
Microbiome Signatures and Inflammatory Biomarkers in Culture-Negative Neonatal Sepsis

 , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
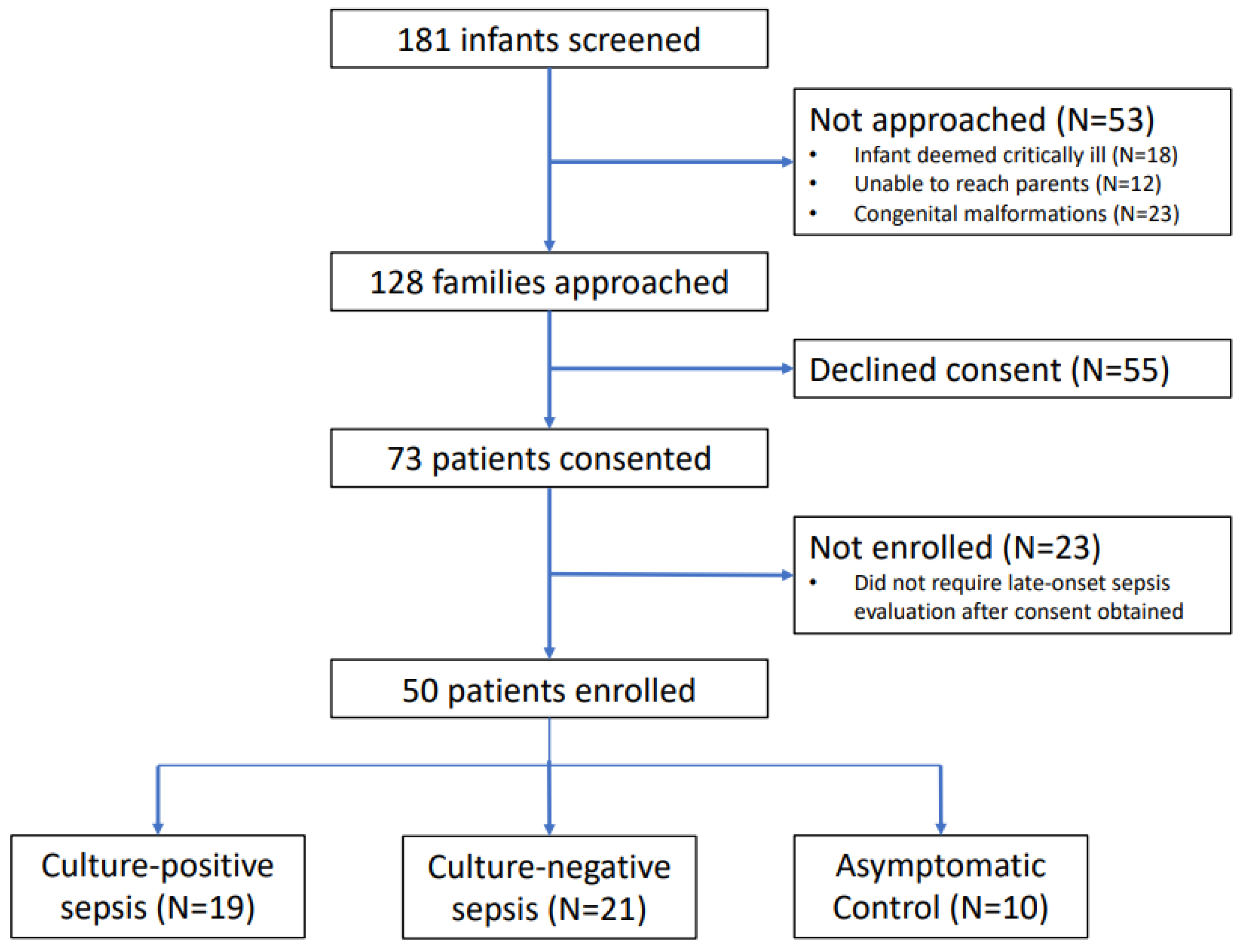
2.1. Study Design and Participants
2.2. Bacterial and Fungal Microbiome Evaluation
2.3. Virome Evaluation
2.4. Evaluation of Cytokine and Chemokine Profiles
2.5. Statistical Analyses and Data Interpretation
3. Results
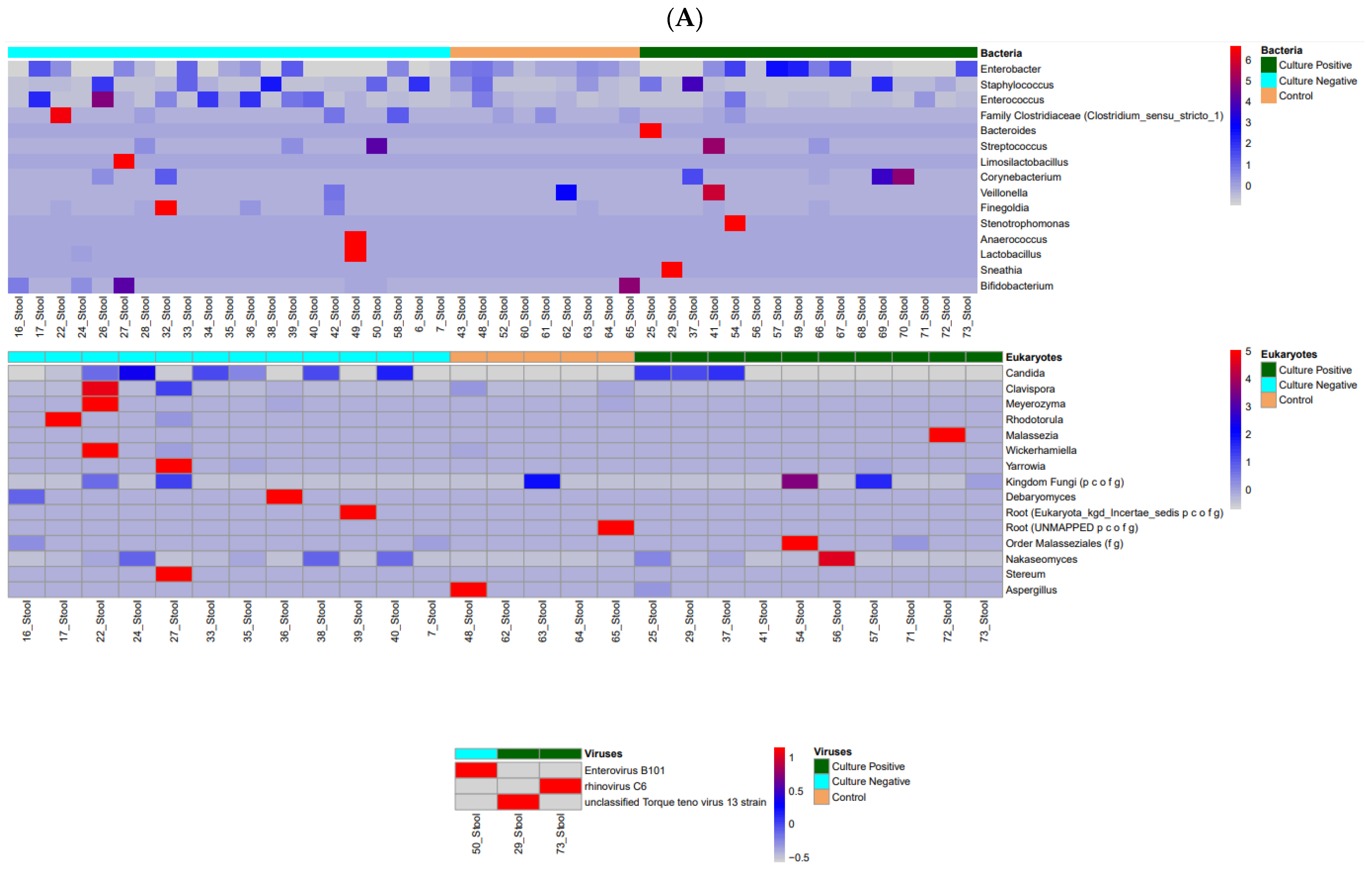
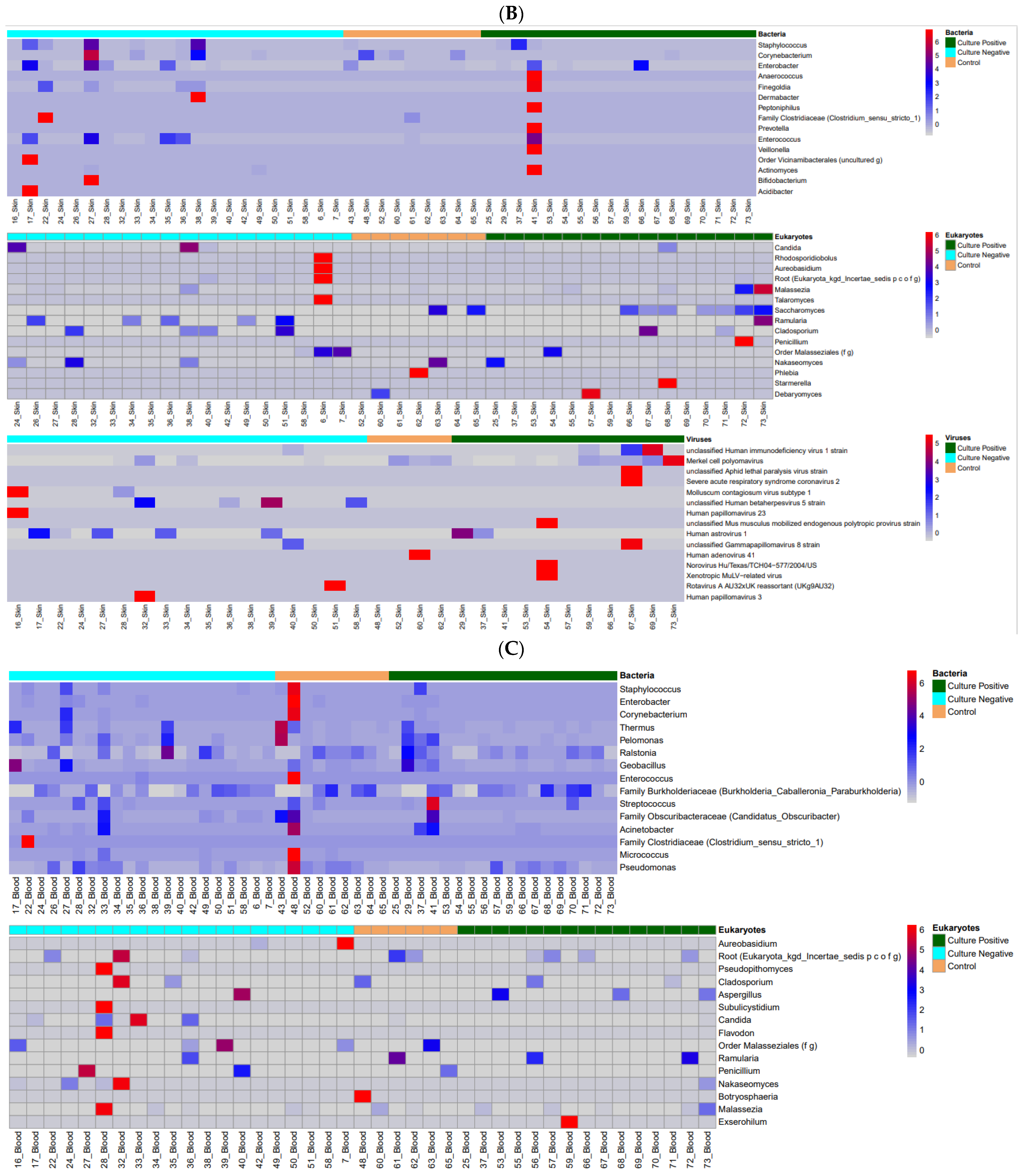
3.1. Microbiome Profiles in Preterm Infants
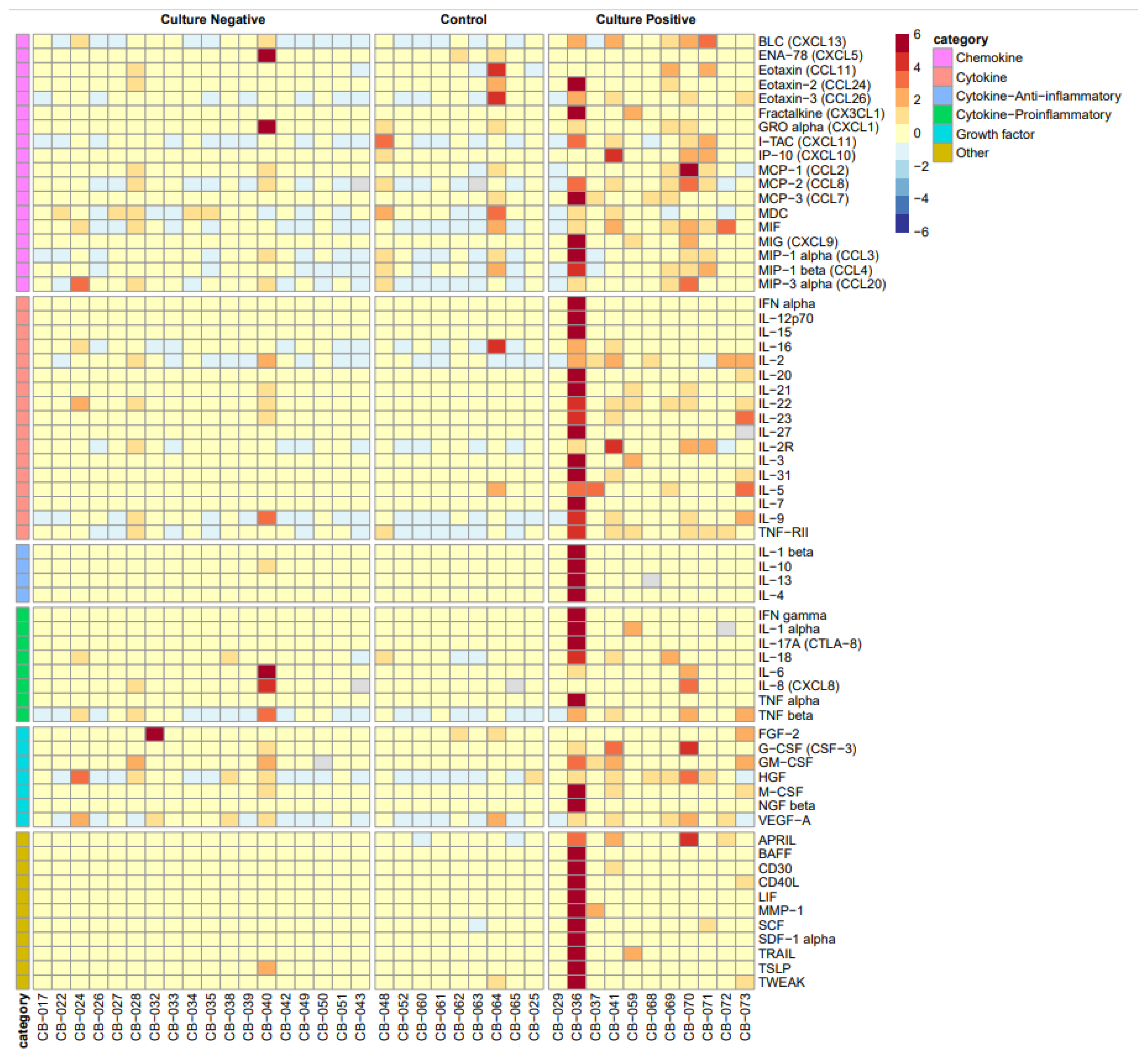
3.2. Systemic Cytokine and Chemokine Biomarkers
3.3. Preterm Sepsis Groups and Neonatal Outcomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 16S | 16S ribosomal RNA (used for bacterial microbiome analysis) |
| ANOVA | Analysis of Variance |
| ATIMA | Agile Toolkit for Incisive Microbial Analyses |
| BCL | Binary base call |
| BPD | Bronchopulmonary dysplasia |
| cDNA | Complementary DNA |
| CP | Culture-positive sepsis |
| CMMR | Center for Metagenomics and Microbiome Research |
| CN | Culture-negative sepsis |
| CO | Control group (asymptomatic preterm neonates) |
| CXCL13 | B lymphocyte chemoattractant |
| DNA | Deoxyribonucleic acid |
| ITS2 | Internal transcribed spacer 2 (used for fungal microbiome analysis) |
| LOS | Length of stay |
| MaAsLin2 | Microbiome Multivariable Association with Linear Models 2 |
| NEC | Necrotizing enterocolitis |
| NGS | Next-generation sequencing |
| NICU | Neonatal Intensive Care Unit |
| ROP | Retinopathy of prematurity |
| RNA | Ribonucleic acid |
| SDI | Shannon Diversity Index |
| TSS | Total Sum Scaling |
References
- Celik, I.H.; Hanna, M.; Canpolat, F.E.; Mohan, P. Diagnosis of Neonatal Sepsis: The Past, Present and Future. Pediatr. Res. 2022, 91, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Cantey, J.B.; Baird, S.D. Ending the Culture of Culture-Negative Sepsis in the Neonatal ICU. Pediatrics 2017, 140, e20170044. [Google Scholar] [CrossRef]
- Cantey, J.B.; Wozniak, P.S.; Pruszynski, J.E.; Sánchez, P.J. Reducing Unnecessary Antibiotic Use in the Neonatal Intensive Care Unit (SCOUT): A Prospective Interrupted Time-Series Study. Lancet Infect. Dis. 2016, 16, 1178–1184. [Google Scholar] [CrossRef] [PubMed]
- Fjalstad, J.W.; Stensvold, H.J.; Bergseng, H.; Simonsen, G.S.; Salvesen, B.; Rønnestad, A.E.; Klingenberg, C. Early-Onset Sepsis and Antibiotic Exposure in Term Infants: A Nationwide Population-Based Study in Norway. Pediatr. Infect. Dis. J. 2016, 35, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ting, J.Y.; Synnes, A.; Roberts, A.; Deshpandey, A.; Dow, K.; Yoon, E.W.; Lee, K.-S.; Dobson, S.; Lee, S.K.; Shah, P.S.; et al. Association Between Antibiotic Use and Neonatal Mortality and Morbidities in Very Low-Birth-Weight Infants Without Culture-Proven Sepsis or Necrotizing Enterocolitis. JAMA Pediatr. 2016, 170, 1181–1187. [Google Scholar] [CrossRef]
- Esaiassen, E.; Fjalstad, J.W.; Juvet, L.K.; van den Anker, J.N.; Klingenberg, C. Antibiotic Exposure in Neonates and Early Adverse Outcomes: A Systematic Review and Meta-Analysis. J. Antimicrob. Chemother. 2017, 72, 1858–1870. [Google Scholar] [CrossRef]
- Wensel, C.R.; Pluznick, J.L.; Salzberg, S.L.; Sears, C.L. Next-Generation Sequencing: Insights to Advance Clinical Investigations of the Microbiome. J. Clin. Investig. 2022, 132, e154944. [Google Scholar] [CrossRef]
- Brodin, P. Immune-Microbe Interactions Early in Life: A Determinant of Health and Disease Long Term. Science 2022, 376, 945–950. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction Between Microbiota and Immunity in Health and Disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Handley, S.A. The Virome: A Missing Component of Biological Interaction Networks in Health and Disease. Genome Med. 2016, 8, 32. [Google Scholar] [CrossRef]
- Begum, N.; Harzandi, A.; Lee, S.; Uhlen, M.; Moyes, D.L.; Shoaie, S. Host–Mycobiome Metabolic Interactions in Health and Disease. Gut Microbes 2022, 14, 2121576. [Google Scholar] [CrossRef]
- Dominguez-Bello, M.G.; Costello, E.K.; Contreras, M.; Magris, M.; Hidalgo, G.; Fierer, N.; Knight, R. Delivery Mode Shapes the Acquisition and Structure of the Initial Microbiota Across Multiple Body Habitats in Newborns. Proc. Natl. Acad. Sci. USA 2010, 107, 11971–11975. [Google Scholar] [CrossRef]
- Bezirtzoglou, E. The Intestinal Microflora During the First Weeks of Life. Anaerobe 1997, 3, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Nolt, D.; O’lEary, S.T.; Aucott, S.W. Risks of Infectious Diseases in Newborns Exposed to Alternative Perinatal Practices. Pediatrics 2022, 149, e2021055554. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Zhao, C.; Zhang, H.; Mattei, L.; Sherrill-Mix, S.; Bittinger, K.; Kessler, L.R.; Wu, G.D.; Baldassano, R.N.; DeRusso, P.; et al. The Stepwise Assembly of the Neonatal Virome Is Modulated by Breastfeeding. Nature 2020, 581, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.S.; Zhou, Y.; Zhao, G.; Bauer, I.K.; Droit, L.; Ndao, I.M.; Warner, B.B.; Tarr, P.I.; Wang, D.; Holtz, L.R. Early Life Dynamics of the Human Gut Virome and Bacterial Microbiome in Infants. Nat. Med. 2015, 21, 1228–1234. [Google Scholar] [CrossRef]
- Liu, X.; Liu, X.; He, G.; Lan, Y.; Guo, W.; Liu, X.; Li, J.; Liu, A.; He, M.; Liu, X.; et al. Virome and Metagenomic Analysis Reveal the Distinct Distribution of Microbiota in Human Fetal Gut During Gestation. Front. Immunol. 2022, 13, 1079294. [Google Scholar] [CrossRef]
- Pammi, M.; O’bRien, J.L.; Ajami, N.J.; Wong, M.C.; Versalovic, J.; Petrosino, J.F.; Wilson, B.A. Development of the Cutaneous Microbiome in the Preterm Infant: A Prospective Longitudinal Study. PLoS ONE 2017, 12, e0176669. [Google Scholar] [CrossRef]
- Pammi, M.; Thapa, S.; Balderas, M.; Runge, J.K.; Venkatachalam, A.; Luna, R.A.; Pai, N. Microbiome Signatures in Neonatal Central Line-Associated Bloodstream Infections. PLoS ONE 2020, 15, e0227967. [Google Scholar] [CrossRef]
- The Human Microbiome Project Consortium. Structure, Function and Diversity of the Healthy Human Microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef]
- The Human Microbiome Project Consortium. A Framework for Human Microbiome Research. Nature 2012, 486, 215–221. [Google Scholar] [CrossRef]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Ladau, J.; Locey, K.J.; Prill, R.J.; Tripathi, A.; Gibbons, S.M.; Ackermann, G.; et al. A Communal Catalogue Reveals Earth’s Multiscale Microbial Diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef]
- Bushnell, B. 9th Annual Genomics of Energy & Environment Meeting; Lawrence Berkeley National Lab: Berkeley, CA, USA, 2014. [Google Scholar]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly Accurate OTU Sequences from Microbial Amplicon Reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-Species Living Tree Project (LTP)” Taxonomic Frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Larsson, K.-H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L.; et al. The UNITE Database for Molecular Identification of Fungi: Handling Dark Taxa and Parallel Taxonomic Classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for Large-Scale Multiple Sequence Alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Mallick, H.; Rahnavard, A.; McIver, L.J.; Ma, S.; Zhang, Y.; Nguyen, L.H.; Tickle, T.L.; Weingart, G.; Ren, B.; Schwager, E.H.; et al. Multivariable Association Discovery in Population-Scale Meta-Omics Studies. PLoS Comput. Biol. 2021, 17, e1009442. [Google Scholar] [CrossRef]
- Tisza, M.; Cregeen, S.J.; Avadhanula, V.; Zhang, P.; Ayvaz, T.; Feliz, K.; Hoffman, K.L.; Clark, J.R.; Terwilliger, A.; Ross, M.C.; et al. Wastewater Sequencing Reveals Community and Variant Dynamics of the Collective Human Virome. Nat. Commun. 2023, 14, 6878. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Liu, C.; Cui, Y.; Li, X.; Yao, M. Microeco: An R Package for Data Mining in Microbial Community Ecology. FEMS Microbiol. Ecol. 2021, 97, fiaa255. [Google Scholar] [CrossRef]
- Schwartz, D.J.; Shalon, N.; Wardenburg, K.; DeVeaux, A.; Wallace, M.A.; Hall-Moore, C.; Ndao, I.M.; Sullivan, J.E.; Radmacher, P.; Escobedo, M.; et al. Gut Pathogen Colonization Precedes Bloodstream Infection in the Neonatal Intensive Care Unit. Sci. Transl. Med. 2023, 15, eadg5562. [Google Scholar] [CrossRef]
- Tarr, P.I.; Warner, B.B. Gut Bacteria and Late-Onset Neonatal Bloodstream Infections in Preterm Infants. Semin. Fetal Neonatal Med. 2016, 21, 388–393. [Google Scholar] [CrossRef] [PubMed]
- El Saie, A.; Fu, C.; Grimm, S.L.; Robertson, M.J.; Hoffman, K.; Putluri, V.; Ambati, C.S.R.; Putluri, N.; Shivanna, B.; Coarfa, C.; et al. Metabolome and Microbiome Multi-Omics Integration from a Murine Lung Inflammation Model of Bronchopulmonary Dysplasia. Pediatr. Res. 2022, 92, 1580–1589. [Google Scholar] [CrossRef]
- Arıkan, M.; Muth, T. Integrated Multi-Omics Analyses of Microbial Communities: A Review of the Current State and Future Directions. Mol. Omics 2023, 19, 607–623. [Google Scholar] [CrossRef]
- Virgin, H.W. The Virome in Mammalian Physiology and Disease. Cell 2014, 157, 142–150. [Google Scholar] [CrossRef]
- Neil, J.A.; Cadwell, K. The Intestinal Virome and Immunity. J. Immunol. 2018, 201, 1615–1620. [Google Scholar] [CrossRef]
- Santus, W.; Devlin, J.R.; Behnsen, J. Crossing Kingdoms: How the Mycobiota and Fungal-Bacterial Interactions Impact Host Health and Disease. Infect. Immun. 2021, 89, 1110–1128. [Google Scholar] [CrossRef] [PubMed]
- De Sordi, L.; Lourenço, M.; Debarbieux, L. “I Will Survive”: A Tale of Bacteriophage-Bacteria Coevolution in the Gut. Gut Microbes 2019, 10, 92–99. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Group | p-Value | ||
|---|---|---|---|---|
| CN (n = 21) | CO (n = 10) | CP (n = 19) | ||
| Birth weight (g) 2 | 900 (690, 1140) | 1250 (1040, 1620) | 860 (665, 1250) | 0.028 * |
| Gestational age (weeks) 2 | 27.1 (24.1, 29.0) | 30.5 (27.9, 31.3) | 25.9 (24.4, 28.1) | 0.026 * |
| Male sex 1 | 8 (38.1) | 4 (40.0) | 12 (63.2) | 0.287 |
| Race 1 | 0.064 | |||
| Asian | 0 (0.0) | 0 0.00 | 1 5.26 | |
| Black | 6 (28.6) | 2 20.00 | 6 31.58 | |
| Hispanic | 9 (42.9) | 0 (0.0) | 4 (21.1) | |
| White | 6 (28.6) | 8 (80.0) | 8 (42.1) | |
| Vaginal delivery 1 | 9 (42.9) | 0 (0.0) | 6 (31.6) | 0.048 * |
| Multiple births 1 | 5 (23.8) | 5 (50.0) | 2 (10.5) | 0.058 |
| Prenatal steroids 1 | 17 (81.0) | 10 (100.0) | 16 (84.2) | 0.442 |
| Antibiotics at birth 1 | 15 (71.4) | 5 (50.0) | 13 (68.4) | 0.507 |
| Apgar score at 1 min 2 | 7.0 (4.0, 7.0) | 8.0 (7.0, 8.0) | 4.0 (3.0, 7.0) | 0.094 |
| Apgar score at 5 min 2 | 8.0 (6.0, 8.0) | 9.0 (8.0, 9.0) | 7.0 (5.0, 9.0) | 0.024 * |
| Age at sepsis evaluation (days) 2 | 20.0 (14.0, 29.0) | 16.0 (12.0, 22.0) | 37.0 (13.0, 52.0) | 0.087 |
| CN vs. CO | Coef | Qval | ||
|---|---|---|---|---|
| 16S | Blood | Corynebacterium | −1.929 | 0.282 |
| Pseudomonas | −1.106 | 0.282 | ||
| Staphylococcus | −1.766 | 0.282 | ||
| Pelomonas | −0.768 | 0.700 | ||
| Family Burkholderiaceae | −0.210 | 0.837 | ||
| Geobacillus | 0.689 | 0.775 | ||
| Thermus | −0.930 | 0.775 | ||
| Streptococcus | −0.312 | 0.923 | ||
| Ralstonia | −0.090 | 0.934 | ||
| Halomonas | 0.035 | 0.951 | ||
| Skin | Staphylococcus | 2.514 | 0.009 * | |
| Haemophilus | 2.092 | 0.128 * | ||
| Streptococcus | 1.486 | 0.226 * | ||
| Enterobacter | 1.330 | 0.513 | ||
| Corynebacterium | 0.185 | 0.949 | ||
| Pelomonas | −0.051 | 0.949 | ||
| Pseudomonas | 0.174 | 0.949 | ||
| Stool | Enterobacter | −0.496 | 0.000 * | |
| Streptococcus | 4.875 | 0.002 * | ||
| Corynebacterium | 2.489 | 0.049 * | ||
| Enterococcus | 1.485 | 0.229 * | ||
| Family Clostridiaceae | 1.200 | 0.541 | ||
| Staphylococcus | 0.485 | 0.541 | ||
| ITS2 | Skin | Candida | 9.525 | 0.000 * |
| Saccharomyces | −4.810 | 0.001 * | ||
| Malassezia | 0.847 | 0.458 | ||
| Stool | Candida | 3.922 | 0.000 * | |
| Nakaseomyces | 3.468 | 0.041 * | ||
| Saccharomyces | 3.016 | 0.052 * | ||
| Malassezia | −0.204 | 0.919 |
| Cytokine/Chemokine | CN Median (IQR) | CO Median (IQR) | CP Median (IQR) | p-Value |
|---|---|---|---|---|
| APRIL | 86.7 (44.6, 275.4) A | 25.6 (10.5, 76.1) A | 416.6 (232.8, 1266.4) B | 0.001 * |
| BAFF | 37.4 (30.5, 44.1) | 55.7 (48.9, 64.7) | 41.7 (32.5, 48.4) | 0.131 |
| BLC-CXCL13 | 47.2 (41.6, 72.2) A | 35.5 (29.4, 55.6) A | 184.8 (84.7, 418.7) B | 0.001 * |
| CD30 | 918.2 (568.3, 1164.7) | 587.0 (570.4, 981.1) | 1102.4 (773.2, 1729.0) | 0.120 |
| CD40L | 0.0 (0.0, 0.0) A | 0.0 (0.0, 11.8) A,B | 15.0 (3.7, 52.2) B | 0.009 * |
| ENA78-CXCL5 | 93.3 (79.1, 205.6) | 218.6 (136.9, 252.4) | 73.2 (46.6, 164.4) | 0.114 |
| Eotaxin-CCL11 | 7.0 (5.5, 8.3) | 6.5 (6.0, 8.1) | 11.5 (5.4, 16.7) | 0.642 |
| Eotaxin 2-CCL24 | 103.6 (91.5, 184.3) | 126.1 (76.8, 142.8) | 240.2 (205.4, 837.0) | 0.046 * |
| Eotaxin 3-CCL26 | 0.1 (0.1, 0.2) | 0.1 (0.1, 0.2) | 0.2 (0.2, 0.3) | 0.058 |
| FGF 2 | 0.0 (0.0, 0.0) | 0.0 (0.0, 5.1) | 0.0 (0.0, 1.8) | 0.533 |
| Fractalkine-CX3CL1 | 0.0 (0.0, 0.0) A | 0.0 (0.0, 0.0) A | 4.1 (0.0, 6.2) B | 0.003 * |
| G-CSF | 11.5 (6.2, 14.0) A | 11.5 (10.1, 14.0) A,B | 38.7 (20.9, 220.5) B | 0.017 * |
| GM-CSF | 0.0 (0.0, 0.0) A,B | 0.0 (0.0, 0.0) A | 4.3 (0.0, 25.2) B | 0.015 * |
| GRO-alpha-CXCL1 | 6.5 (5.8, 9.0) | 7.1 (5.5, 21.0) | 13.5 (6.7, 34.1) | 0.210 |
| HGF | 134.7 (77.5, 211.8) A,B | 68.3 (48.1, 69.1) A | 426.5 (176.2, 495.7) B | 0.002 * |
| ITAC-CXCL11 | 3.9 (3.3, 10.4) | 3.3 (2.8, 9.3) | 11.4 (4.8, 21.1) | 0.051 |
| IFN-alpha | 0.0 (0.0, 0.0) A | 0.0 (0.0, 0.0) A | 0.9 (0.0, 1.8) B | 0.001 * |
| IFN-gamma | 0.0 (0.0, 0.8) A | 0.0 (0.0, 0.0) A | 4.7 (2.4, 9.5) B | 0.007 * |
| IL1-alpha | 0.0 (0.0, 0.0) | 0.0 (0.0, 0.0) | 4.2 (0.0, 5.5) | 0.076 |
| IL-1-beta | 0.0 (0.0, 0.0) A | 0.0 (0.0, 0.0) A | 6.2 (1.7, 14.4) B | 0.005 * |
| IL-10 | 0.3 (0.0, 2.2) A,B | 0.0 (0.0, 0.7) A | 4.4 (2.6, 8.7) B | 0.016 * |
| IL-12p70 | 1.6 (1.4, 1.9) A,B | 1.7 (1.3, 1.9) A | 2.6 (1.9, 3.6) B | 0.014 * |
| IL-13 | 0.0 (0.0, 0.0) | 0.0 (0.0, 0.0) | 0.0 (0.0, 2.7) | 0.181 |
| IL-15 | 5.8 (4.7, 10.2) | 9.8 (8.7, 12.4) | 8.8 (5.1, 14.4) | 0.454 |
| IL-16 | 313.3 (216.8, 479.9) A | 194.8 (129.6, 298.0) A | 720.1 (644.7, 910.8) B | 0.005 * |
| IL-17A-CTLA 8 | 0.3 (0.0, 0.9) A | 0.5 (0.0, 3.8) A | 3.2 (1.9, 6.3) B | 0.036 * |
| IL-18 | 37.1 (22.3, 49.6) A | 25.5 (12.7, 29.9) A | 81.2 (61.2, 189.3) B | 0.002 * |
| IL-2 | 2.2 (0.0, 4.9) | 1.5 (0.0, 5.3) | 11.0 (3.3, 18.6) | 0.098 |
| IL-20 | 2.8 (2.1, 4.1) | 2.0 (1.4, 6.4) | 9.2 (6.1, 16.3) | 0.038 * |
| IL-21 | 0.0 (0.0, 5.5) A | 0.0 (0.0, 0.0) A | 12.3 (1.8, 27.0) B | 0.007 * |
| IL-22 | 0.0 (0.0, 15.6) | 0.0 (0.0, 0.0) | 26.8 (0.0, 48.9) | 0.045 * |
| IL-23 | 0.0 (0.0, 6.8) A,B | 0.0 (0.0, 0.0) A | 10.4 (0.0, 42.9) B | 0.040 * |
| IL-27 | 0.0 (0.0, 0.0) | 0.0 (0.0, 0.0) | 0.0 (0.0, 0.0) | 0.400 |
| IL-2R | 11548.5 (9430.1, 20,497.2) A,B | 9131.5 (7430.5, 11,656.5) A | 18153.4 (12,519.6, 38,817.7) B | 0.022 * |
| IL-3 | 0.0 (0.0, 0.0) | 0.0 (0.0, 0.0) | 0.0 (0.0, 12.3) | 0.116 |
| IL-31 | 0.0 (0.0, 0.0) A | 0.0 (0.0, 0.0) A | 1.1 (0.0, 23.7) B | 0.003 * |
| IL-4 | 6.0 (2.0, 11.3) A,B | 2.4 (1.0, 3.4) A | 17.8 (7.4, 26.5) B | 0.010 |
| IL-5 | 0.0 (0.0, 0.0) A | 0.0 (0.0, 0.8) A | 1.9 (1.0, 4.5) B | 0.000 * |
| IL-6 | 0.0 (0.0, 7.9) A | 0.0 (0.0, 0.0) A | 43.6 (6.5, 132.0) B | 0.005 * |
| IL-7 | 0.5 (0.5, 0.6) A | 0.6 (0.4, 0.6) A,B | 0.9 (0.8, 1.3) B | 0.024 * |
| IL-8-CXCL8 | 9.7 (5.6, 42.0) A,B | 5.3 (3.4, 11.5) A | 48.7 (19.8, 106.0) B | 0.007 * |
| IL-9 | 0.4 (0.0, 2.7) A | 0.0 (0.0, 3.6) A | 6.5 (4.1, 14.1) A,B | 0.013 * |
| IP-10-CXCL10 | 13.6 (7.9, 19.8) | 7.8 (6.5, 10.0) | 15.2 (11.7, 124.2) | 0.054 |
| LIF-15 | 3.2 (2.9, 3.4) A,B | 2.7 (2.5, 4.1) A | 4.8 (3.9, 9.0) B | 0.029 * |
| M-CSF | 3.2 (1.6, 9.3) A,B | 0.0 (0.0, 0.8) A | 12.4 (9.3, 41.4) B | 0.008 * |
| MCP-1-CCL2 | 140.3 (123.6, 206.6) | 132.0 (93.0, 173.2) | 327.4 (208.4, 585.5) | 0.124 |
| MCP-2-CCL8 | 3.0 (2.1, 5.0) | 2.6 (2.4, 7.0) | 4.1 (3.2, 18.6) | 0.108 |
| MCP-3-CCL7 | 0.0 (0.0, 1.4) | 0.0 (0.0, 2.9) | 2.5 (0.0, 11.5) | 0.104 |
| MDC | 284.1 (214.1, 409.0) | 257.3 (241.3, 343.2) | 318.0 (263.1, 351.7) | 0.875 |
| MIF | 13.0 (7.8, 20.4) A | 5.1 (4.8, 6.8) A | 57.3 (18.6, 110.9) B | 0.002 * |
| MIG-CXCL9 | 0.0 (0.0, 0.0) A | 0.0 (0.0, 0.0) A,B | 17.6 (0.0, 73.6) B | 0.007 * |
| MIP-1alpha-CCL3 | 5.1 (3.2, 7.7) A | 3.0 (2.6, 12.1) A,B | 10.9 (7.6, 19.8) B | 0.025 * |
| MIP-1beta-CCL4 | 36.9 (18.6, 45.1) | 22.2 (14.6, 68.3) | 73.2 (41.4, 126.1) | 0.099 |
| MIP-3alpha-CCL20 | 50.0 (29.9, 154.9) | 20.4 (15.5, 24.1) | 75.0 (38.8, 249.9) | 0.047 * |
| MMP-1 | 10.3 (8.7, 17.1) | 8.3 (2.2, 24.1) | 16.1 (11.8, 19.4) | 0.374 |
| NGF-beta | 0.4 (0.0, 2.3) A | 0.0 (0.0, 0.5) A | 3.4 (2.6, 4.7) B | 0.004 * |
| SCF | 31.9 (16.6, 48.3) A,B | 16.1 (12.9, 21.9) A | 70.2 (42.1, 91.0) B | 0.011 |
| SDF-1-alpha | 337.2 (179.5, 684.3) A | 171.9 (149.6, 195.4) A | 889.1 (578.6, 1311.5) B | 0.002 * |
| TNF-alpha | 4.4 (2.9, 7.1) A,B | 3.8 (1.8, 5.0) A | 12.5 (6.3, 20.5) B | 0.030 * |
| TNF-beta | 0.2 (0.0, 1.8) | 0.5 (0.0, 1.7) | 3.8 (1.7, 10.6) | 0.031 * |
| TNF-RII | 155.7 (113.1, 213.0) A | 80.1 (77.6, 101.8) A | 273.2 (193.9, 340.2) B | <0.001 * |
| TRAIL | 0.0 (0.0, 6.4) A | 0.0 (0.0, 0.0) A | 11.9 (1.3, 21.8) B | 0.012 * |
| TSLP | 2.1 (1.6, 2.5) A,B | 1.9 (1.5, 2.1) A | 3.2 (2.2, 3.9) B | 0.017 * |
| TWEAK | 169.9 (119.8, 191.5) A | 156.2 (113.5, 184.3) A,B | 323.9 (192.5, 445.4) B | 0.040 * |
| VEGF-A | 189.5 (109.2, 298.5) A,B | 81.7 (78.3, 294.1) A | 387.9 (290.6, 515.8) B | 0.022 * |
| Outcomes | Group | p-Value | ||
|---|---|---|---|---|
| CN (n = 21) | CO (n = 10) | CP (n = 19) | ||
| NEC 1 | 3 (14.3) | 0 (0.0) | 5 (26.3) | 0.190 |
| Any ROP 1 | 16 (76.2) A,B | 4 (40.0) A | 16 (84.2) B | 0.039 * |
| BPD 1† | 16 (76.2) B | 3 (30.0) A | 17 (89.5) B | 0.002* |
| BPD severity 3 | n = 16 | n = 3 | n = 17 | 0.070 |
| Mild | 4 (25.0) | 2 (66.7) | 1 (5.9) | |
| Moderate | 3 (18.9) | 1 (33.3) | 3 (17.7) | |
| Severe | 9 (56.3) | 0 (0.0) | 13 (76.5) | |
| Severe ROP or ROP surgery 1 | 2 (9.1) | 0 (0.0) | 4 (22.2) | 0.229 |
| Any IVH 1 | 10 (47.6) | 1 (10.0) | 5 (26.3) | 0.087 |
| Severe IVH or PVL 1 | 3 (14.3) | 0 (0.0) | 1 (5.3) | 0.515 |
| Length of stay (days) 2 | 119.0 A (76.0, 160.0) | 50.5 B (46.0, 90.0) | 166.0 C (144.0, 264.0) | <0.001 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanna, M.; Huang, S.; Ross, M.; Reyes, A.; Perera, D.; Surathu, A.; Javornik Cregeen, S.; Hagan, J.; Pammi, M. Microbiome Signatures and Inflammatory Biomarkers in Culture-Negative Neonatal Sepsis. Appl. Microbiol. 2025, 5, 57. https://doi.org/10.3390/applmicrobiol5030057
Hanna M, Huang S, Ross M, Reyes A, Perera D, Surathu A, Javornik Cregeen S, Hagan J, Pammi M. Microbiome Signatures and Inflammatory Biomarkers in Culture-Negative Neonatal Sepsis. Applied Microbiology. 2025; 5(3):57. https://doi.org/10.3390/applmicrobiol5030057
Chicago/Turabian StyleHanna, Morcos, Shixia Huang, Matthew Ross, Anaid Reyes, Dimuthu Perera, Anil Surathu, Sara Javornik Cregeen, Joseph Hagan, and Mohan Pammi. 2025. "Microbiome Signatures and Inflammatory Biomarkers in Culture-Negative Neonatal Sepsis" Applied Microbiology 5, no. 3: 57. https://doi.org/10.3390/applmicrobiol5030057
APA StyleHanna, M., Huang, S., Ross, M., Reyes, A., Perera, D., Surathu, A., Javornik Cregeen, S., Hagan, J., & Pammi, M. (2025). Microbiome Signatures and Inflammatory Biomarkers in Culture-Negative Neonatal Sepsis. Applied Microbiology, 5(3), 57. https://doi.org/10.3390/applmicrobiol5030057