. Group 12 Elements
Zinc
Cadmium
Mercury(I)
Mercury(II)
3.4. Group 13 Elements
Aluminum, Gallium Indium, and Thallium(III)
3.5. d10s2 Metal Ions in Groups 13, 14, and 15
3.5.1. Thallium(I)
3.5.2. Tin(II)
3.5.3. Lead(II)
3.5.4. Bismuth(III)
4. Summary

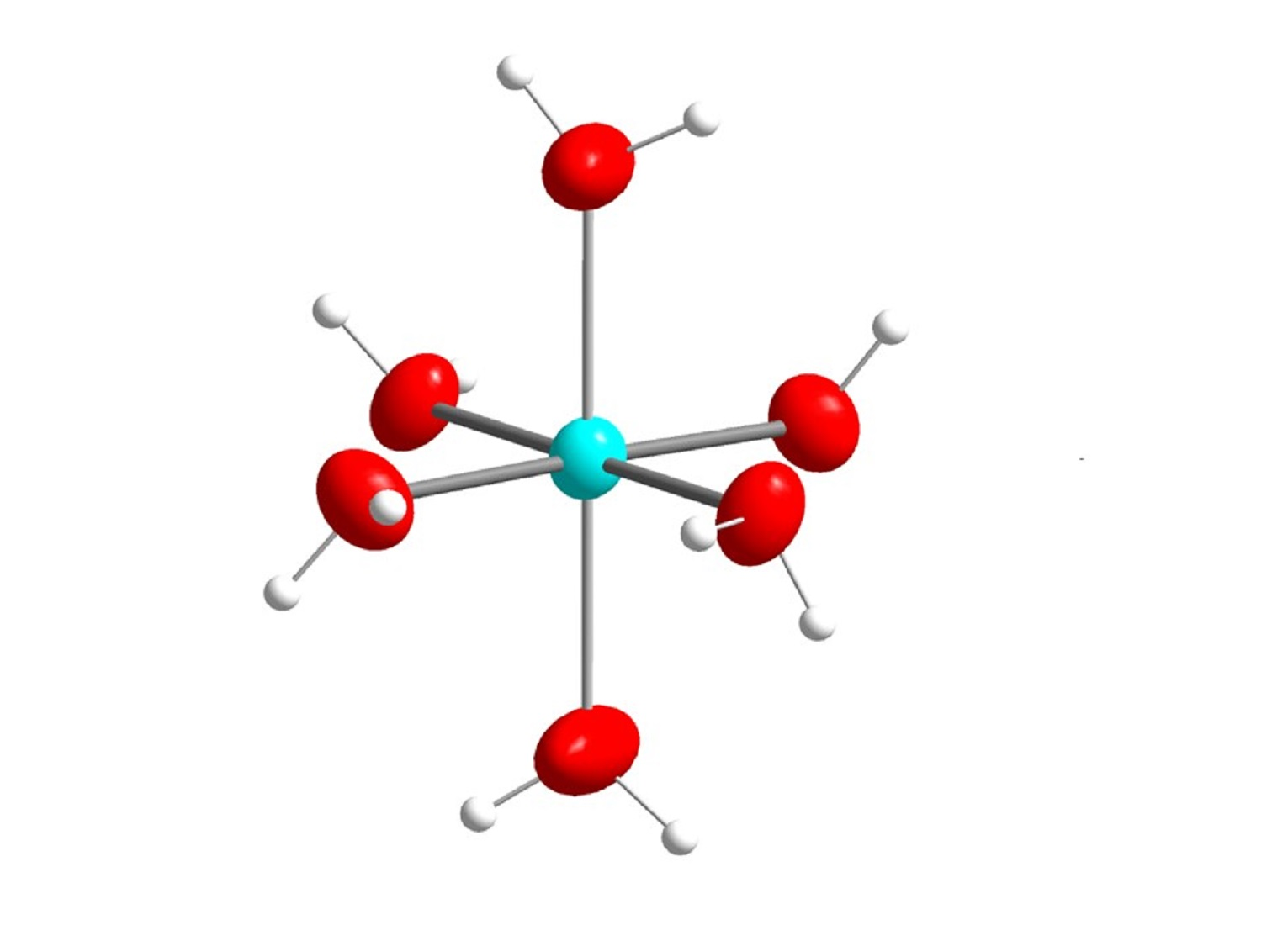{kind=link}
| Metal Ion | Solid State | Aqueous Solution |
|---|---|---|
| Lithium(I) | [Li(H2O)4]+, Li(H2O)5]+, [Li(H2O)6]+, [Li2(H2O)6]2+, [Li2(H2O)7]2+, [Li3(H2O)6]3+,[Li3(H2O)8]3+ | [Li(H2O)4]+ |
| Sodium(I) | [Na(H2O)2]+, [Na(H2O)4]+, [Na(H2O)5]+, [Na(H2O)6]+, [Na(H2O)8]+, [Na2(H2O)6]n2+, [Na2(H2O)7]n2+, [Na2(H2O)8]n2+, [Na2(H2O)8]2+, [Na2(H2O)9]2+, [Na2(H2O)10]2+, [Na2(H2O)13]2+, [Na4(H2O)14]n4+, [Na4(H2O)14]n4+, [Na4(H2O)18]n4+, [Na5(H2O)14]5+, [Na8(H2O)24]n8+ | [Na(H2O)6]+ |
| Potassium(I) | [K(H2O)6]+, [K(H2O)7]+, [K(H2O)8]+, [K2(H2O)9]n2+ | [K(H2O)7]+ |
| Rubidium(I) | [Rb(H2O)9]+ | [Rb(H2O)8]+ |
| Cesium(I) | [Cs(H2O)10]+ | [Cs(H2O)8]+ |
| Beryllium(II) | [Be(H2O)4]2+ | [Be(H2O)4]2+ |
| Magnesium(II) | [Mg(H2O)6]2+ | [Mg(H2O)6]2+ |
| Calcium(II) | [Ca(H2O)6]2+, [Ca(H2O)7]2+, [Ca(H2O)8]2+, [Ca2(H2O)10]4+, [Ca2(H2O)11]4+, [Ca2(H2O)14]4+ | [Ca(H2O)8]2+ |
| Strontium(II) | [Sr(H2O)6]2+, [Sr(H2O)8]2+, [Sr(H2O)9]2+, [Sr2(H2O)14]4+ | [Sr(H2O)8]2+ |
| Barium(II) | [Ba(H2O)8]2+, [Ba(H2O)9]2+, [Ba2(H2O)14]2+, [Ba2(H2O)18]4+ | [Ba(H2O)8–9]2+ |
| Radium(II) | [Ra(H2O)≈10]2+ | |
| Scandium(III) | [Sc(H2O)6]3+, [Sc(H2O)7]3+, [Sc(H2O)8.0]3+ | [Sc(H2O)8.0]3+ |
| Yttrium(III) | [Y(H2O)8]3+, [Y(H2O)9]3+ | [Y(H2O)8]3+ |
| Lanthanum(III) | [La(H2O)6]3+, [La(H2O)8]3+, [La(H2O)9]3+ | [La(H2O)9]3+ |
| Cerium(III) | [Ce(H2O)8]3+, [Ce(H2O)9]3+ | [Ce(H2O)9]3+ |
| Cerium(IV) | [Ce(H2O)8]4+ | |
| Praseodymium(III) | [Pr(H2O)8]3+, [Pr(H2O)9]3+ | [Pr(H2O)9]3+ |
| Neodymium(III) | [Nd(H2O)8]3+, [Nd(H2O)9]3+ | [Nd(H2O)9]3+ |
| Prometium(III) | [Pm(H2O)9]3+ | |
| Samarium(III) | [Sm(H2O)8]3+, [Sm(H2O)9]3+ | [Sm(H2O)9]3+ |
| Europium(II) | [Eu(H2O)7]2+ | |
| Europium(III) | [Eu(H2O)8]3+, [Su(H2O)9]3+ | [Eu(H2O)9]3+ |
| Gadolinium(III) | [Gd(H2O)8]3+, [Gd(H2O)9]3+ | [Gd(H2O)9]3+ |
| Terbium(III) | [Tb(H2O)8]3+, [Tb(H2O)9]3+ | [Tb(H2O)9]3+ |
| Dysprosium(III) | [Dy(H2O)8]3+, [Dy(H2O)9]3+ | [Dy(H2O)9]3+ |
| Holmium(III) | [Ho(H2O)8]3+, [Ho(H2O)8.91]3+, [Ho(H2O)9]3+ | [Ho(H2O)8.91]3+ |
| Erbium(III) | [Er(H2O)8]3+, [Er(H2O)8.95]3+, [Er(H2O)9]3+ | [Er(H2O)8.95]3+ |
| Thulium(III) | [Tm(H2O)8]3+, [Tm(H2O)8.8]3+, [Tm(H2O)9]3+ | [Tm(H2O)8.8]3+ |
| Ytterbium(II) | [Yb(H2O)8]2+ | |
| Ytterbium(III) | [Yb(H2O)8]3+, [YbH2O)8.7]3+, [Yb(H2O)9]3+ | [Yb(H2O)8.7]3+ |
| Lutetium(III) | [Lu(H2O)8]3+, [Lu(H2O)8.2]3+, [Lu(H2O)9]3+ | [Lu(H2O)8.2]3+ |
| Actinium(III) | [Ac(H2O)11]3+ | |
| Thorium(IV) | [Th(H2O)8]4+, [Th(H2O)9]4+, [Th(H2O)10]4+ | [Th(H2O)9]4+ |
| Uranium(III) | [U(H2O)9]3+ | [U(H2O)9]3+ |
| Uranium(IV) | [U(H2O)9]4+ | |
| Uranium(VI) | [UO2(H2O)5]2+ | [UO2(H2O)5]2+ |
| Neptunium(III) | [Np(H2O)9]3+ | [Np(H2O)9]3+ |
| Neptunium(IV) | [Np(H2O)9]4+ | |
| Neptunium(V) | [NpO2(H2O)5]+ | |
| Neptunium(VI) | [NpO2(H2O)5]2+ | [NpO2(H2O)5]2+ |
| Plutonium(III) | [Pu(H2O)9]3+ | [Pu(H2O)9]3+ |
| Plutonium(IV) | [Pu(H2O)9]4+ | |
| Plutonium(V) | [PuO2(H2O)5]+ | |
| Plutonium(VI) | [PuO2(H2O)5]2+ | [PuO2(H2O)5]2+ |
| Americium(III) | [Am(H2O)9]3+ | [Am(H2O)9]3+ |
| Curium(III) | [Cm(H2O)9]3+ | [Am(H2O)9]3+ |
| Berkelium(III) | [Bk(H2O)9]3+ | |
| Californium(III) | [Cf(H2O)9]3+ | [Cf(H2O)9]3+ |
| Einsteinium(III) | [Es(H2O)9]3+ | |
| Fermium(III) | [Fm(H2O)9]3+ | |
| Medelevium(III) | [Md(H2O)9]3+ | |
| Nobelium(III) | [No(H2O)9]3+ | |
| Lawrentium(III) | [Lr(H2O)9]3+ | |
| Titanium(III) | [Ti(H2O)6]3+ | [Ti(H2O)6]3+ |
| Titanium(IV) | [TiO(H2O)5]2+ | |
| Zirconium(IV) | [Zr(H2O)8]4+ | |
| Hafnium(IV) | [Hf(H2O)8]4+ | |
| Vanadium(II) | [V(H2O)6]2+ | [V(H2O)6]2+ |
| Vanadium(III) | [V(H2O)6]3+ | [V(H2O)6]3+ |
| Vanadium(IV) | [VO(H2O)6]2+ | [VO(H2O)5]2+ |
| Vanadium(V) | cis-[VO2(H2O)4]+ | |
| Chromium(II) | [Cr(H2O)6]2+ | [Cr(H2O)6]2+ |
| Chromium(III) | [Cr(H2O)6]3+ | [Cr(H2O)6]3+ |
| Molybdenum(II) | [Mo2(H2O)4]4+ | |
| Molybdenum(III) | [Mo(H2O)6]3+ | [Mo(H2O)6]3+ |
| Manganese(II) | [Mn(H2O)6]2+ | [Mn(H2O)6]2+ |
| Manganese(III) | [Mn(H2O)6]3+ | [Mn(H2O)6]3+ |
| Iron(II) | [Fe(H2O)6]2+ | [Fe(H2O)6]2+ |
| Iron(III) | [Fe(H2O)6]3+ | [Fe(H2O)6]3+ |
| Ruthenium(II) | [Ru(H2O)6]2+ | [Ru(H2O)6]2+ |
| Ruthenium(III) | [Ru(H2O)6]3+ | [Ru(H2O)6]3+ |
| Cobalt(II) | [Co(H2O)6]2+, [Co2(H2O)10]4+ | [Co(H2O)6]2+ |
| Cobalt(III) | [Co(H2O)6]3+ | [Co(H2O)6]3+ |
| Rhodium(III) | [Rh(H2O)6]3+ | [Rh(H2O)6]3+ |
| Iridium(III) | [Ir(H2O)6]3+ | [Ir(H2O)6]3+ |
| Nickel(II) | [Ni(H2O)6]2+, [Ni2(H2O)10]4+ | [Ni(H2O)6]2+ |
| Palladium(II) | [Pd(H2O)4]2+ | [Pd(H2O)5/6]2+ |
| Platinum(II) | [Pt(H2O)4]2+ | [Pt(H2O)5/6]2+ |
| Copper(I) | [Cu(H2O)4]+ | |
| Copper(II) | [Cu(H2O)4]2+, [Cu(H2O)5]2+, [Cu(H2O)6]2+ | [Cu(H2O)6]2+ |
| Silver(I) | [Ag(H2O)2]+, [Ag(H2O)3]+ | [Ag(H2O)2]+ |
| Gold(III) | [Au(H2O)4]3+ | [Au(H2O)4]3+ |
| Zinc(II) | [Zn(H2O)4]2+, [Zn(H2O)5]2+, [Zn(H2O)6]2+ | [Zn(H2O)6]2+ |
| Cadmium(II) | [Cd(H2O)6]2+, [Cd2(H2O)10]4+ | [Cd(H2O)6]2+ |
| Mercury(I) | [Hg2(H2O)2]2+ | [Hg2(H2O)6]2+ |
| Mercury(II) | [Hg(H2O)6]2+ | [Hg(H2O)6]2+ |
| Aluminum(III) | [Al(H2O)6]3+ | [Al(H2O)6]3+ |
| Gallium(III) | [Ga(H2O)6]3+ | [Ga(H2O)6]3+ |
| Indium(III) | [In(H2O)6]3+ | [In(H2O)6]3+ |
| Thallium(I) | [Tl(H2O)n]+ | |
| Thallium(III) | [Tl(H2O)6]3+ | [Tl(H2O)6]3+ |
| Tin(II) | [Sn(H2O)3]2+ | [Sn(H2O)3]2+ |
| Lead(II) | [Pb(H2O)3]2+, [Pb(H2O)4]2+ | [Pb(H2O)5–7]2+ |
| Bismuth(III) | [Bi(H2O)9]3+ | [Bi(H2O)8]3+ |
| Metal Ion | Solid State | Aqueous Solution |
|---|---|---|
| Li(H2O)4+ | 1.941–Td (Table S1a) | 1.95–Td [10,11,12,13,14,15,16] |
| Na(H2O)6+ | 2.417–Oh (Table S1b) | 2.43–Oh [10,21,22,23,24,25] |
| Na(H2O)53+ | 2.341–D3h (Table S1b) | |
| K(H2O)7+ | 2.81–7 [10,28,29,30,32] | |
| Rb(H2O)8+ | 2.98–D4h(8) [33,34] | |
| Cs(H2O)8+ | 3.10–D4h(8) [10,33,36,37] | |
| Be(H2O)42+ | 1.613–Td (Table S2a) | 1.62–Td a |
| Mg(H2O)62+ | 2.066–Oh (Table S2b) | 2.07–Oh [28,29] |
| Ca(H2O)82+ | 2.476–D4h(8) (Table S2c) | 2.48–D4h(8) [28,29,43,44,45,46,47,48,49,50,51,52] |
| Ca(H2O)73+ | 2.401–C3v(7) (Table S2c) | |
| Ca(H2O)63+ | 2.324–Oh (Table S2c) | |
| Sr(H2O)82+ | 2.613–D4h (Table S2d) | 2.61–D4h(8) [58,59,60,61,62,63,64,65,66,67] |
| Ba(H2O)82+ | 2.80–D4h (Table S2e) | 2.81–D4h(8) [58,69,74,75,76] |
| Ra(H2O)102+ | 2.93–10 [74] | |
| Sc(H2O)83+ | 2.170 + 2.459–D3h(9) (Table S3a) | 2.17 + 2.33–D3h(9) [81] |
| Y(H2O)83+ | 2.353–D4h(8) (Table S3b) | 2.367–D4h(8) [82] |
| La(H2O)93+ | 2.528 + 2.611–D3h(9) (Table S4a) | 2.53 + 2.60–D3h(9) [88] |
| La(H2O)83+ | 2.495–D4h(8) (Table S4a) | |
| Ce(H2O)93+ | 2.491 + 2.597–D3h(9) (Table S4b) | 2.49 + 2.62–D3h(9) [88] |
| Ce(H2O)83+ | 2.485–D4h(8) (Table S4b) | |
| Pr(H2O)93+ | 2.475 + 2.568–D3h(9) (Table S4c) | 2.48 + 2.58–D3h(9) [88] |
| Pr(H2O)83+ | 2.476–D4h(8) (Table S4c) | |
| Nd(H2O)93+ | 2.458 + 2.565–D3h(9) (Table S4d) | 2.47 + 2.60–D3h(9) [88] |
| Nd(H2O)83+ | 2.439–D4h(8) (Table S4d) | |
| Sm(H2O)93+ | 2.436 + 2.545–D3h(9) (Table S4e) | 2.43 + 2.53–D3h(9) [88] |
| Sm(H2O)83+ | 2.430–D4h(8) (Table S4e) | |
| Eu(H2O)82+ | 2.584–D4h(8) [66] | |
| Eu(H2O)93+ | 2.414 + 2.535–D3h(9) (Table S4f) | 2.42 + 2.57–D3h(9) [88] |
| Eu(H2O)83+ | 2.416–D4h(8) (Table S4f) | |
| Gd(H2O)93+ | 2.404 + 2.542–D3h(9) (Table S4g) | 2.41 + 2.53–D3h(9) [88] |
| Gd(H2O)83+ | 2.403–D4h(8) (Table S4g) | |
| Tb(H2O)93+ | 2.388 + 2.514–D3h(9) (Table S4h) | 2.40 + 2.53–D3h(9) [88] |
| Tb(H2O)83+ | 2.381–D4h(8) (Table S4h) | |
| Dy(H2O)93+ | 2.368 + 2.519–D3h(9) (Table S4i) | 2.38 + 2.50–D3h(9) [88] |
| Dy(H2O)83+ | 2.371–D4h(8) (Table S4i) | |
| Ho(H2O)93+ | 2.375 + 2.494–D3h(9) (Table S4j) | 2.37 + 2.50–D3h(9) [88] |
| Ho(H2O)83+ | 2.356–D4h(8) (Table S4j) | |
| Er(H2O)8.93+ | 2.351 + 2.515–D3h(9) (Table S4k) | 2.36 + 2.48–D3h(9) [88] |
| Er(H2O)83+ | 2.345–D4h(8) (Table S4k) | |
| Tm(H2O)8.93+ | 2.331 + 2.513–D3h(9) (Table S4l) | 2.35 + 2.47–D3h(9) [88] |
| Tm(H2O)83+ | 2.339–D4h(8) (Table S4l) | |
| Yb(H2O)8.73+ | 2.314 + 2.501–D3h(9) (Table S4m) | 2.32 + 2.43–D3h(9) [88] |
| Yb(H2O)83+ | 2.326–D4h(8) (Table S4m) | |
| Lu(H2O)8.23+ | 2.296 + 2.506–D3h(9) (Table S4n) | 2.28 + 2.37–D3h(9) [88] |
| Lu(H2O)83+ | 2.318–D4h(8) (Table S4n) | |
| Ac(H2O)113+ | 2.63–11 [116] | |
| Th(H2O)104+ | 2.498–10 2.345–D4h(8) (Table S4k) | |
| Th(H2O)94+ | 2.448–9 (Table S5a) | 2.45–9 [119,122] |
| U(H2O)93+ | 2.509 + 2.596–D3h(9) (Table S5b) | 2.50 + 2.58–D3h(9) [110] |
| U(H2O)94+ | 2.41–9 (Table S5b) | 2.42–9 [123,124] |
| U(H2O)84+ | 2.395–D4h(8) (Table S5b) | |
| UO2(H2O)52+ | 1.760 + 2.444–D5h (Table S5b) | 1.77 + 2.42–D5h [111,122] |
| Np(H2O)93+ | 2.491 + 2.571–D3h (Table S5c) | 2.48 + 2.56–D3h [111] |
| Np(H2O)94+ | 2.40–9 [110] | |
| Np(H2O)84+ | 2.335–D4h(8) (Table S5c) | |
| NpO2(H2O)52+ | 1.744 + 2.416–D5h (Table S5c) | 1.83 + 2.48–D5h [111,126] |
| Pu(H2O)3+ | 2.475 + 2.571–D3h (Table S5d) | 2.45 + 2.55–D3h(9) [110,111,112,114,115] |
| Pu(H2O)84+ | 2.39–9 (Table S5d) | 2.395–D4h(8) [114,115] |
| PuO2(H2O)5+ | 1.81 + 2.47–D5h [114,115] | |
| PuO2(H2O)52+ | 1.732 + 2.409–D3h (Table S5d) | 1.75 + 2.41–D5h [114,115] |
| Am(H2O)93+ | 2.466 + 2.578–D3h(9) (Table S5e) | 2.43 + 2.54–D3h(9) [108,110,113] |
| Cm(H2O)93+ | 2.453 + 2.555–D3h(9) (Table S5e) | 2.40 + 2.53–D3h(9) [108,110,113] |
| Bk(H2O)93+ | 2.38 + 2.50–D3h(9) [110] | |
| Cf(H2O)93+ | 2.423 + 2.550–D3h(9) (Table S5e) | 2.37 + 2.49–D3h(9) [110] |
| Ti(H2O)63+ | 2.02–Oh (Table S6a) | 2.03–Oh a |
| TiO(H2O)52+ | 1.644 + 2.034 + 2.233 a (Table S6a) | |
| Zr(H2O)84+ | 2.19–D4h(8) [132] | |
| Hf(H2O)84+ | 2.16–D4h(8) [132] | |
| V(H2O)62+ | 2.131–Oh (Table S7) | 2.13–Oh a |
| V(H2O)63+ | 1.994–Oh (Table S7) | 1.99–Oh [133] |
| VO(H2O)52+ | 1.580 + 2.026 + 2.187 (Table S7) | 1.60 + 2.03 + 2.20 [134] |
| VO2(H2O)4+ | 1.62 + 1.99 + 2.22 [134] | |
| Cr(H2O)62+ | 2.083 + 2.333–C4v (Table S8) | 2.08 + ~2.3–C4v [133] |
| Cr(H2O)63+ | 1.965–Oh (Table S8) | 1.97–Oh [28,29,135] |
| Mo(H2O)63+ | 2.089–Oh (Table S8) | 2.09–Oh [138] |
| Mn(H2O)62+ | 2.174–Oh (Table S9) | 2.17–Oh [28,29,139,140] |
| Mn(H2O)63+ | 2.088–Oh (Table S9) | |
| Fe(H2O)62+ | 2.120–Oh (Table S10a) | 2.12–Oh [28,29,145,146] |
| Fe(H2O)63+ | 1.995–Oh (Table S10a) | 2.00–Oh [28,29,145] |
| Ru(H2O)62+ | 2.111–Oh (Table S10b) | 2.11–Oh a |
| Ru(H2O)63+ | 2.021–Oh (Table S10b) | 2.03–Oh a |
| Co(H2O)62+ | 2.087–Oh (Table S11a) | 2.09–Oh [28,29,147,148,149,150] |
| Co(H2O)63+ | 1.873–Oh (Table S11a) | |
| Rh(H2O)63+ | 2.014–Oh (Table S11b) | 2.04–Oh [152,153,154] |
| Ir(H2O)63+ | 2.04–Oh (Table S11b) | |
| Ni(H2O)62+ | 2.055–Oh (Table S12a) | 2.055–Oh [28,29,155] |
| Pd(H2O)62+ | 2.01 + 2.8–D4h(6) [156,157,158,159,160] | |
| Pd(H2O)42+ | 2.029–D4h(4) (Table S12b) | |
| Pt(H2O)62+ | 2.02 + 2.8–D4h(6) [156,157,158,159,160] | |
| Pt(H2O)42+ | 2.012–D4h(4) (Table S12b) | |
| Cu(H2O)4+ | 2.14–Td b [161] | |
| Cu(H2O)62+ | 1.980 + 2.322–C4v(6) (Table S13a) | 1.96 + 2.32–D4h(6) [163,167] |
| Cu(H2O)62+ | 1.944 + 2.248–C4v(5) (Table S13a) | |
| Ag(H2O)4+ | 2.32 + 2.54–2 + 2 [181] | |
| Ag(H2O)2+ | 2.129–Dꝏh (Table S13b) | 2.34–2 [182] |
| Au((H2O)43+ | 2.155–D4h(4) (Table S13c) | |
| Zn(H2O)62+ | 2.088–Oh (Table S14a) | 2.09–Oh [28,29] |
| Zn(H2O)52+ | 2.017–D3h(5) (Table S14a) | |
| Cd(H2O)62+ | 2.266–Oh (Table S14b) | 2.27–Oh [184] |
| Hg2(H2O)62+ | 2.26–Td [185] | |
| Hg2(H2O)22+ | 2.14–Td (Table S14c) | |
| Hg(H2O)72+ | 2.32–7 [188] | |
| Hg(H2O)62+ | 2.342–Oh (Table S14c) | 2.34–Oh [187] |
| Al(H2O)63+ | 1.883–Oh (Table S15a) | 1.89–Oh [28,29] |
| Ga(H2O)6 3+ | 1.946–Oh (Table S15b) | 1.96–Oh [28,29,135] |
| In(H2O)63+ | 2.124–Oh (Table S15c) | 2.14–Oh [135] |
| Ti(H2O)n+ | 2.73 + 3.18–2 + 2 [202] | |
| Tl(H2O) 63+ | 2.23–Oh (Table S15d) | 2.23–Oh [193,194] |
| Sn(H2O)32+ | 2.204–C3v (Table S16a) | 2.21–C3v [203] |
| Pb(H2O)62+ | 2.53–6 [204,205] | |
| Bi(H2O)93+ | 2.440 + 2.582–D3h(9) (Table S16c) | |
| Bi(H2O)83+ | 2.41–D4h(8) [207] |
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Bunker, G. Introduction XAFS: A Pratical Guide to X-ray Absortion Fine Structure Spectroscopy; Cambridge University Press: Cambridge, UK, 2010; ISBN 978-0-521-76775-0. [Google Scholar]
- Rehr, J.J.; Albers, R.C. Theoretical approaches to X-ray absorption fine structure. Rev. Mod. Phys. 2000, 72, 621–654. [Google Scholar] [CrossRef]
- Stern, E.A.; Sayers, D.E.; Lytle, F.W. Extended X-ray Absorption Fine-Structure Technique. III. Determination of Physical Parameters. Phys. Rev. B 1975, 11, 4836–4846. [Google Scholar] [CrossRef]
- Van Bokhaven, J. X-ray Absorption and X-ray Emission Spectroscopy—Theory and Applications; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2016; ISBN 9781118844236. [Google Scholar]
- Levy, H.A.; Danford, M.D.; Narten, A.H. Data Collection and Evaluation with an X-ray Diffractometer Designed for the Study of Liquid Structure; Technical Report ORNL-3960; Oak Ridge National Laboratory: Oak Ridge, TN, USA, 1966. Available online: https://www.osti.gov/servlets/purl/4524253 (accessed on 27 August 2022).
- Magini, M.; Licheri, G.; Paschina, G.; Piccaluga, G.; Pinna, G. X-ray Diffraction of Ions in Aqueous Solutions: Hydration and Complex Formation; CRC Press: Boca Raton, FL, USA, 2018; Chapter 1; ISBN 978-1-351-08622-6. [Google Scholar]
- Neilson, G.W. Diffraction studies of aqueous electrolyte solutions. Pure Appl. Chem. 1988, 60, 1797–1806. [Google Scholar] [CrossRef]
- Linert, W.; Bridge, M.; Ohtaki, H. Comprehensive Coordination Chemistry II; Elsevier Science: Amsterdam, Netherlands, 2003; Chapter 2.46 Solvation. [Google Scholar]
- Stangret, J.; Gampe, T. Hydration sphere of tetrabutylammonium cation. FTIR studies of HDO spectra. J. Phys. Chem. B 1999, 103, 3778–3783. [Google Scholar] [CrossRef]
- Mähler, J.; Persson, I. A study of the hydration of the alkali metal ions in aqueous solution. Inorg. Chem. 2012, 51, 425–438. [Google Scholar] [CrossRef]
- Smirnov, P.R.; Trostin, V.N. Structure of the nearest surrounding of the Li+ ion in aqueous solutions of its salts. Russ. J. Gen. Chem. 2006, 76, 175–182. [Google Scholar] [CrossRef]
- Marcus, Y. Effect of ions on the structure of water: Structure making and breaking. Chem. Rev. 2009, 109, 1346–1370. [Google Scholar] [CrossRef]
- Persson, I. Hydrated metal ions in aqueous solution: How regular are their structures? Pure Appl. Chem. 2010, 82, 1901–1917. [Google Scholar] [CrossRef]
- Vinogradov, E.V.; Smirnov, P.R.; Trostin, V.N. Structure of hydrated complexes formed by metal ions of Groups I—III of the Periodic Table in aqueous electrolyte solutions under ambient conditions. Russ. Chem. Bull. 2003, 52, 1253–1271. [Google Scholar] [CrossRef]
- Marcus, Y. Ionic radii in aqueous solutions. Chem. Rev. 1988, 88, 1475–1498. [Google Scholar] [CrossRef]
- Mason, P.E.; Ansell, S.; Neilson, G.W.; Rempe, S.B. Neutron Scattering Studies of the Hydration Structure of Li+. J. Phys. Chem. B 2015, 119, 2003–2009. [Google Scholar] [CrossRef]
- Loeffler, H.; Rode, B. The hydration structure of the lithium ion. J. Chem. Phys. 2002, 117, 110–117. [Google Scholar] [CrossRef]
- Bouazizi, S.; Nasr, S. Local order in aqueous lithium chloride solutions as studied by X-ray scattering and molecular dynamics simulations. J. Mol. Struct. 2007, 837, 206–213. [Google Scholar] [CrossRef]
- Rudolph, W.; Brooker, M.; Pye, C. Hydration of lithium ion in aqueous solutions. J. Phys. Chem. 1995, 99, 3793–3797. [Google Scholar] [CrossRef]
- Du, H.; Rasaiah, J.C.; Miller, J.D. Structural and dynamic properties of concentrated alkali halide solutions: A molecular dynamics simulation study. J. Phys. Chem. B 2007, 111, 209–217. [Google Scholar] [CrossRef]
- Smirnov, P.R.; Trostin, V.N. Structure of the nearest surrounding of the Na+ ion in aqueous solutions of its salts. Russ. J. Gen. Chem. 2007, 77, 844–850. [Google Scholar] [CrossRef]
- Smirnov, P.R. Structure of the nearest environment of Na+, K+, Rb+, and Cs+ ions in oxygen-containing solvents. Russ. J. Gen. Chem. 2020, 90, 1693–1702. [Google Scholar] [CrossRef]
- Azam, S.S.; Hofer, T.S.; Randolf, B.R.; Rode, B.M. Hydration of sodium(I) and potassium(I) revisited: A comparative QM/MM and QMCF MD simulation study of weakly hydrated ions. J. Phys. Chem. A 2009, 113, 1827–1834. [Google Scholar] [CrossRef]
- Rowley, N.C.; Roux, B. The solvation structure of Na+ and K+ in liquid water determined from high level ab initio molecular dynamics simulations. J. Chem. Theor. Comput. 2012, 8, 3526–3535. [Google Scholar] [CrossRef]
- Bankura, A.; Carnevale, V.; Klein, M.L. Structure and spectroscopy of hydrated sodium ions at different temperatures and the cluster stability rules. J. Chem. Phys. 2013, 138, 014501. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Beattie, J.K.; Best, S.P.; Skelton, B.W.; White, A.H. Structural studies on the caesium alums, CsMIII[SO4]2·12H2O. J. Chem. Soc. Dalton Trans. 1981, 2105–2111. [Google Scholar] [CrossRef]
- Ohtaki, H.; Radnai, T. Structure and dynamics of hydrated ions. Chem. Rev. 1993, 93, 1157–1204. [Google Scholar] [CrossRef]
- Johansson, G. Structures of complexes in solution derived from X-ray diffraction measurements. Adv. Inorg. Chem. 1992, 39, 159–232. [Google Scholar]
- Smirnov, P.R.; Trostin, V.N. Structures of the nearest surroundings of the K+, Rb+, and Cs+ ions in aqueous solutions of their salt. Russ. J. Gen. Chem. 2007, 77, 2101–2107. [Google Scholar] [CrossRef]
- Tongaar, A.; Leidl, K.R.; Rode, B.M. Born–Oppenheimer ab initio QM/MM dynamics simulations of Na+ and K+ in water: From structure making to structure breaking effects. J. Phys. Chem. A 1998, 103, 10340–10347. [Google Scholar] [CrossRef]
- Glezakou, V.-A.; Chen, Y.; Fulton, J.L.; Schenter, G.K.; Dang, L.X. Electronic structure, statistical mechanical simulations, and EXAFS spectroscopy of aqueous potassium. Theor. Chem. Acc. 2006, 115, 86–99. [Google Scholar] [CrossRef]
- Caralampio, D.Z.; Martínez, J.M.; Pappalardo, R.R.; Sánchez Marcos, E. The hydration structure of the heavy-alkalines Rb+ and Cs+ through molecular dynamics and X-ray absorption spectroscopy: Surface clusters and eccentricity. Phys. Chem. Chem. Phys. 2017, 19, 28993–29004. [Google Scholar] [CrossRef]
- D’Angelo, P.; Persson, I. Structure of the hydrated and dimethyl sulfoxide solvated rubidium ions in solution. Inorg. Chem. 2004, 43, 3543–3549. [Google Scholar] [CrossRef]
- Audebrand, N.; Jeanneau, E.; Bataille, T.; Raite, S.; Louër, D. A family of microporous mixed oxalates with isotypic-framework structures based on eight-coordinate metals. Solid State Sci. 2004, 6, 579–591. [Google Scholar] [CrossRef]
- Pálinkás, G.; Radnai, T.; Hajdu, H. Ion-solvent and solvent-solvent interactions. X-ray study of aqueous alkali chloride solutions. Z. Naturforsch. Ser. A 1980, 35, 107–114. [Google Scholar] [CrossRef]
- Bertagnolli, H.; Weidner, J.-U.; Zimmermann, H. Röntgenstrukturuntersuchung wäßriger Caesiumfluorid-Lösungen. Ber. Bunsen-Ges. Phys. Chem. 1974, 78, 2–19. [Google Scholar]
- Yamaguchi, T.; Ohtaki, H.; Spohr, E.; Pálinkás, G.; Heinzinger, K.; Probst, M.M. Molecular dynamics and X-ray diffraction study of aqueous beryllium(II) chloride solutions. Z. Naturforsch. Ser. A 1986, 41, 1175–1185. [Google Scholar] [CrossRef]
- Di Tommaso, S.; de Leeuw, N.H. Structure and dynamics of the hydrated magnesium ion and of the solvated magnesium carbonates: Insights from first principles simulations. Phys. Chem. Chem. Phys. 2010, 12, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Boero, M.; Terakura, K. Hydration properties of magnesium and calcium ions from constrained first principles molecular dynamics. J. Chem. Phys. 2007, 127, 074503. [Google Scholar] [CrossRef] [PubMed]
- Duboué-Dijon, E.; Mason, P.; Fischer, H.; Jungwirth, P. Hydration and ion pairing in Aqueous Mg2+ and Zn2+ solutions: Force-field description aided by neutron scattering experiments and ab Initio molecular dynamics simulations. J. Phys. Chem. B 2017, 122, 3296–3306. [Google Scholar] [CrossRef] [PubMed]
- Delgado, A.A.A.; Sethio, D.; Kraka, E. Assessing the intrinsic strengths of ion–solvent and solvent–solvent interactions for hydrated Mg2+ clusters. Inorganics 2021, 9, 31. [Google Scholar] [CrossRef]
- Smirnov, P.; Yamagami, M.; Wakita, H.; Yamaguchi, T. An X-ray diffraction study on concentrated aqueous calcium nitrate solutions at subzero semperatures. J. Mol. Liq. 1997, 73–74, 305–316. [Google Scholar] [CrossRef]
- Jalilehvand, F.; Spångberg, D.; Lindqvist-Reis, P.; Hermansson, K.; Persson, I.; Sandström, M. Hydration of the calcium ion. An EXAFS, large-angle X-ray scattering, and molecular dynamics simulation study. J. Am. Chem. Soc. 2001, 123, 431–441. [Google Scholar] [CrossRef]
- Fulton, J.L.; Heald, S.M.; Badyal, Y.S.; Simonson, J.M. Understanding the effects of concentration on the solvation structure of Ca2+ in aqueous solution. I: The perspective on local structure from EXAFS and XANES. J. Phys. Chem. A 2003, 107, 4688–4696. [Google Scholar] [CrossRef]
- Badyal, Y.S.; Barnes, A.C.; Cuello, G.J.; Simonson, J.M. Understanding the effects of concentration on the solvation structure of Ca2+ in aqueous solutions. II: Insights into longer range order from neutron diffraction isotope substitution. J. Phys. Chem. A 2004, 108, 11819–11827. [Google Scholar] [CrossRef]
- D’Angelo, P.; Petit, P.E.; Pavel, N.V. Double-electron excitation channels at the Ca2+ K-edge of hydrated calcium ion. J. Phys. Chem. B 2004, 108, 11857–11865. [Google Scholar] [CrossRef]
- Gaspar, A.M.; Marques, M.A.; Cabaco, M.I.; Marques, M.I.D.; Buslaps, T.; Honkimaki, V.; European Mol Liquid Grp; Japenese Mol Liquid Grp. X-ray diffraction investigations of concentrated aqueous solutions of calcium halides. J. Mol. Liq. 2004, 110, 15–22. [Google Scholar] [CrossRef]
- Fulton, J.L.; Chen, Y.; Heald, S.M.; Balasubramanian, M. High-pressure, High-temperature X-ray absorption fine structure transmission cell for the study of aqueous ions with low absorption-edge energies. Rev. Sci. Instrum. 2004, 75, 5228–5231. [Google Scholar] [CrossRef]
- Megyes, T.; Grosz, T.; Radnai, T.; Bako, I.; Palinkas, G. Solvation of calcium ion in polar solvents: An X-ray diffraction and ab initio study. J. Phys. Chem. A 2004, 108, 7261–7271. [Google Scholar] [CrossRef]
- Megyes, T.; Bako, I.; Balint, S.; Grosz, T.; Radnai, T. Ion pairing in aqueous calcium chloride solution: Molecular dynamics simulation and diffraction studies. J. Mol. Liq. 2006, 129, 63–74. [Google Scholar] [CrossRef]
- Bucher, D.; Kuyucak, S. Polarization of water in the first hydration shell of K+ and Ca2+ ions. J. Phys. Chem. B 2008, 112, 10786–10790. [Google Scholar] [CrossRef]
- Pham, V.-T.; Fulton, J.L. Ion-pairing in Aqueous CaCl2 and RbBr Solutions: Simultaneous Structural Refinement of XAFS and XRD Data. J. Chem. Phys. 2013, 138, 044201. [Google Scholar] [CrossRef]
- Rudolph, W.; Irmer, G. Hydration of calcium(II) ion in aqueous solution of common anions (ClO4−, Cl−, Br− and NO3−). Dalton Trans. 2013, 37, 3919–3935. [Google Scholar] [CrossRef]
- Yoo, J.; Wilson, J.; Aksimentiev, A. Improved model of hydrated calcium ion for molecular dynamics simulations using classical biomolecular force fields. Biopolymers 2016, 105, 752–763. [Google Scholar] [CrossRef]
- Delgado, A.A.A.; Sethio, D.; Munar, I.; Aviyente, V.; Kraka, E. Local vibrational mode analysis of ion-solvent and solvent-solvent interactions for hydrated Ca2+ clusters. J. Chem. Phys. 2020, 153, 224303. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Zhao, Z.; Huang, X.; Wang, P.; Su, Y.; Sai, L.; Liang, X.; Han, H.; Zhao, J. Ground-state structures of hydrated calcium ion clusters from comprehensive genetic algorithm search. Front. Chem. 2021, 9, 637750. [Google Scholar] [CrossRef] [PubMed]
- Persson, I.; Sandström, M.; Yokoyama, H.; Chaudhry, M. Structures of the solvated barium and strontium ions in aqueous, dimethyl sulfoxide and pyridine solution, and crystal structure of strontium and barium hydroxide octahydrate. Z. Naturforsch. Sect. B. 1995, 50, 21–37. [Google Scholar] [CrossRef]
- D’Angelo, P.; Migliorati, V.; Sessa, F.; Mancini, G.; Persson, I. XANES reveals the flexible nature of hydrated strontium in aqueous solution. J. Phys. Chem. B 2016, 120, 4114–4124. [Google Scholar] [CrossRef]
- O’Day, P.A.; Newvillre, M.; Neuhoff, P.S.; Sahai, N.; Carroll, S.A. X-ray absorption spectroscopy of strontium (II) coordination: I. Static and thermal disorder in crystalline, hydrated, and precipitated solids and in aqueous solution. J. Colloid Interface Sci. 2000, 222, 184–197. [Google Scholar]
- Axe, L.; Bunker, G.B.; Anderson, P.R.; Tyson, T.A. An XAFS analysis of strontium at the hydrous ferric oxide surface. J. Colloid Interface Sci. 1998, 199, 44–52. [Google Scholar] [CrossRef]
- Caminiti, R.; Musinu, A.; Paschina, G.; Pinna, G. X-ray diffraction study of aqueous SrCl2 solutions. J. Appl. Crystallogr. 1982, 15, 482–487. [Google Scholar] [CrossRef]
- Albright, J.N. X-ray diffraction studies of aqueous alkaline-earth chloride solutions. J. Chem. Phys. 1972, 56, 3783–3786. [Google Scholar] [CrossRef]
- Ramos, S.; Neilson, G.W.; Barnes, A.C.; Capitán, M.J. Anomalous X-ray diffraction studies of Sr2+ hydration in aqueous solution. J. Chem. Phys. 2003, 118, 5542–5546. [Google Scholar] [CrossRef]
- Pfund, D.M.; Darab, J.G.; Fulton, J.L.; Ma, Y. An XAFS study of strontium ions and krypton in supercritical water. J. Phys. Chem. 1994, 98, 13102–13107. [Google Scholar] [CrossRef]
- Moreau, G.; Helm, L.; Purans, J.; Merbach, A.E. Structural Investigation of the Aqueous Eu2+ Ion: Comparison with Sr2+ Using the XAFS Technique. J. Phys. Chem. A 2002, 106, 3034–3043. [Google Scholar] [CrossRef]
- Steward, T.M.; Henderson, C.M.B.; Charnock, J.M.; Driesner, T. An EXAFS study of solvation and ion pairing in aqueous strontium solutions to 300 °C. Geochim. Cosmochim. Acta 1999, 63, 2409–2418. [Google Scholar] [CrossRef]
- Harris, D.J.; Brodholt, J.P.; Sherman, D.M. Hydration of Sr2 + in Hydrothermal Solutions from ab Initio Molecular Dynamics. J. Phys. Chem. B 2003, 107, 9056–9058. [Google Scholar] [CrossRef]
- Chaudhari, M.I.; Rempee, S.B. Strontium and barium in aqueous solution and a potassium channel binding site. J. Chem. Phys. 2018, 148, 222831. [Google Scholar] [CrossRef] [PubMed]
- Spohr, E.; Pálinkás, G.; Heinzinger, K.; Bopp, B.; Probst, M.M. Molecular dynamics study of an aqueous strontium chloride solution. J. Phys. Chem. 1988, 92, 6754–6761. [Google Scholar] [CrossRef]
- Palmer, B.J.; Pfund, D.M.; Fulton, J.L. Direct modeling of EXAFS spectra from molecular dynamics simulations. J. Phys. Chem. 1996, 100, 13393–13398. [Google Scholar] [CrossRef]
- Boda, A.; De, S.; Ali, S.M.; Tulishetti, S.; Khan, S.; Singh, J.K. From microhydration to bulk hydration of Sr2+ metal ion: DFT, MP2 and molecular dynamics study. J. Mol. Liq. 2012, 172, 110–118. [Google Scholar] [CrossRef]
- Hofer, T.S.; Randolf, B.R.; Rode, B.M. Sr(II) in water: A labile hydrate with a highly mobile structure. J. Phys. Chem. B 2006, 110, 20409–20417. [Google Scholar] [CrossRef]
- Pappalardo, R.R.; Caralampio, D.Z.; Martínez, J.M.; Sánchez Marcos, E. Hydration of heavy alkaline-earth cations studied by molecular dynamics simulations and X-ray absorption spectroscopy. Inorg. Chem. 2021, 60, 13578–13587. [Google Scholar] [CrossRef] [PubMed]
- Hofer, T.S.; Rode, B.M.; Randolf, B.R. Structure and dynamics of solvated Ba(II) in dilute aqueous solution—An ab initio QM/MM MD approach. Chem. Phys. 2005, 312, 81–88. [Google Scholar] [CrossRef]
- Stack, A.G.; Rustad, J.R. Structure and dynamics of water on aqueous barium ion and the {001} barite surface. J. Phys. Chem. C 2007, 111, 16387–16391. [Google Scholar] [CrossRef]
- Hoogerstraete, T.V.; Brooks, N.R.; Onghena, B.; Van Meervelt, L.; Binnemans, K. Crystal structures of hydrated rare-earth bis(trifluoromethylsulfonyl)imide salts. CrystEngComm 2015, 17, 7142–7149. [Google Scholar] [CrossRef]
- Waller, F.J.; Barrett, A.G.M.; Braddock, D.C.; Ramprasad, D.; McKinnell, R.M.; White, A.J.P.; Williams, D.J.; Ducray, R. Tris(trifluoromethanesulfonyl)methide (“Triflide”) anion: Convenient preparation, X-ray crystal structures, and exceptional catalytic activity as a counterion with ytterbium(III) and scandium(III). J. Org. Chem. 1999, 64, 2910–2913. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, A.; Lindqvist-Reis, P.; Eriksson, L.; Sandström, D.; Lidin, S.; Persson, I.; Sandström, M. Highly Hydrated cations: Deficiency, mobility, and coordination of water in crystalline nonahydrated scandium(III), yttrium(III) and lanthanoid(III) trifluoromethanesulfonates. Chem. Eur. J. 2005, 11, 4065–4077. [Google Scholar] [CrossRef] [PubMed]
- Castellani, C.B.; Carugo, O.; Giusti, M.; Sardone, N. Crystal structure of scandium(III) triflate enneahydrated. Eur. J. Solid State Inorg. Chem. 1995, 32, 1089–1099. [Google Scholar]
- Lindqvist-Reis, P.; Persson, I.; Sandström, M. The hydration of the scandium(III) ion in aqueous solution and crystalline hydrates studied by xafs spectroscopy, large angle X-ray scattering and crystallography. Dalton Trans. 2006, 3868–3878. [Google Scholar] [CrossRef]
- Lindqvist-Reis, P.; Lamble, K.; Pattanaik, S.; Sandström, M.; Persson, I. Hydration of the yttrium(III) ion in aqueous solution. an X-ray diffraction and XAFS structural study. J. Phys. Chem. Sect. B 2000, 104, 402–408. [Google Scholar] [CrossRef]
- Van de Voorde, M.; Geboes, B.; Tom Vander Hoogerstraete, T.; Van Hecke, K.; Cardinaels, T.; Binnemans, K. Stability of europium(II) in aqueous nitrate solutions. Dalton Trans. 2019, 48, 14758–14768. [Google Scholar] [CrossRef]
- Szostak, M.; Spain, M.; Procter, D.J. Preparation of samarium(II) iodide: Quantitative evaluation of the effect of water, oxygen, and peroxide content, preparative methods, and the activation of samarium metal. J. Org. Chem. 2012, 77, 3049–3059. [Google Scholar] [CrossRef]
- Tsuneo, I.; Yasuhiro, K.; Susumu, H. Cerium(IV) trifluoromethanesulfonate as a strong oxidizing agent. Chem. Lett. 1990, 19, 1445–1446. [Google Scholar]
- Marsac, R.; Réal, F.; Lal Banik, N.; Pédrot, M.; Pourret, O.; Vallet, V. Aqueous chemistry of Ce(IV): Estimations using actinide analogues. Dalton Trans. 2017, 46, 13553–13561. [Google Scholar] [CrossRef] [PubMed]
- Ikeda-Ohno, A.; Tsushima, S.; Hennig, C.; Yaitab, T.; Bernharda, G. Dinuclear complexes of tetravalent cerium in an aqueous perchloric acid solution. Dalton Trans. 2012, 41, 7190–7192. [Google Scholar] [CrossRef] [PubMed]
- Persson, I.; D’Angelo, P.; De Panfilis, S.; Sandström, M.; Eriksson, L. Hydration of the lanthanoid(III) ions in aqueous solution and crystalline hydrates studied by EXAFS spectroscopy and crystallography. The myth of the “Gadolinium Break”. Chem. Eur. J. 2008, 14, 3056–3066. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, D.; Persson, I.; Eriksson, L.; D’Angelo, P.; de Panfilis, S. Structural study of the N,N’-dimethylpropyleneurea solvated lanthanoid(III) ions in solution and solid state with an analysis of the ionic radii of lanthanoid(III) Ions. Inorg. Chem. 2010, 49, 4420–4432. [Google Scholar] [CrossRef]
- Spedding, F.H.; Pikal, M.J.; Ayers, B.O. Apparent molal volumes of some aqueous rare earth chloride and nitrate solutions at 25°. J. Phys. Chem. 1966, 70, 2440–2449. [Google Scholar] [CrossRef]
- Spedding, F.H.; Jones, K.C. Heat capacities of aqueous rare earth chloride solutions at 25 °C. J. Phys. Chem. 1966, 70, 2450–2455. [Google Scholar] [CrossRef]
- Spedding, F.H.; Pikal, M.J. Relative viscosities of some aqueous rare earth chloride solutions at 25 °C. J. Phys. Chem. 1966, 70, 2430–2440. [Google Scholar] [CrossRef]
- Marsh, K. 125. The dimethyl phosphates of the rare-earth metals. J. Chem. Soc. 1939, 61, 554–558. [Google Scholar] [CrossRef]
- Templeton, D.H.; Dauben, C.H. Lattice parameters of some rare earth compounds and a set of crystal radii. J. Am. Chem. Soc. 1954, 76, 5237–5239. [Google Scholar] [CrossRef]
- Cossy, C.; Helm, L.; Merbach, A.E. Oxygen-17 nuclear magnetic resonance kinetic study of water exchange on the lanthanide(III) aqua ions. Inorg. Chem. 1988, 27, 1973–1979. [Google Scholar] [CrossRef]
- Cossy, C.; Helm, L.; Merbach, A.E. High-pressure NMR study. 38. Water-exchange mechanisms on the terbium to thulium octaaqualanthanide (III) ions: A variable-pressure oxygen-17 NMR study. Inorg. Chem. 1989, 28, 2699–2703. [Google Scholar] [CrossRef]
- Micskei, K.; Powell, D.H.; Helm, L.; Brucher, E.; Merbach, A.E. Water exchange on Gd(H2O)8]3+ and [Gd(PDTA)(H2O)2]− in aqueous solution: A variable-pressure, -temperature and -magnetic field 17O NMR study. Magn. Reson. Chem. 1993, 31, 1011–1020. [Google Scholar] [CrossRef]
- Powell, D.H.; Merbach, A.E. Water exchange on the light lanthanide aqua ions [Pr(H2O)9]3+ and [Nd(H2O)9]3+: A variable temperature and magnetic field 17O NMR study. Magn. Reson. Chem. 1994, 32, 739–745. [Google Scholar] [CrossRef]
- Fay, D.; Litchinski, D.; Purdie, N. Ultrasonic absorption in aqueous salts of the lanthanides. J. Phys. Chem. 1969, 73, 544–552. [Google Scholar] [CrossRef]
- Garza, V.L.; Purdie, N. Ultrasonic absorption in aqueous salts of the lanthanides. II. Acetates. J. Phys. Chem. 1970, 74, 275–280. [Google Scholar] [CrossRef]
- Peppard, D.F.; Mason, G.W.; Lewey, S. A tetrad effect in the liquid-liquid extraction ordering of lanthanides(III). J. Inorg. Nucl. Chem. 1969, 31, 2271–2272. [Google Scholar] [CrossRef]
- Schwarzenbach, G.; Gut, R.; Anderegg, G. Komplexone XXV. Die polarographische Untersuchung von Austauschgleichgewichten. Neue Daten der Bildungskonstanten von Metallkomplexen der Äthylendiamin-tetraessigsäure und der 1, 2-Diaminocyclohexan-tetraessigsäure. Helv. Chim. Acta 1954, 37, 937–957. [Google Scholar] [CrossRef]
- Wheelwright, E.; Spedding, F.H.; Schwarzenbach, G. The stability of the rare earth complexes with ethylenediaminetetraacetic acid. J. Am. Chem. Soc. 1953, 75, 4196–4201. [Google Scholar] [CrossRef]
- Peppard, D.F.; Bloomquist, C.A.A.; Horwitz, E.P.; Lewey, S.; Mason, G.W. Analogous actinide (III) and lanthanide (III) tetrad effects. J. Inorg. Nucl. Chem. 1970, 32, 339–343. [Google Scholar] [CrossRef]
- Lundberg, D.; Persson, I. The size of actinoid(III) ions—Structural analysis vs. common misinterpretations. Coord. Chem. Rev. 2016, 318, 131–134. [Google Scholar] [CrossRef]
- Apostolidis, C.; Schimmelpfennig, B.; Magnani, N.; Lindqvist-Reis, P.; Walter, O.; Sykora, R.; Morgenstern, A.; Colineau, E.; Caciuffo, R.; Klenze, R.; et al. [An(H2O)9](CF3SO3)3 (An = U–Cm, Cf): Exploring their stability, structural chemistry, and magnetic behavior by experiment and theory. Angew. Chem. Int. Ed. 2010, 49, 6343–6347. [Google Scholar] [CrossRef] [PubMed]
- Matonic, J.H.; Scott, B.L.; Neu, M.P. High-yield synthesis and single-crystal X-ray structure of a plutonium(III) aquo complex: [Pu(H2O)9][CF3SO3]3. Inorg. Chem. 2001, 40, 2638–2639. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist-Reis, P.; Apostolidis, C.; Rebizant, J.; Morgenstern, A.; Klenze, R.; Walter, O.; Fanghänel, T.; Haire, R.G. The structures and optical spectra of hydrated transplutonium ions in the solid state and in solution. Angew. Chem. Int. Ed. 2007, 46, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Skanthakumar, S.; Antonio, M.R.; Wilson, R.E.; Soderholm, L. The curium aqua ion. Inorg. Chem. 2007, 46, 3485–3491. [Google Scholar] [CrossRef]
- D’Angelo, P.; Martelli, F.; Spezia, R.; Filipponi, A.; Denecke, M.A. Hydration properties and ionic radii of actinide(III) ions in aqueous solution. Inorg. Chem. 2013, 52, 10318–10324. [Google Scholar] [CrossRef]
- Allen, P.G.; Bucher, J.J.; Shuh, D.K.; Edelstein, N.M.; Reich, T. Investigation of aquo and chloro complexes of UO22+, NpO2+, Np4+, and Pu3+ by X-ray absorption fine structure spectroscopy. Inorg. Chem. 1997, 36, 4676–4783. [Google Scholar] [CrossRef]
- Wilson, R.E.; Almond, P.M.; Burns, P.C.; Soderholm, L. The structure and synthesis of plutonium(III) chlorides from aqueous solution. Inorg. Chem. 2006, 45, 8483–8485. [Google Scholar] [CrossRef]
- Allen, P.G.; Bucher, J.J.; Shuh, D.K.; Edelstein, N.M.; Craig, I. Coordination chemistry of trivalent lanthanide and actinide ions in dilute and concentrated chloride solutions. Inorg. Chem. 2000, 39, 595–601. [Google Scholar] [CrossRef]
- Conradson, S.D.; Abney, K.D.; Begg, B.D.; Brady, E.D.; Clark, D.L.; den Auwer, C.; Ding, M.; Dorhout, P.K.; Espinosa-Faller, F.J.; Gordon, P.L.; et al. Higher order speciation effects on plutonium L3 X-ray absorption near edge spectra. Inorg. Chem. 2004, 43, 116–131. [Google Scholar] [CrossRef]
- Conradson, S.D.; Clark, D.L.; Neu, M.P.; Runde, W.H.; Tait, C.D. Characterizing the plutonium aquo ions by XAFS spectroscopy. Los Alamos Sci. 2000, 26, 418–421. [Google Scholar]
- Ferrier, M.G.; Stein, B.W.; Batista, E.R.; Berg, J.M.; Birnbaum, E.R.; Engle, J.W.; John, K.D.; Kozimor, S.A.; Lezama Pacheco, J.S.; Redman, L.N. Synthesis and characterization of the actinium aquo ion. ACS Central Sci. 2017, 3, 176–185. [Google Scholar] [CrossRef]
- Deblonde, G.J.-P.; Zavarin, M.; Kersting, A.B. The coordination properties and ionic radius of actinium: A 120-year old enigma. Coord. Chem. Rev. 2021, 446, 214130. [Google Scholar] [CrossRef]
- Wilson, R.E.; Skanthakumar, S.; Burns, P.C.; Soderholm, L. Structure of the homoleptic thorium(IV) aqua ion [Th(H2O)10]Br4. Angew. Chem, Int. Ed. 2007, 46, 8043–8045. [Google Scholar] [CrossRef]
- Torapava, N.; Persson, I.; Eriksson, L.; Lundberg, D. Hydration and hydrolysis of thorium(IV) in aqueous solution and the structures of two crystalline thorium(IV) hydrates. Inorg. Chem. 2009, 48, 11712–11723. [Google Scholar] [CrossRef]
- Pohl, R.W.H.; Wiebke, J.; Klein, A.; Dolg, M.; Maggiarosa, N. New 5,5’-bitetrazole thorium(IV) compound: Synthesis, crystal structure and quantum chemical investigation. Eur. J. Inorg. Chem. 2009, 2009, 2472–2476. [Google Scholar] [CrossRef]
- Thuery, P. Second-sphere complexation of thorium(IV) by cucurbit[6]uril with included perrhenate counterions—Crystal structure and Hirshfeld surface analysis. Eur. J. Inorg. Chem. 2015, 2015, 2037–2040. [Google Scholar] [CrossRef]
- Hennig, C.; Schmeide, K.; Brendler, V.; Moll, H.; Tsushima, S.; Scheinost, A.C. EXAFS investigation of U(VI), U(IV), and Th(IV) sulfato complexes in aqueous solution. Inorg. Chem. 2007, 46, 5882–5892. [Google Scholar] [CrossRef]
- Moll, H.; Denecke, M.A.; Jalilehvand, F.; Sandström, M.; Grenthe, I. Structure of the aqua ions and fluoride complexes of uranium (IV) and thorium (IV) in aqueous solution an EXAFS study. Inorg. Chem. 1999, 38, 1795–1799. [Google Scholar] [CrossRef]
- Ikeda-Ohno, A.; Hennig, C.; Tsushima, S.; Scheinost, A.C.; Bernhard, G.; Yaita, T. Speciation and structural study of U(IV) and-(VI) in perchloric and nitric acid solutions. Inorg. Chem. 2009, 48, 7201–7210. [Google Scholar] [CrossRef]
- Ikeda-Ohno, A.; Hennig, C.; Rossberg, A.; Fune, H.; Scheinost, A.C.; Bernhard, G.; Yaita, T. Electrochemical and complexation behavior of neptunium in aqueous perchlorate and nitrate solutions. Inorg. Chem. 2008, 47, 8294–8305. [Google Scholar] [CrossRef]
- Skanthakumar, S.; Antonio, M.R.; Soderholm, L. A comparison of neptunyl(V) and neptunyl(VI) solution coordination: The stability of cation-cation interactions. Inorg. Chem. 2008, 47, 4591–4595. [Google Scholar] [CrossRef]
- Miyanaga, T.; Watanabe, I.; Ikeda, S. Structures of hydrated titanium and vanadium ions in aqueous-solutions studied by X-ray absorption-spectroscopy. Bull. Chem. Soc. Jpn. 1990, 63, 3282–3287. [Google Scholar] [CrossRef]
- Tachikawa, H.; Ichikawa, T.; Yoshida, H. Hydration structure of titanium(III) ion. Electron spin resonance and electron spin-echo study. J. Am. Chem. Soc. 1990, 112, 977–982. [Google Scholar] [CrossRef]
- Persson, I.; Lundberg, D. Structure of a hydrated sulfonatotitanyl(IV) complex in aqueous solution and the dimethylsulfoxide solvated titanyl(IV) ion in solution and solid state. J. Solution Chem. 2017, 46, 476–487. [Google Scholar]
- Rabe, S.; Muller, U. Oxotitan-Addukte mit Dimethylsulfoxid: [TiO(OSMe2)5]Cl2 und [Ti4O6(OSMe2)12]Cl4·5Me2SO·½H2O/Oxotitanium compounds with dimethylsulfoxide: [TiO(OSMe2)5]Cl2 and [Ti4O6(OSMe2)12]Cl4·5Me2SO·½H2O. Z. Naturforsch. Teil B 1997, 52, 1291–1295. [Google Scholar] [CrossRef]
- Enders, M.; Rudolph, R.; Pritzkow, H. Synthese und kristallstruktur von pentakis(dimethylsulfoxid)-oxo-titan(IV)chlorid/synthesis and crystal structure of pentakis(dimethylsulfoxide)-oxo-titanium(IV) chloride. Z. Naturforsch. Teil B 1997, 52, 496–499. [Google Scholar] [CrossRef]
- Hagfeldt, C.; Kessler, V.; Persson, I. Structure of the hydrated, hydrolysed and solvated zirconium(IV) and hafnium(IV) ions in water and aprotic oxygen donor solvents. A crystallographic, EXAFS spectroscopic and large angle X-ray scattering study. Dalton Trans. 2004, 2142–2151. [Google Scholar] [CrossRef] [PubMed]
- Miyanaga, T.; Watanabe, I.; Ikeda, S. Amplitude in EXAFS and ligand-exchange reaction of hydrated 3D transition-metal complexes. Chem. Lett. 1988, 17, 1073–1076. [Google Scholar] [CrossRef]
- Krakowiak, J.; Lundberg, D.; Persson, I. A coordination chemistry study of hydrated and solvated cationic vanadium ions in oxidation states +III, +IV, and +V in solution and solid state. Inorg. Chem. 2012, 51, 9598–9609. [Google Scholar] [CrossRef]
- Lindqvist-Reis, P.; Díaz-Moreno, S.; Munoz-Páez, A.; Pattanaik, S.; Persson, I.; Sandström, M. On the structure of hydrates gallium, indium and chromium(III) ions in aqueous solutions. A large angle X-ray scattering and EXAFS study. Inorg. Chem. 1998, 37, 6675–6683. [Google Scholar] [CrossRef]
- Torapava, N.; Radkevich, A.; Davydov, D.; Titov, I.; Persson, I. Composition and structure of polynuclear chromium(III) hydroxo complexes. Inorg. Chem. 2009, 48, 10383–10388. [Google Scholar] [CrossRef]
- Cramer, S.P.; Eidem, P.K.; Pafett, M.T.; Winkler, J.R.; Dori, Z.; Gray, H.B. X-ray absorption edge and EXAFS spectroscopic studies of molybdenum ions in aqueous solution. J. Am. Chem. Soc. 1983, 105, 799–802. [Google Scholar] [CrossRef]
- Brorson, M.; Gajhede, M. Crystal structure of cesium molybdenum alum, Cs[Mo(H2O)6](SO4)2·6H2O, at 110 K. Inorg. Chem. 1987, 26, 2109–2112. [Google Scholar] [CrossRef]
- Konieczna, H.; Lundberg, D.; Persson, I. Solvation and coordination chemistry of manganese(II) ion in solvents with solvation properties. A transfer thermodynamic, complex formation, EXAFS spectroscopic and crystallographic study. Polyhedron 2021, 195, 114961. [Google Scholar] [CrossRef]
- Chen, Y.; Fulton, J.L.; Partenheimer, W. A XANES and EXAFS study of hydration and ion pairing in ambient aqueous MnBr2 solutions. J. Solut. Chem. 2005, 34, 993–1007. [Google Scholar] [CrossRef]
- Caminiti, R.; Cucca, G.; Pintori, G. Hydration and ion-pairing in concentrated aqueous Mn(NO3)2 solutions. An X-ray and Raman spectroscopy study. Chem. Phys. 1984, 88, 155–161. [Google Scholar] [CrossRef]
- Chukanov, N.V.; Zubkova, N.V.; Pautov, L.A.; Göttlicher, J.; Kasatkin, A.V.; Van, K.V.; Ksenofontov, D.A.; Pekov, I.V.; Vozchikova, S.A.; Pushcharovsky, D.Y. Jouravskite: Refined data on the crystal structure, chemical composition and spectroscopic properties. Phys. Chem. Miner. 2019, 46, 417–4257. [Google Scholar] [CrossRef]
- Allen, F.H. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Crystallogr. Sect. B 2002, 58, 380–388, Cambridge Structure Database (Conquest 2022.1.0). [Google Scholar] [CrossRef]
- Inorganic Crystal Structure Database 1.4.6, Release 2021-2; FIZ Karlsruhe: Eggenstein-Leopoldshafen, Germany, 2021.
- Lundberg, D.; Ullström, A.-S.; D’Angelo, P.; Persson, I. A structural study of the hydrated and the dimethylsulfoxide, N,N’-dimethylpropyleneurea, and N,N-dimethylthioformamide solvated iron(II) and iron(III) ions in solution and solid state. Inorg. Chim. Acta 2007, 360, 1809–1818. [Google Scholar] [CrossRef]
- Kálmán, E.; Radnai, T.; Pálinkás, G.; Hajdus, F.; Vertes, A. Hydration of iron(II) ion in aqueous solutions. Hydration of iron(II) ion in aqueous solution. Electrochim. Acta 1988, 33, 1223–1228. [Google Scholar] [CrossRef]
- D’Angelo, P.; Barone, V.; Chillemi, G.; Sanna, N.; Meyer-Klauche, W.; Pavel, N.V. Hydrogen and higher shell contributions in Zn2+, Ni2+, and Co2+ aqueous solutions: An X-ray absorption fine structure and molecular dynamics study. J. Am. Chem. Soc. 2002, 124, 1958–1967. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Hayes, K. X-ray absorption spectroscopy investigation of aqueous Co(II) and Sr(II) sorption at clay–water interfaces. Geochim. Cosmochim. Acta 1999, 63, 3205–3215. [Google Scholar] [CrossRef]
- Spezia, R.; Duvail, M.; Vitorge, P.; Cartailler, T.; Tortajada, J.; Chillemi, G.; D’Angelo, P. A coupled Car-Parrinello molecular dynamics and EXAFS data analysis investigation of aqueous Co2+. J. Phys. Chem. A 2006, 110, 13081–13088. [Google Scholar] [CrossRef]
- Caralampio, D.Z.; Reeves, B.; Beccia, M.R.; Martínez, J.M.; Pappalardo, R.R.; Christophe den Auwer, C.; Sánchez Marcos, E. Revisiting the cobalt(II) hydration from molecular dynamics and X-ray absorption spectroscopy. Mol. Phys. 2019, 117, 3320–3328. [Google Scholar] [CrossRef]
- Magini, M.; Giubileo, G. Solute Structuring in Concentrated A Co(II) and Ni(II) Perchlorate Solutions. Gazz. Chim. Ital. 1981, 111, 449–454. [Google Scholar]
- Caminiti, R.; Atzei, D.; Cucca, P.; Anedda, A.; Bongiovanni, G. Structure of Rhodium(III) Nitrate Aqueous Solutions. An Investigation by X-ray Diffraction and Raman Spectroscopy. J. Phys. Chem. 1986, 90, 238–243. [Google Scholar]
- Caminiti, R.; Cucca, P. An X-ray diffraction study of Rh(III) coordination in a dilute aqueous-solution of Rh(ClO4)3. Phys. Chem. Lett. 1984, 108, 51–57. [Google Scholar] [CrossRef]
- Read, M.C.; Sandström, M. 2nd-sphere hydration of rhodium(III) and chromium(III) in aqueous-solution—A large-angle X-ray-scattering and EXAFS study. Acta Chem. Scand. 1992, 46, 1177–1182. [Google Scholar] [CrossRef]
- Kristiansson, O.; Persson, I.; Bobicz, D.; Xu, D. A structural study of the hydrated and the dimethylsulfoxide, N,N’-dimethylpropyleneurea, acetonitrile, pyridine and N,N-dimethylthioformamide solvated nickel(II) ion solution and solid state. Inorg. Chim. Acta 2003, 344, 15–27. [Google Scholar] [CrossRef]
- Purans, J.; Fourest, B.; Cannes, C.; Sladkov, V.; David, F.; Venault, L.; Lecomte, M. Structural investigation of Pd(II) in concentrated nitric and perchloric acid solutions by XAFS. J. Chem. Phys. B 2005, 109, 11074–11082. [Google Scholar] [CrossRef]
- Hofer, T.S.; Randolf, B.R.; Ali Shah, A.; Rode, B.M.; Persson, I. Structure and dynamics of the hydrated palladium(II) ion in aqueous solution. A QMCF MD simulation and EXAFS spectroscopic study. Chem. Phys. Lett. 2007, 445, 193–197. [Google Scholar] [CrossRef]
- Beret, E.C.; Pappalardo, R.R.; Doltsinis, N.L.; Marx, D.; Marcos, E.S. Aqueous PdII and PtII: Anionic hydration revealed by Car–Parrinello simulations. ChemPhysChem 2008, 9, 237–240. [Google Scholar] [CrossRef]
- Jalilehvand, F.; Laffin, L.J. Structure of the hydrated platinum(II) ion and the cis-diammineplatinum(II) complex in acidic aqueous solution: An EXAFS study. Inorg. Chem. 2008, 47, 3248–3254. [Google Scholar] [CrossRef]
- Hofer, T.S.; Randolf, B.R.; Rode, B.M.; Persson, I. The hydrated platinum(II) ion in aqueous solution—A combined theoretical and EXAFS spectroscopic study. Dalton Trans. 2009, 1512–1515. [Google Scholar] [CrossRef]
- Persson, I.; Penner-Hahn, J.E.; Hodgson, K.O. An EXAFS spectroscopic study of solvates of copper(I) and copper(II) in acetonitrile, dimethyl sulfoxide, pyridine and tetrahydrothiophene solution and a large angle X-ray scattering study of the copper(II) acetonitrile solvate in solution. Inorg. Chem. 1993, 32, 2497–2501. [Google Scholar] [CrossRef]
- Persson, I.; Persson, P.; Sandström, M.; Ullström, A.-S. Structure of Jahn-Teller distorted solvated copper(II) ions in solution, and in solids with apparently regular octahedral coordination geometry. J. Chem. Soc. Dalton Trans. 2002, 1256–1265. [Google Scholar] [CrossRef]
- Persson, I.; Lundberg, D.; Bajnoczi, E.G.; Klementiev, K.; Just, J.; Sigfridsson Clauss, K.G.V. EXAFS study on the coordination chemistry of the solvated copper(II) ion in a series of oxygen donor solvents. Inorg. Chem. 2020, 59, 9538–9550. [Google Scholar] [CrossRef]
- Peisach, J.; Mims, W.B. Deviations from centrosymmetry in some simple Cu2+ complexes. Chem. Phys. Lett. 1976, 37, 307–310. [Google Scholar] [CrossRef]
- Peisach, J.; Mims, W.B. Linear electric-field effect in stellacyanin, azurin and in some simple-model compounds. Eur. J. Biochem. 1978, 84, 207–214. [Google Scholar] [CrossRef]
- Pasquarello, A.; Petri, I.; Salmon, P.S.; Parisel, O.; Car, R.; Toth, E.; Powell, D.H.; Fischer, H.E.; Helm, L.; Merbach, A.E. First solvation shell of the Cu(II) aqua ion: Evidence for fivefold coordination. Science 2001, 291, 856–859. [Google Scholar] [CrossRef]
- Benfatto, M.; D’Angelo, P.; Della Longa, S.; Pavel, N.V. Evidence of distorted fivefold coordination of the Cu2+ aqua ion from an X-ray-absorption spectroscopy quantitative analysis. Phys. Rev. B 2002, 65, 174205. [Google Scholar] [CrossRef]
- Franck, P.; Benfatto, M.; Szilagyi, R.K.; D’Angelo, P.; Della Longa, S.; Hodgson, K.O. The solution structure of [Cu(aq)]2+ and its implications for rack-induced bonding in blue copper protein active sites. Inorg. Chem. 2005, 44, 1922–1933. [Google Scholar] [CrossRef] [PubMed]
- Chaboy, J.; Munoz-Paez, A.; Marcos, E.S. The interplay of the 3d9 and 3d10 L electronic configurations in the copper K-edge XANES spectra of Cu(II) compounds. J. Synchrotron Rad. 2006, 13, 471–476. [Google Scholar] [CrossRef]
- Bryantsev, V.S.; Diallo, M.S.; van Duin, A.C.T.; Goddard III, W.A. Hydration of copper(II): New insights from density functional theory and the COSMO solvation model. J. Phys. Chem. A 2008, 112, 9104–9112. [Google Scholar] [CrossRef]
- De Almeida, K.J.; Murugan, N.A.; Rinkevicius, Z.Z.; Hugosson, H.-W.; Vahtras, O.; Ågren, H.; Cesar, A. Conformations, structural transitions and visible near-infrared absorption spectra of four-, five- and six-coordinated Cu(II) aqua complexes. Phys. Chem. Chem. Phys. 2009, 11, 508–519. [Google Scholar] [CrossRef]
- Liu, X.; Lu, X.; Meijer, E.J.; Wang, R. Hydration mechanisms of Cu2+: Tetra-, penta- or hexa-coordinated? Phys. Chem. Chem. Phys. 2010, 12, 10801–10804. [Google Scholar] [CrossRef]
- Gomez-Salces, S.; Aguado, F.; Valiente, R.; Rodrigues, F. Unraveling the coordination geometry of copper(II) ions in aqueous solution through absorption intensity. Angew. Chem. Int. Ed. 2012, 51, 9335–9338. [Google Scholar] [CrossRef]
- Bowron, D.T.; Amboage, M.; Boada, R.; Freeman, A.; Hayama, S.; Diaz-Moreno, S. The hydration structure of Cu2+: More tetrahedral than octahedral? RSC Adv. 2013, 3, 17803–17812. [Google Scholar] [CrossRef]
- Moin, S.T.; Hofer, T.S.; Weiss, A.K.H.; Rode, B.M. Dynamics of ligand exchange mechanism at Cu(II) in water: An ab initio quantum mechanical charge field molecular dynamics study with extended quantum mechanical region. J. Chem. Phys. 2013, 139, 014503. [Google Scholar] [CrossRef]
- Bowron, D.T.; Diaz-Moreno, S. The structure of water in solutions containing di- and trivalent cations by empirical potential structure refinement. J. Phys. Condens. Matter 2013, 25, 454213. [Google Scholar] [CrossRef]
- Frank, P.; Benfatto, M.; Qayyam, M.; Hedman, B.; Hogdson, K.O. A high-resolution XAS study of aqueous Cu(II) in liquid and frozen solutions: Pyramidal, polymorphic, and non-centrosymmetric. J. Chem. Phys. 2015, 142, 084310. [Google Scholar] [CrossRef]
- Chillemi, G.; Pace, E.; D’Abramo, M.; Benfatto, M. Equilibrium between 5- and 6-fold coordination in the first hydration shell of Cu(II). J. Phys. Chem. A 2016, 120, 3958–3965. [Google Scholar] [CrossRef]
- Galván-Garcáa, E.A.; Agacino-Valdés, E.; Franco-Pérez, M.; Gómez-Balderas, R. [Cu(H2O)n]2+ (n = 1–6) complexes in solution phase: A DFT hierarchical study. Theo. Chem. Acc. 2017, 136, 29. [Google Scholar] [CrossRef]
- Frank, P.; Benfatto, M.; Qayyum, M. [Cu(aq)]2+ is structurally plastic and the axially elongated octahedron goes missing. J. Chem. Phys. 2018, 148, 204302. [Google Scholar] [CrossRef]
- Persson, I.; Nilsson, K.B. Coordination chemistry of the solvated silver(I) ion in oxygen donor solvents water, dimethyl sulfoxide and N,N’-dimethylpropyleneurea. Inorg. Chem. 2006, 45, 7428–7434. [Google Scholar] [CrossRef]
- Busato, M.; Melchior, A.; Migliorati, V.; Colella, A.; Persson, I.; Mancini, G.; Veclaniz, D.; D’Angelo, P. The elusive coordination of the Ag+ ion in aqueous solution: Evidence for a linear structure. Inorg. Chem. 2020, 59, 10291–10302. [Google Scholar] [CrossRef]
- Lin, R.-L.; Dong, Y.-P.; Tang, M.; Liu, Z.; Tao, Z.; Liu, J.-X. Selective Recovery and Detection of Gold with Cucurbit[n]urils (n = 5–7). Inorg. Chem. 2020, 59, 3850–3855. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, P.; Chillemi, G.; Barone, V.; Mancini, G.; Sanna, N.; Persson, I. Experimental evidence for a variable first coordination shell of the cadmium(II) ion in aqueous, dimethyl sulfoxide and N,N’-dimethylpropyleneurea solution. J. Phys. Chem. B 2005, 109, 9178–9185. [Google Scholar] [CrossRef]
- Rosdahl, J.; Persson, I.; Kloo, L.; Ståhl, K. On the solvation of the mercury(I) ion. A Structural, vibration spectroscopic and quantum chemical study. Inorg. Chim. Acta 2004, 357, 2624–2634. [Google Scholar] [CrossRef]
- Hofer, T.S.; Randolf, B.R.; Rode, B.M. The hydration of the mercury(I)-dimer—A quantum mechanical charge field molecular dynamics study. Chem. Phys. 2008, 349, 210–218. [Google Scholar] [CrossRef]
- Persson, I.; Eriksson, L.; Lindqvist-Reis, P.; Persson, P.; Sandström, M. An EXAFS spectroscopic, large-angle X-ray scattering, and crystallographic study of hexahydrated, dimethyl sulfoxide and pyridine 1-oxide hexasolvated mercury(II) Ions. Chem. Eur. J. 2008, 14, 6687–6696. [Google Scholar] [CrossRef] [PubMed]
- Chillemi, G.; Mancini, G.; Sanna, N.; Barone, V.; Della Longa, S.; Benfatto, M.; Pavel, N.V.; D’Angelo, P. Evidence for sevenfold coordination in the first solvation shell of Hg(II) aqua ion. J. Am. Chem. Soc. 2007, 129, 5430–5436. [Google Scholar] [CrossRef] [PubMed]
- Sandström, M.; Persson, I. Crystal and molecular structure of hexakis(dimethylsulfoxide)mercury(II) perchlorate, [Hg((CH3)2SO)6](ClO4)2. Acta Chem. Scand. Ser. A 1978, 32, 95–100. [Google Scholar] [CrossRef]
- Strömberg, D.; Sandström, M.; Wahlgren, U. Theoretical calculations on the structure of the hexahydrated divalent zinc, cadmium and mercury ions. Chem. Phys. Lett. 1990, 172, 49–54. [Google Scholar] [CrossRef]
- Bersuker, I.B. The Jahn–Teller Effect and Vibronic Interactions in Modern Chemistry; Plenum Press: New York, NY, USA, 1984; Chapters 1–5. [Google Scholar]
- Bersuker, I.B.; Polinger, V.Z. Vibronic Interactions in Molecules and Crystals; Springer: Berlin, Germany, 1989; Chapters 3 and 4. [Google Scholar]
- Glaser, J.; Johansson, G.; Jennische, P.; Wahlberg, A.; Bastiansen, O.; Braathen, G.; Fernholt, L.; Gundersen, G.; Nielsen, C.J.; Cyvin, B.N.; et al. On the Structures of the thallium(III) ion and Its bromide complexes in aqueous solution. Acta Chem. Scand. A 1982, 36, 125–135. [Google Scholar] [CrossRef][Green Version]
- Blixt, J.; Glaser, J.; Mink, J.; Persson, I.; Persson, P.; Sandström, M. On the structure of thallium(III) chloride, bromide and cyanide complexes in aqueous solution. J. Am. Chem. Soc. 1995, 117, 5089–5104. [Google Scholar] [CrossRef]
- Shimoni-Livny, L.; Glusker, J.P.; Bock, C.W. Lone pair functionality in divalent lead compounds. Inorg. Chem. 1998, 37, 1853–1867. [Google Scholar] [CrossRef]
- Gillespie, R.J.; Nyholm, R.S. Inorganic stereochemistry. Q. Rev. London 1957, 11, 339–380. [Google Scholar] [CrossRef]
- Gillespie, R.J.; Hargittai, I. The VSEPR Model of Molecular Geometry; Allyn and Bacon: Boston, MA, USA, 1991; ISBN 978-0-205-12369-8. [Google Scholar]
- Mudring, A.-V.; Rieger, F. Lone Pair Effect in Thallium(I) macrocyclic compounds. Inorg. Chem. 2005, 44, 6240–6243 and references therein. [Google Scholar] [CrossRef]
- Walsh, G.; Watson, G.A. Influence of the anion on lone pair formation in Sn(II) monochalcogenides: A DFT study. J. Phys. Chem. B 2005, 109, 18868–18875. [Google Scholar] [CrossRef]
- Stoltzfus, M.W.; Woodward, P.M.; Seshadri, R.; Klepeis, J.H.; Bursten, B. Structure and bonding in SnWO4, PbWO4, and BiVO4: Lone pairs vs inert pairs. Inorg. Chem. 2007, 46, 3839–3850. [Google Scholar] [CrossRef]
- Walsh, A.; Payne, D.J.; Edgell, R.G.; Watson, G.A. Stereochemistry of post-transition metal oxides: Revision of the classical lone pair model. Chem. Soc. Rev. 2011, 40, 4455–4463. [Google Scholar] [CrossRef]
- Persson, I.; Jalilehvand, F.; Sandström, M. Structure of the solvated thallium(I) ion in aqueous, dimethyl sulfoxide, N,N’-dimethylpopyleneurea and N,N-dimethylthioformamide solution. Inorg. Chem. 2002, 41, 192–197. [Google Scholar] [CrossRef]
- Persson, I.; D’Angelo, P.; Lundberg, D. Hydrated and solvated tin(II) ions in solution and the solid state, and a coordination chemistry overview of the d10s2. Chem. Eur. J. 2016, 22, 18583–18592. [Google Scholar] [CrossRef]
- Swift, T.J.; Sayre, W.G. Determination of hydration numbers of cations in aqueous solution by means of proton NMR. J. Chem. Phys. 1966, 44, 3567–3574. [Google Scholar] [CrossRef]
- Bargar, J.R.; Brown, G.E., Jr.; Parks, G.A. Surface complexation of Pb(II) at oxide-water interfaces: I. XAFS and bond-valence determination of mononuclear and polynuclear Pb(II) sorption products on aluminum oxides. Geochim. Cosmochim. Acta 1997, 61, 2617–2637. [Google Scholar] [CrossRef]
- Persson, I.; Lyczko, K.; Lundberg, D.; Eriksson, L.; Płaczek, A. A Coordination chemistry study of hydrated and solvated lead(II) ions in solution and solid state. Inorg. Chem. 2011, 50, 1058–1072. [Google Scholar] [CrossRef]
- Näslund, J.; Persson, I.; Sandström, M. Solvation of the bismuth(III) ion by water, dimethyl sulfoxide, N,N’-dimethylpropyleneurea, and N,N-dimethylthioformamide. An EXAFS, large-angle X-ray scattering, and crystallographic structural study. Inorg. Chem. 2000, 39, 4012–4021. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Persson, I. Structures of Hydrated Metal Ions in Solid State and Aqueous Solution. Liquids 2022, 2, 210-242. https://doi.org/10.3390/liquids2030014
Persson I. Structures of Hydrated Metal Ions in Solid State and Aqueous Solution. Liquids. 2022; 2(3):210-242. https://doi.org/10.3390/liquids2030014
Chicago/Turabian StylePersson, Ingmar. 2022. "Structures of Hydrated Metal Ions in Solid State and Aqueous Solution" Liquids 2, no. 3: 210-242. https://doi.org/10.3390/liquids2030014
APA StylePersson, I. (2022). Structures of Hydrated Metal Ions in Solid State and Aqueous Solution. Liquids, 2(3), 210-242. https://doi.org/10.3390/liquids2030014