
Respiratory Microbiota Associations with Asthma Across American and Emirati Adults: A Comparative Analysis


Abstract
1. Introduction
2. Materials and Methods
3. Results
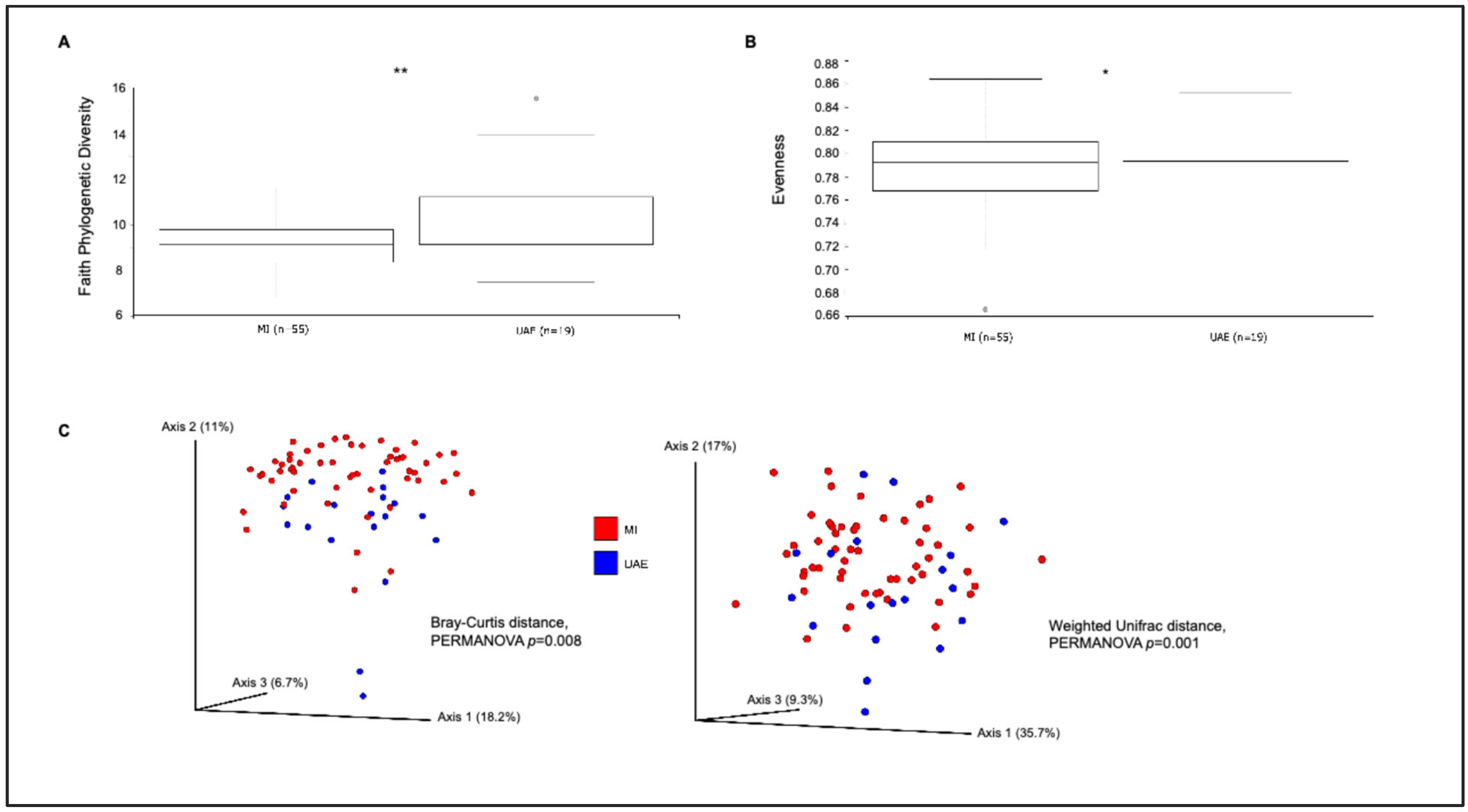
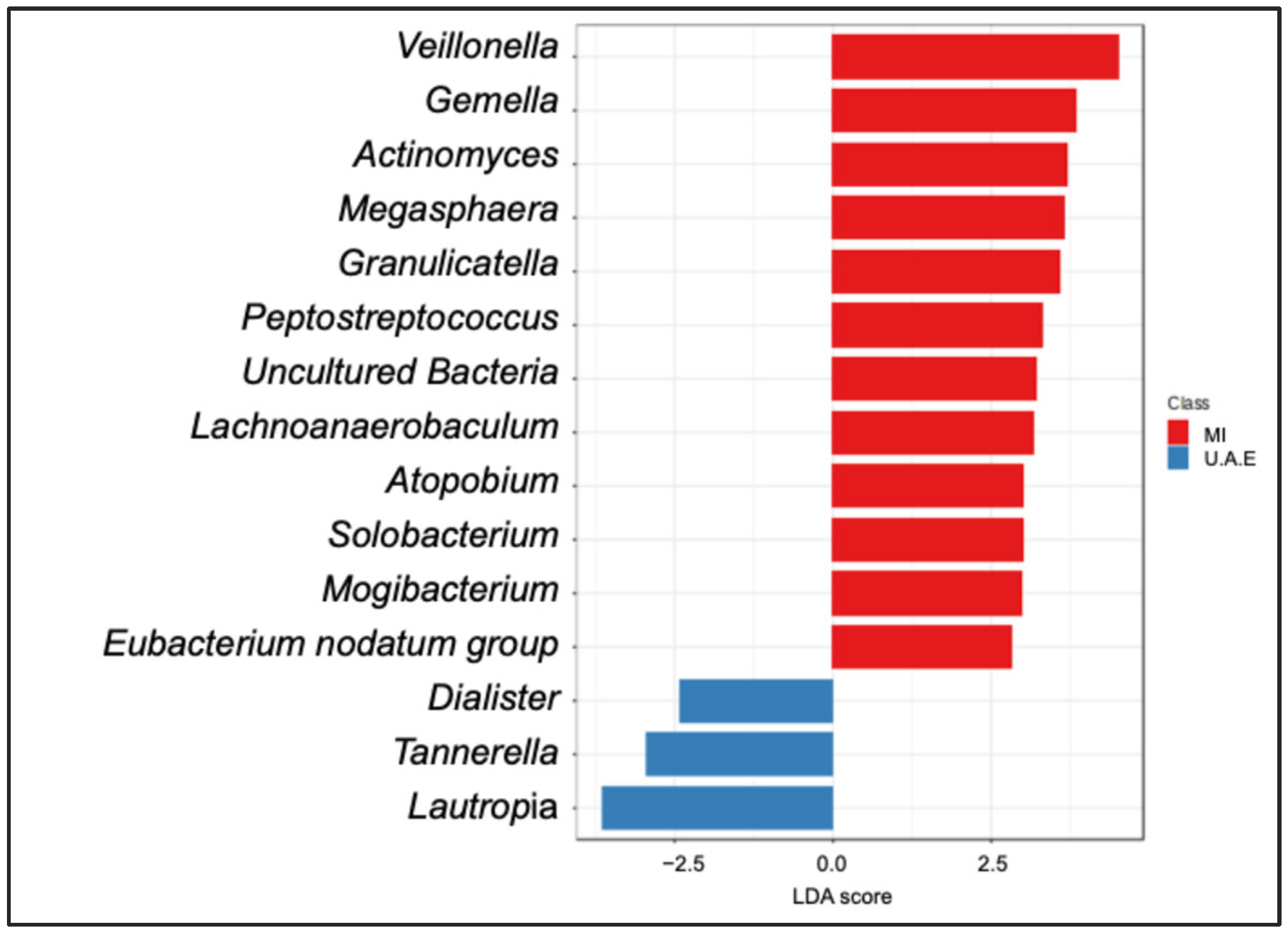
3.1. Sputum Bacterial Community Composition by Study Site
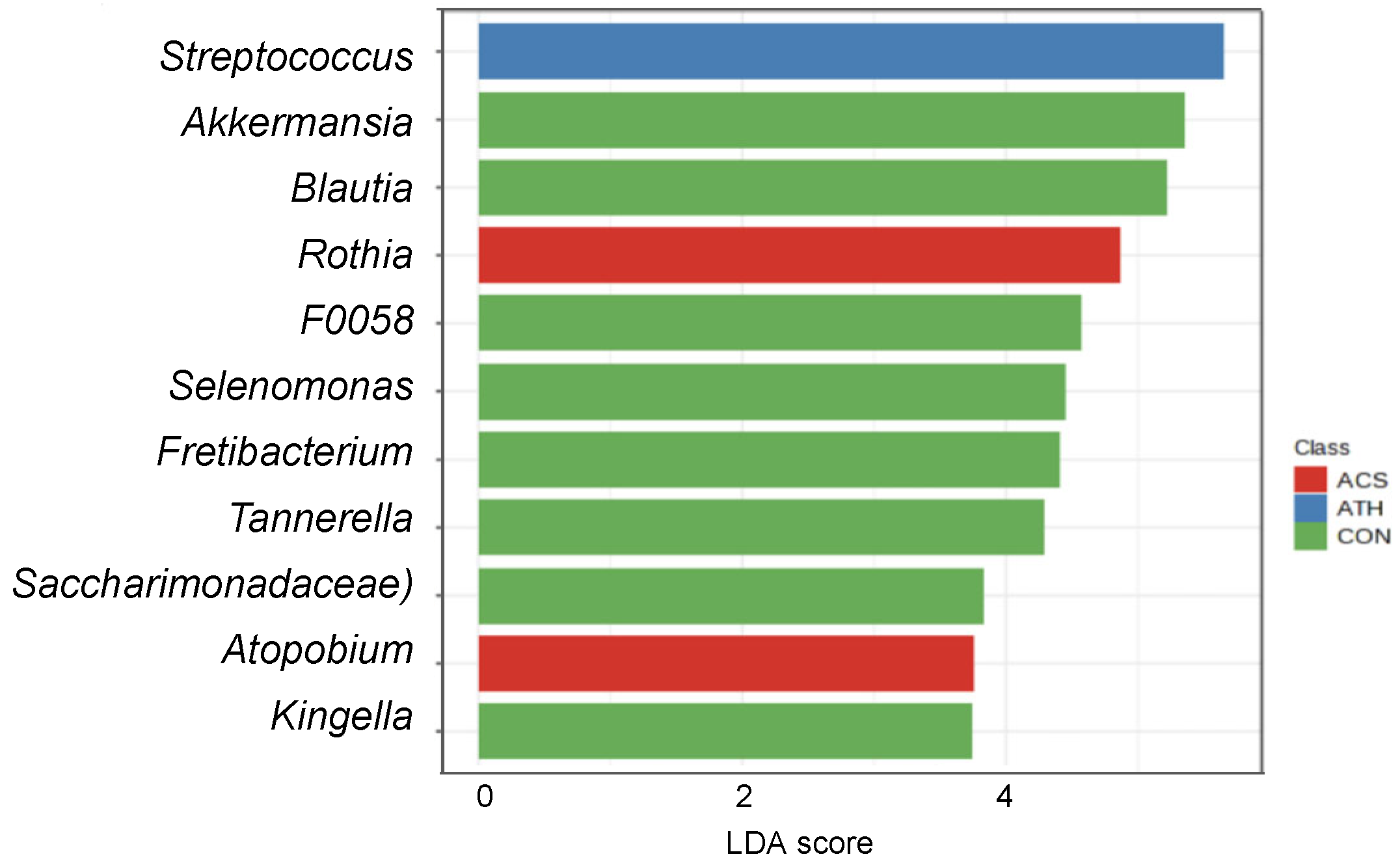
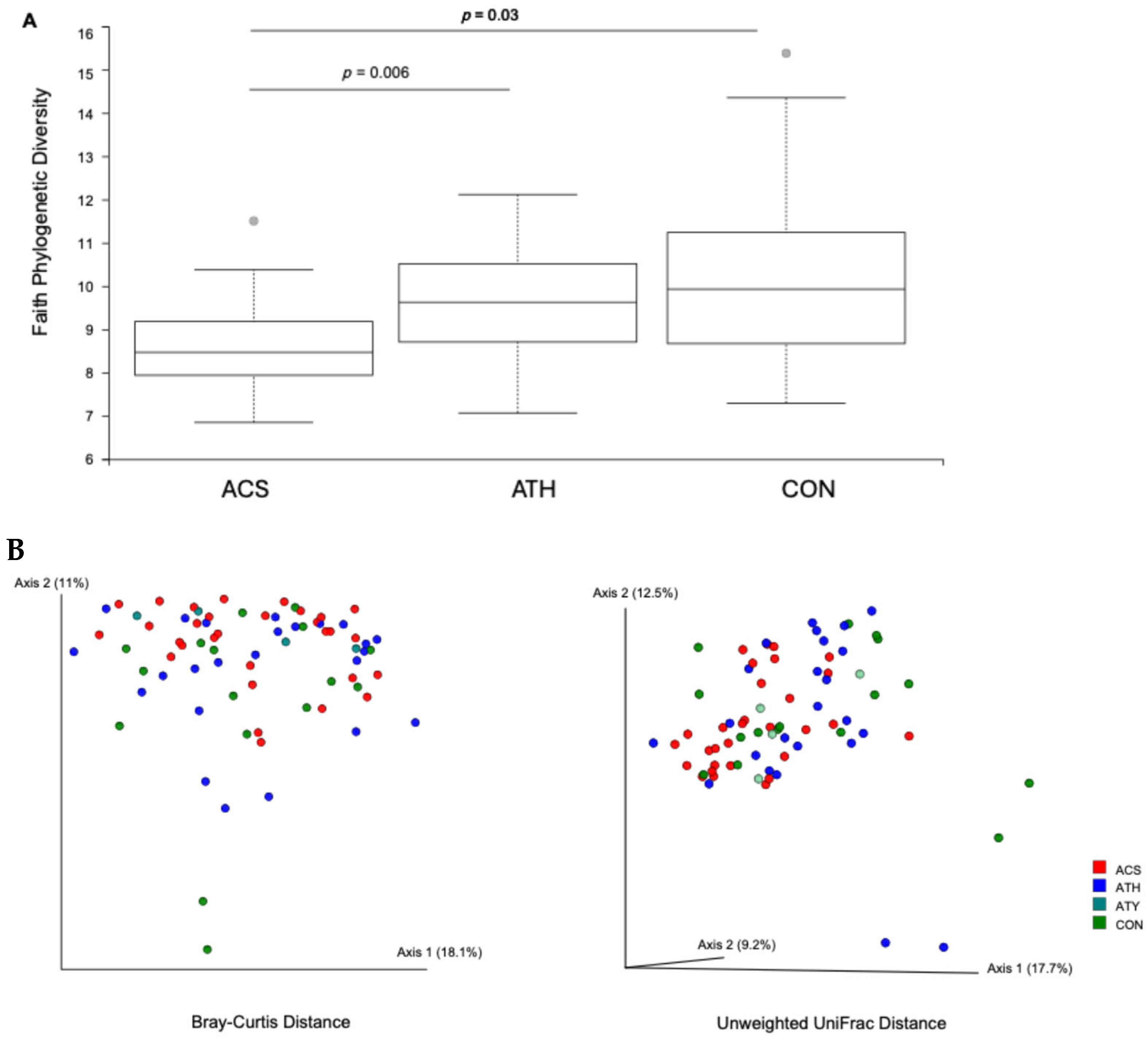
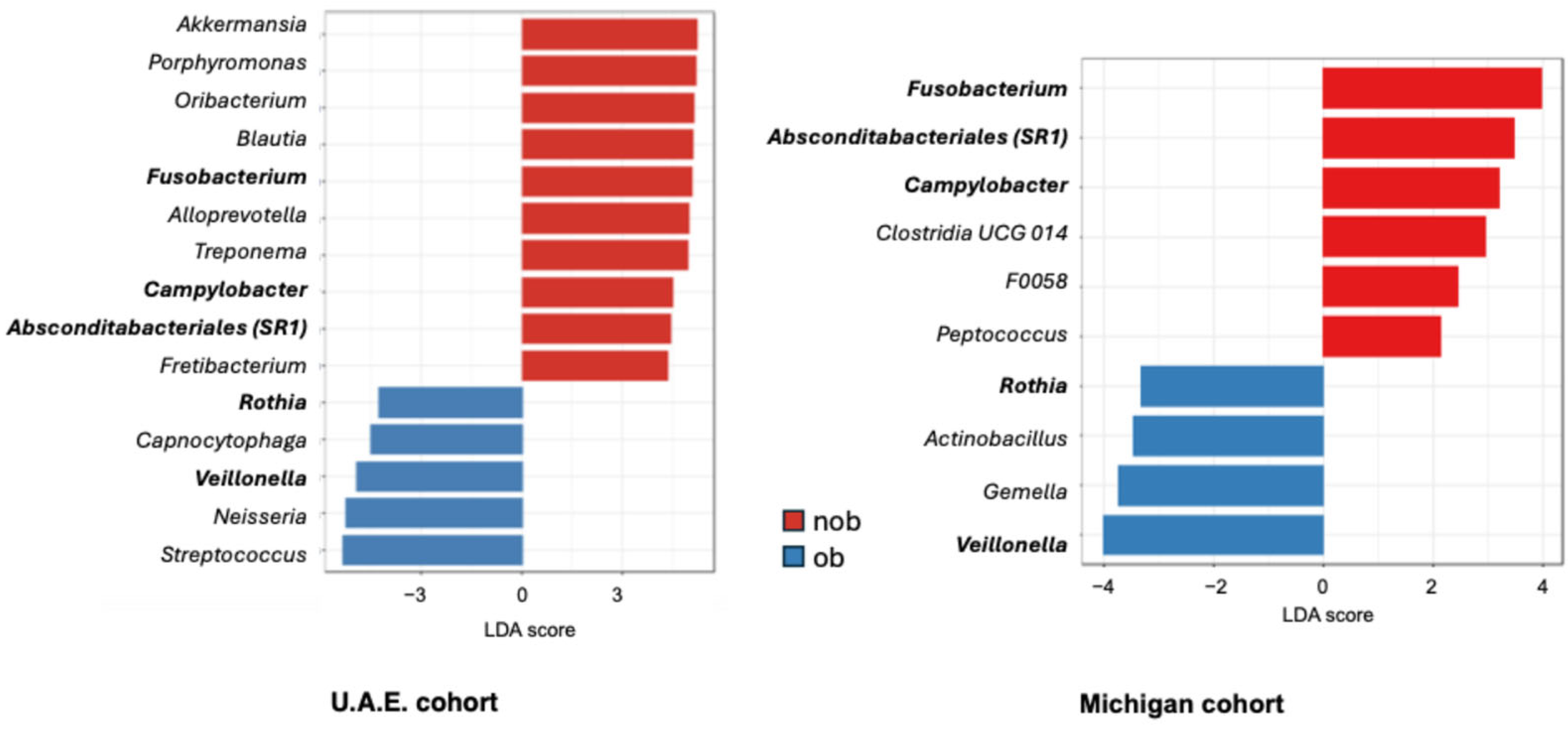
3.2. Sputum Bacterial Community Composition and Asthma Status Across Both Cohorts
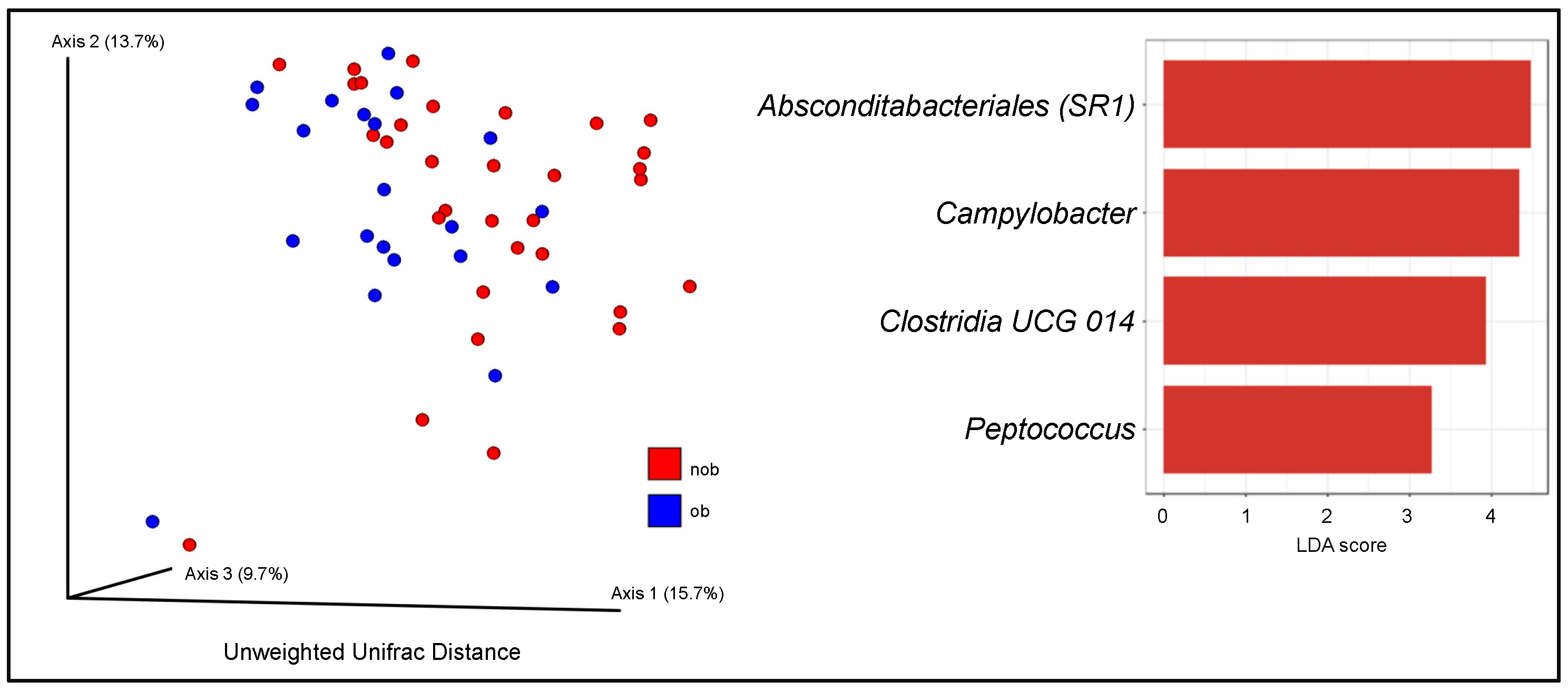
3.3. Sputum Bacterial Community Composition and Obesity-Associated Asthma
3.4. Airway Microbiota Associations with Asthma Clinical Factors in the U.A.E. Cohort
4. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACS | asthma on inhaled corticosteroid treatment |
| ATH | asthma not on inhaled corticosteroid treatment |
| Faith PD | Faith phylogenetic diversity index |
| ICS | inhaled corticosteroid |
| LEfSe | linear discriminant analysis effect size |
| MaAsLin2 | Microbiome Multivariate Association with Linear Models |
| MI | Michigan |
| nob | non-obese |
| ob | obese |
| QIIME2 | Quantitative Insights Into Microbial Ecology |
| U.A.E. | United Arab Emirates |
Appendix A
References
- Wang, Z.; Li, Y.; Gao, Y.; Fu, Y.; Lin, J.; Lei, X.; Zheng, J.; Jiang, M. Global, regional, and national burden of asthma and its attributable risk factors from 1990 to 2019: A systematic analysis for the Global Burden of Disease Study 2019. Respir. Res. 2023, 24, 169. [Google Scholar] [CrossRef] [PubMed]
- Fahy, J.V. Type 2 inflammation in asthma–present in most, absent in many. Nat. Rev. Immunol. 2015, 15, 57–65. [Google Scholar] [CrossRef]
- Abdel-Aziz, M.I.; Brinkman, P.; Vijverberg, S.J.H.; Neerincx, A.H.; Riley, J.H.; Bates, S.; Hashimoto, S.; Kermani, N.Z.; Chung, K.F.; Djukanovic, R.; et al. Sputum microbiome profiles identify severe asthma phenotypes of relative stability at 12 to 18 months. J. Allergy Clin. Immunol. 2021, 147, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Durack, J.; Christian, L.S.; Nariya, S.; Gonzalez, J.; Bhakta, N.R.; Ansel, K.M.; Beigelman, A.; Castro, M.; Dyer, A.M.; Israel, E.; et al. Distinct associations of sputum and oral microbiota with atopic, immunologic, and clinical features in mild asthma. J. Allergy Clin. Immunol. 2020, 146, 1016–1026. [Google Scholar] [CrossRef] [PubMed]
- Durack, J.; Lynch, S.V.; Nariya, S.; Bhakta, N.R.; Beigelman, A.; Castro, M.; Dyer, A.M.; Israel, E.; Kraft, M.; Martin, R.J.; et al. Features of the bronchial bacterial microbiome associated with atopy, asthma, and responsiveness to inhaled corticosteroid treatment. J. Allergy Clin. Immunol. 2017, 140, 63–75. [Google Scholar] [CrossRef]
- Huang, Y.J.; Nariya, S.; Harris, J.M.; Lynch, S.V.; Choy, D.F.; Arron, J.R.; Boushey, H. The airway microbiome in patients with severe asthma: Associations with disease features and severity. J. Allergy Clin. Immunol. 2015, 136, 874–884. [Google Scholar] [CrossRef]
- Huang, Y.J.; Nelson, C.E.; Brodie, E.L.; Desantis, T.Z.; Baek, M.S.; Liu, J.; Woyke, T.; Allgaier, M.; Bristow, J.; Wiener-Kronish, J.P.; et al. Airway microbiota and bronchial hyperresponsiveness in patients with suboptimally controlled asthma. J. Allergy Clin. Immunol. 2011, 127, 372–381.e1-3. [Google Scholar] [CrossRef]
- Kozik, A.J.; Begley, L.A.; Lugogo, N.; Baptist, A.; Erb-Downward, J.; Opron, K.; Huang, Y.J. Airway microbiota and immune mediator relationships differ in obesity and asthma. J. Allergy Clin. Immunol. 2023, 151, 931–942. [Google Scholar] [CrossRef]
- Denner, D.R.; Sangwan, N.; Becker, J.B.; Hogarth, D.K.; Oldham, J.; Castillo, J.; Sperling, A.I.; Solway, J.; Naureckas, E.T.; Gilbert, J.A.; et al. Corticosteroid therapy and airflow obstruction influence the bronchial microbiome, which is distinct from that of bronchoalveolar lavage in asthmatic airways. J. Allergy Clin. Immunol. 2016, 137, 1398–1405.e3. [Google Scholar] [CrossRef]
- Goleva, E.; Jackson, L.P.; Harris, J.K.; Robertson, C.E.; Sutherland, E.R.; Hall, C.F.; Good, J.T., Jr.; Gelfand, E.W.; Martin, R.J.; Leung, D.Y. The effects of airway microbiome on corticosteroid responsiveness in asthma. Am. J. Respir. Crit. Care Med. 2013, 188, 1193–1201. [Google Scholar] [CrossRef]
- Opron, K.; Begley, L.A.; Erb-Downward, J.R.; Freeman, C.; Madapoosi, S.; Alexis, N.E.; Barjaktarevic, I.; Barr, R.G.; Bleecker, E.R.; Bowler, R.P.; et al. Lung microbiota associations with clinical features of COPD in the SPIROMICS cohort. npj Biofilms Microbiomes 2021, 7, 14. [Google Scholar] [CrossRef]
- Opron, K.; Begley, L.A.; Erb-Downward, J.R.; Li, G.; Alexis, N.E.; Barjaktarevic, I.; Barr, R.G.; Bleecker, E.R.; Boucher, R.; Bowler., R.P.; et al. Loss of Airway Phylogenetic Diversity Is Associated with Clinical and Pathobiological Markers of Disease Development in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2024, 210, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Tang, G.; Fu, Y.; Deng, P.; Yao, R. The impact of environmental factors on respiratory tract microbiome and respiratory system diseases. Eur. J. Med. Res. 2025, 30, 236. [Google Scholar] [CrossRef] [PubMed]
- Adamko, D.J.; Hildebrand, K.J. The changing epidemiology of paediatric childhood asthma and allergy in different regions of the world. Front. Allergy 2025, 6, 1584928. [Google Scholar] [CrossRef]
- Halpin, D.M.G.; Balmes, J.; Han, M.K.; Papi, A.; Martinez, F.J.; Montes de Oca, M.; Ozoh, O.B.; Salvi, S.; Sin, D.D.; Zheng, J.; et al. Climate Change & COPD: A GOLD Science Committee Review. Am. J. Respir. Crit. Care Med. 2025, 211, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Bunyavanich, S. Microbial influencers: The airway microbiome’s role in asthma. J. Clin. Investig. 2025, 135, e184316. [Google Scholar] [CrossRef] [PubMed]
- McCauley, K.E.; Flynn, K.; Calatroni, A.; DiMassa, V.; LaMere, B.; Fadrosh, D.W.; Lynch, K.V.; Gill, M.A.; Pongracic, J.A.; Khurana Hershey, G.K.; et al. National Institute of Allergy and Infectious Diseases–sponsored Inner-City Asthma Consortium. Seasonal airway microbiome and transcriptome interactions promote childhood asthma exacerbations. J. Allergy Clin. Immunol. 2022, 150, 204–213. [Google Scholar] [CrossRef]
- Makrufardi, F.; Peng, S.W.; Chung, K.F.; Chadeau-Hyam, M.; Lee, K.Y.; Hsiao, T.C.; Ho, K.F.; Rusmawatiningtyas, D.; Murni, I.K.; Arguni, E.; et al. Extreme temperatures modulate gene expression in the airway epithelium of the lungs in mice and asthma patients. Front. Med. 2025, 12, 1531154. [Google Scholar] [CrossRef] [PubMed]
- Boniardi, L.; Solazzo, G.; Favero, C.; Campo, L.; Ferrari, L.; Fustinoni, S. Short-term personal exposure to multiple air pollutants affects nasal microbiota in school-age children. Sci. Total Environ. 2025, 981, 179588. [Google Scholar] [CrossRef] [PubMed]
- Al Bataineh, M.T.; Hamoudi, R.A.; Dash, N.R.; Ramakrishnan, R.K.; Almasalmeh, M.A.; Sharif, H.A.; Al-Hajjaj, M.S.; Hamid, Q. Altered respiratory microbiota composition and functionality associated with asthma early in life. BMC Infect. Dis. 2020, 20, 697. [Google Scholar] [CrossRef]
- Mahboub, B.H.; Al-Hammadi, S.; Rafique, M.; Sulaiman, N.; Pawankar, R.; Al Redha, A.I.; Mehta, A.C. Population prevalence of asthma and its determinants based on European Community Respiratory Health Survey in the United Arab Emirates. BMC Pulm. Med. 2012, 12, 4. [Google Scholar] [CrossRef]
- Hassan Mahboub, B.H.; Santhakumar, S.; Soriano, J.B.; Pawankar, R. Asthma insights and reality in the United Arab Emirates. Ann. Thorac. Med. 2010, 5, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Tarraf, H.; Aydin, O.; Mungan, D.; Albader, M.; Mahboub, B.; Doble, A.; Lahlou, A.; Tariq, L.; Aziz, F.; El Hasnaoui, A. Prevalence of asthma among the adult general population of five Middle Eastern countries: Results of the SNAPSHOT program. BMC Pulm. Med. 2018, 18, 68. [Google Scholar] [CrossRef]
- Sharma, A.; Laxman, B.; Naureckas, E.T.; Hogarth, D.K.; Sperling, A.I.; Solway, J.; Ober, C.; Gilbert, J.A.; White, S.R. Associations between fungal and bacterial microbiota of airways and asthma endotypes. J. Allergy Clin. Immunol. 2019, 144, 1214–1227.e7. [Google Scholar] [CrossRef] [PubMed]
- Soccio, P.; Quarato, C.M.I.; Tondo, P.; Lacedonia, D.; Hoxhallari, A.; Foschino Barbaro, M.P.; Scioscia, G. Breath and Sputum Analyses in Asthmatic Patients: An Overview. Cells 2024, 13, 1355. [Google Scholar] [CrossRef]
- Lacy, P.; Lee, J.L.; Vethanayagam, D. Sputum analysis in diagnosis and management of obstructive airway diseases. Ther. Clin. Risk Manag. 2005, 1, 169–179. [Google Scholar] [PubMed] [PubMed Central]
- Su, L.; Qiao, Y.; Luo, J.; Huang, R.; Li, Z.; Zhang, H.; Zhao, H.; Wang, J.; Xiao, Y. Characteristics of the sputum microbiome in COPD exacerbations and correlations between clinical indices. J. Transl. Med. 2022, 20, 76. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mac Aogáin, M.; Dicker, A.J.; Mertsch, P.; Chotirmall, S.H. Infection and the microbiome in bronchiectasis. Eur. Respir. Rev. 2024, 33, 240038. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Reddel, H.K.; Bacharier, L.B.; Bateman, E.D.; Brightling, C.E.; Brusselle, G.G.; Buhl, R.; Cruz, A.A.; Duijts, L.; Drazen, J.M.; FitzGerald, J.M.; et al. Global Initiative for Asthma Strategy 2021: Executive Summary and Rationale for Key Changes. Am. J. Respir. Crit. Care Med. 2022, 205, 17–35. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Glöckner, F.O.; Yilmaz, P.; Quast, C.; Gerken, J.; Beccati, A.; Ciuprina, A.; Bruns, G.; Yarza, P.; Peplies, J.; Westram, R.; et al. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J. Biotechnol. 2017, 261, 169–176. [Google Scholar] [CrossRef]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhou, G.; Ewald, J.; Pang, Z.; Shiri, T.; Xia, J. MicrobiomeAnalyst 2.0: Comprehensive statistical, functional and integrative analysis of microbiome data. Nucleic Acids Res. 2023, 51, W310–W318. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome. Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Huang, S.; Chen, J.; Cui, Z.; Ma, K.; Wu, D.; Luo, J.; Li, F.; Xiong, W.; Rao, S.; Xiang, Q.; et al. Lachnospiraceae-derived butyrate mediates protection of high fermentable fiber against placental inflammation in gestational diabetes mellitus. Sci. Adv. 2023, 9, eadi7337. [Google Scholar] [CrossRef]
- Li, W.; Wang, B.; Tan, M.; Song, X.; Xie, S.; Wang, C. Analysis of sputum microbial metagenome in COPD based on exacerbation frequency and lung function: A case control study. Respir. Res. 2022, 23, 321. [Google Scholar] [CrossRef]
- Han, W.; Wang, N.; Han, M.; Liu, X.; Sun, T.; Xu, J. Identification of microbial markers associated with lung cancer based on multi-cohort 16 s rRNA analyses: A systematic review and meta-analysis. Cancer Med. 2023, 12, 19301–19319. [Google Scholar] [CrossRef] [PubMed]
- Mallick, H.; Rahnavard, A.; McIver, L.J.; Ma, S.; Zhang, Y.; Nguyen, L.H.; Tickle, T.L.; Weingart, G.; Ren, B.; Schwager, E.H.; et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. 2021, 17, e1009442. [Google Scholar] [CrossRef] [PubMed]
- Michalovich, D.; Rodriguez-Perez, N.; Smolinska, S.; Pirozynski, M.; Mayhew, D.; Uddin, S.; Van Horn, S.; Sokolowska, M.; Altunbulakli, C.; Eljaszewicz, A.; et al. Obesity and disease severity magnify disturbed microbiome-immune interactions in asthma patients. Nat. Commun. 2019, 10, 5711. [Google Scholar] [CrossRef]
- Yaghoubi, M.; Adibi, A.; Safari, A.; FitzGerald, J.M.; Sadatsafavi, M. The projected economic and health burden of uncontrolled asthma in the United States. Am. J. Respir. Crit. Care Med. 2019, 200, 1102–1112. [Google Scholar] [CrossRef] [PubMed]
- Nurmagambetov, T.; Kuwahara, R.; Garbe, P. The economic burden of asthma in the United States, 2008–2013. Ann. Am. Thorac. Soc. 2018, 15, 348–356. [Google Scholar] [CrossRef]
- Al Mazrouei, K.; Almannaei, A.I.; Nur, F.M.; Bachnak, N.; Alzaabi, A. Direct and Indirect Costs of Asthma Burden in Abu Dhabi: A Retrospective Analysis of Insurance Claims Data from 2015 to 2018. Clin. Outcomes Res. 2021, 13, 969–980. [Google Scholar] [CrossRef]
- Ibrahim, N.M.; Almarzouqi, F.I.; Al Melaih, F.A.; Farouk, H.; Alsayed, M.; AlJassim, F.M. Prevalence of asthma and allergies among children in the United Arab Emirates: A cross-sectional study. World Allergy Organ. J. 2021, 14, 100588. [Google Scholar] [CrossRef]
- Hilty, M.; Burke, C.; Pedro, H.; Cardenas, P.; Bush, A.; Bossley, C.; Davies, J.; Ervine, A.; Poulter, L.; Pachter, L.; et al. Disordered microbial communities in asthmatic airways. PLoS ONE 2010, 5, e8578. [Google Scholar] [CrossRef]
- Sverrild, A.; Kiilerich, P.; Brejnrod, A.; Pedersen, R.; Porsbjerg, C.; Bergqvist, A.; Erjefält, J.S.; Kristiansen, K.; Backer, V. Eosinophilic airway inflammation in asthmatic patients is associated with an altered airway microbiome. J. Allergy Clin. Immunol. 2017, 140, 407–417.e11. [Google Scholar] [CrossRef]
- Zhang, Q.; Cox, M.; Liang, Z.; Brinkmann, F.; Cardenas, P.A.; Duff, R.; Bhavsar, P.; Cookson, W.; Moffatt, M.; Chung, K.F. Airway microbiota in severe asthma and relationship to asthma severity and phenotypes. PLoS ONE 2016, 11, e0152724. [Google Scholar] [CrossRef]
- Carney, S.M.; Clemente, J.C.; Cox, M.J.; Dickson, R.P.; Huang, Y.J.; Kitsios, G.D.; Kloepfer, K.M.; Leung, J.M.; LeVan, T.D.; Molyneaux, P.L.; et al. Methods in Lung Microbiome Research. Am. J. Respir. Cell Mol. Biol. 2020, 62, 283–299. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | Status | Age | Sex (% Female) | Race (% White) | Body Mass Index (BMI) | Asthma Control Test Score | Use of Inhaled Corticosteroids |
|---|---|---|---|---|---|---|---|
| Michigan | Asthmatic (n = 44) | 38 | 55% | 75% | 28 | 21 | 59% |
| Healthy (n = 11) | 46 | 64% | 91% | 25 | not applicable | not applicable | |
| U.A.E | Asthmatic (n = 10) | 64 | 64% | 0% | 31 | 18 | 40% |
| Healthy (n = 10) | 41 | 78% | 0% | 24 | not applicable | not applicable |
| Organism | Log 2FC | FDR-Adjusted p |
|---|---|---|
| Prevotella nigrescens | 2.32 | ≤0.001 |
| Prevotella histicola | 2.9 | ≤0.001 |
| Prevotella oulorum | 1.9 | 0.013 |
| Solobacterium moorei | −2.2 | 0.013 |
| Prevotella salivae | 1.4 | 0.029 |
| Prevotella denticola | 1.8 | 0.033 |
| Rothia mucilaginosa | 1.9 | 0.037 |
| Prevotella melaninogenica | 1.2 | 0.037 |
| Uncultured Bacteroidetes | −1.8 | 0.037 |
| Organism | Log 2FC | FDR-Adjusted p |
|---|---|---|
| Alloprevotella | 0.7 | 0.01 |
| F0058 | 2.2 | 0.02 |
| Treponema | 1.2 | 0.03 |
| Tannerella | 0.8 | 0.03 |
| Eubacterium brachy group | 1.1 | 0.05 |
| Eubacterium nodatum group | 0.6 | 0.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozik, A.J.; Henderson, K.; Salameh, L.; Mahboub, B.; Al Bataineh, M.T.; Huang, Y.J. Respiratory Microbiota Associations with Asthma Across American and Emirati Adults: A Comparative Analysis. Appl. Microbiol. 2025, 5, 59. https://doi.org/10.3390/applmicrobiol5030059
Kozik AJ, Henderson K, Salameh L, Mahboub B, Al Bataineh MT, Huang YJ. Respiratory Microbiota Associations with Asthma Across American and Emirati Adults: A Comparative Analysis. Applied Microbiology. 2025; 5(3):59. https://doi.org/10.3390/applmicrobiol5030059
Chicago/Turabian StyleKozik, Ariangela J., Kyra Henderson, Laila Salameh, Bassam Mahboub, Mohammad T. Al Bataineh, and Yvonne J. Huang. 2025. "Respiratory Microbiota Associations with Asthma Across American and Emirati Adults: A Comparative Analysis" Applied Microbiology 5, no. 3: 59. https://doi.org/10.3390/applmicrobiol5030059
APA StyleKozik, A. J., Henderson, K., Salameh, L., Mahboub, B., Al Bataineh, M. T., & Huang, Y. J. (2025). Respiratory Microbiota Associations with Asthma Across American and Emirati Adults: A Comparative Analysis. Applied Microbiology, 5(3), 59. https://doi.org/10.3390/applmicrobiol5030059