Abstract
Bioregenerative life support systems (BLSS) are currently in development to tackle low recovery efficiencies, high energy demands, as well as food, water, and oxygen production challenges through the regeneration of nutrients from waste streams. The MELiSSA pilot plant has been developed as a testbed for regenerative life support system bioreactor operation and characterization. As nitrogen is a vital resource in such systems, we studied the functional composition of a new packed-bed nitrifying bioreactor inoculated with a co-culture of Nitrosomonas europaea (ATCC 25978) and Nitrobacter winogradskyi (ATCC 25391). After 840 days of autotrophic continuous cultivation, the packed-bed was sampled at five vertical positions, each with three horizontal positions, and the biomass at each position was characterized via qPCR, 16S amplicon sequencing, and liquid chromatography tandem mass spectrometry. The total number of cells within the different sections fluctuated around 8.95 ± 5.10 × 107 cells/mL of beads. Based on 16S amplicons and protein content, N. europaea and N. winogradskyi constituted overall 44.07 ± 11.75% and 57.53 ± 12.04% of the nitrifying bioreactor, respectively, indicating the presence of a heterotrophic population that, even after such a long operation time, did not affect the nitrification function of the bioreactor. In addition, DNA-based abundance estimates showed that N. europaea was slightly more abundant than N. winogradskyi, whereas protein-based abundance estimates indicated a much higher abundance of N. europaea. This highlights that single-method approaches need to be carefully interpreted in terms of overall cell abundance and metabolic activity.
1. Introduction
Crewed long-distance spaceflight and prolonged stay in space, such as on a base on the Moon or Mars, can only be possible provided if autonomy from terrestrial resupply missions is realized. However, it is impossible to transport a payload that can sustain a crew for a prolonged duration. A three-year mission to Mars would require an estimated payload mass of ca. 20 tons for food and potable water alone for a crew of four, assuming an estimated 1.83 kg of food and 2.50 kg of water per crew member per day [1]. Such a limitation implies the need for the ability to produce food, water, and a breathable atmosphere on the space vessel from waste streams using highly reliable and robust technology. A life support system for long-distance space travel should be able to meet the metabolic needs of the crew by recovering 98% of nutrients and water from waste streams [2]. NASA’s Environmental Control and Life Support System (ECLSS) is currently operational on the International Space Station (ISS) and is entirely based on physicochemical processes. The ECLSS produces potable water and oxygen from waste streams, but only at efficiencies of 85% and 50%, respectively [3,4]. Moreover, no food is produced and waste streams that contain valuable nutrients (N, P, and K) are discarded [3]. Hence, there is a need for new technologies that can meet the requirements for life support in future space exploration.
Regenerative Life Support Systems (RLSS) are currently in development to tackle low recovery efficiencies, high energy demands, as well as food, water, and oxygen production challenges through regeneration of nutrients from waste streams. In these systems, biological and physicochemical processes are combined in a closed-loop configuration to produce food, water, and oxygen that meet the metabolic needs of the crew [5]. The longest running program that is developing a RLSS is the MELiSSA (Micro-Ecological Life Support System Alternative) program. It is led by the European Space Agency together with an international consortium of 15 partners. MELiSSA is inspired on a natural ecosystem, i.e., a lake, by aiming to reproduce its main functions in specific compartments in a closed-loop configuration using an engineering-based approach [6]. Five compartments make up the loop. The first compartment (CI) digests and liquefies fecal, solid, and urinal wastes with a consortium of bacteria in anaerobic and thermophilic conditions into volatile fatty acids (VFAs), CO2, minerals, and free NH4+ [7,8]. Currently, Rhodospirillum rubrum in compartment II (CII) processes VFAs containing effluent from CI [9]. Compartment III (CIII) uses an autotrophic co-culture of Nitrosomonas europaea and Nitrobacter winogradskyi that grow forming a biofilm to oxidize NH4+ to NO3− in a process called nitrification. NO3− serves as an assimilable nitrogen source for cyanobacterium Limnospira indica and higher plants in compartment IVa (CIVa) and IVb (CIVb), respectively. Here, food and O2 are produced for the final compartment V (CV), the crew [10,11,12]. Waste streams from the crew enter CI, closing the MELiSSA loop [7]. A facility called the MELiSSA Pilot Plant (MPP) has been designed to provide 100% of the respiration needs of a single person with concomitant production of 20–40% of food for nutritional demands [11]. The MPP is used as a testbed for bioreactor operation and characterization of the compartments, both individually and in interconnected configuration while also attempting to demonstrate the feasibility of the MELiSSA loop [13,14]. Recently, the experimental bioreactor for CIII development was successfully connected to CIVa in the liquid phase while CIVa was connected to CV (an animal chamber as a crew mock-up, consisting of three Wistar rats) in the gas phase in long-term operation [15].
Nitrogen is a vital resource in the MELiSSA loop and in any BLSS. Most nitrogen excreted by humans (7–16 g N d−1) is present in urine as urea and accounts for ~90% of all excreted nitrogen) [16,17]. Urea is transformed to NH4+ in CI, which is then converted to NO3− in CIII through nitrification [6,7]. In this compartment, NH4+ is converted to NO2− by N. europaea in a process called nitritation. First, ammonia monooxygenase (AMO) oxidizes NH4+ to NH2OH [18]. In a second step, hydroxylamine oxidoreductase (HAO) was hypothesized to transform NH2OH to NO2− [18]. However, a recent study suggests that NO is produced by HAO and converted to NO2− by a third, unidentified enzyme [19]. Finally, N. winogradskyi oxidizes NO2− to NO3− with nitrite oxidoreductase (NXR) in the so-called nitratation process [20]. Nitrification is only possible in an aerobic environment. In anaerobic conditions, the denitrification process leads to the production of gaseous N2O, NO, and N2 [21,22]. During denitrification, N. winogradskyi’s NXR acts as a nitrate reductase to produce NO2− and a copper-containing nitrite reductase (NirK) can reduce NO2− to NO [23]. In N. europaea, a NirK is also responsible for the ability to transform NO2− to NO. NO can then be used as a substrate by nitric oxide reductase (Nor) to produce N2O. Finally, HAO in N. europaea is also able to act as a reductase by converting NH2OH to produce nitrogen oxides [21]. Production of gaseous nitrogen species result in a loss of nitrogen in the bioreactor and production of potentially toxic volatile compounds that have to be avoided in a space environment. Hence, anaerobic conditions should be prevented during CIII operation [5].
The experimental packed-bed bioreactor used to develop CIII contains a mixed culture of N. europaea and N. winogradskyi immobilized on polystyrene beads. This type of bioreactor has been used for several studies to characterize hydrodynamics, mass transfer and nitrification capacity [24], distribution of the nitrifying species along the bioreactor column after long-term continuous operation through qPCR and mathematical modeling [25], and the development of mathematical models to predict nitrification, biomass, and oxygen concentration in the reactor [26]. Both qPCR and 16S-rRNA gene amplicon sequencing are well-established methods to determine the composition of microbial communities of (de)nitrification bioreactors [25,27,28,29,30,31]. A metaproteomic approach is more often used to characterize the functionality and to identify novel functional proteins and their respective metabolic pathways in those bioreactors [30,31,32]. Moreover, the technique can also be used as a taxonomical identification tool [33]. However, metaproteomics has not yet been applied to determine microbial abundancies in (de)nitrification bioreactors.
The main goal of this study was to acquire knowledge from the first 840 days of an operational test of a new packed-bed nitrifying bioreactor. For this, we used an unprecedented multi-omics approach in order to characterize the microbial population colonizing the bioreactor after such a long test period and improve operation, including better axenicity in future runs. To that purpose, the bioreactor population was assessed by qPCR, 16S amplicon sequencing, and liquid chromatography tandem mass spectrometry (LC-MS/MS) methods. The results from the respective procedures to determine microbial abundancies were compared to determine the reproducibility between the techniques. Finally, the metabolic processes at different locations within the bioreactor were identified.
2. Materials and Methods
2.1. Bacterial Strains and Culture Medium
A co-culture of Nitrosomonas europaea (ATCC 25978) and Nitrobacter winogradskyi (ATCC 25391) was provided from the DSMZ collection (Braunschweig, Germany) and used to inoculate the nitrifying packed-bed bioreactor. The medium used contained NH4+ as nitrogen source and was previously defined (in [24] for medium used until day 435, in [15] for day 435 onwards).
2.2. Nitrifying Packed-Bed Reactor
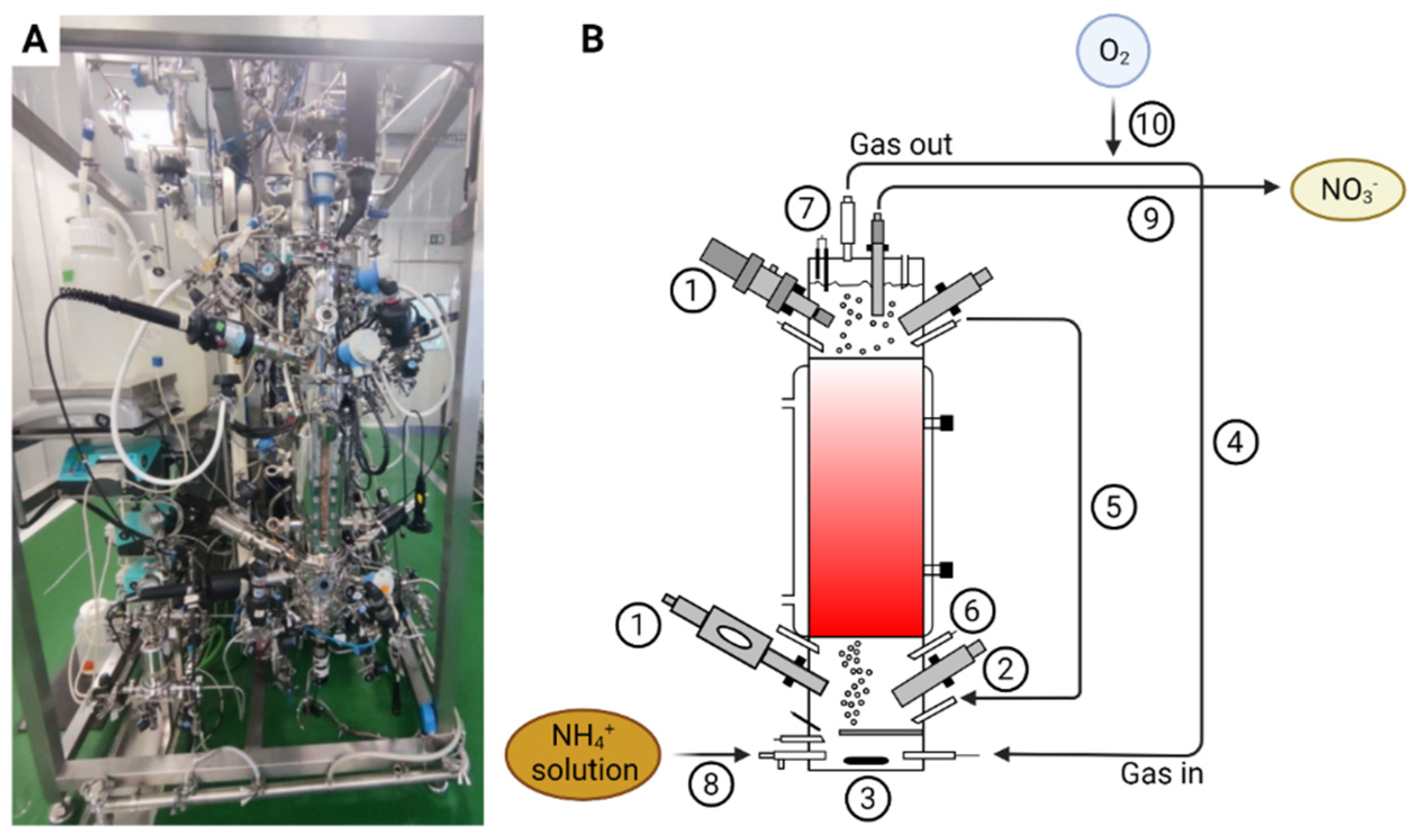
A pilot scale up-flow packed-bed reactor with 0.125 m in diameter and 0.56 m in height, accounting for a total volume of 7 L (SNC-Lavalin, Brussels, Belgium and Bioprocess Technologies, Madrid, Spain) (Figure 1) inoculated with an axenic co-culture (1:1) of N. europaea and N. winogradskyi was used. Biostyr® expanded polystyrene beads (Veolia Water Technologies, Saint-Maurice, France) with a mean diameter of 4.1 mm were used as a biofilm carrier material. The packed-bed section was located at the bioreactor central part. The bioreactor contained a top and bottom section where the instrumentation for liquid phase on-line monitoring of the bioreactor (pH, T, pO2, EC) were located. The bottom section, where fresh liquid feeding, liquid recirculation inlet, and gas sparging took place, was mechanically stirred (marine impeller). Gas-liquid separation took place at the top section where gas, liquid, and recirculation outlets were located. The bioreactor was operated in a recirculation closed gas-loop mode regulated by means of a mass flow-meter (Bronkhorst, F-202D-FA, Bronkhorst, Ruurlo, The Netherlands). The pH was measured by means of two sterilizable glass pH probes connected to a pH amplifier (Mettler Toledo, Inpro 3253, Greifensee, Switzerland). The pH was regulated by the addition of 0.1 M sulfuric acid (H2SO4) and 0.94 M sodium carbonate (Na2CO3) solution. Base solution was changed to 1.9 M KOH after 180 days. The pO2 was measured by means of two Clark amperometric sensors (Mettler Toledo, InPro6950i/12/320, Greifensee, Switzerland). Dissolved oxygen in the top section of the bioreactor was maintained at 80% of the saturation with air (i.e., 6.45 mg O2/L) by adding pulses of pure oxygen by using a proportional–integral–derivative control system.
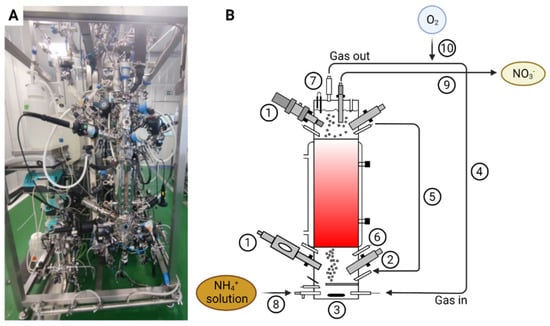
Figure 1.
Picture of the nitrifying bioreactor (A) and a schematic of the nitrifying bioreactor (B) with pO2 sensor (1), pH sensor (2), agitator (3), recirculating gas closed-loop (4), liquid recirculation loop (5), acid and base additions (6), vapor condenser (7), liquid influent line (8), liquid effluent line (9) and pure oxygen inlet line (10).
EC was measured by means of two EC probes connected to an EC amplifier (Mettler Toledo, Inpro 7001, Greifensee, Switzerland). At the outlet, the gas phase was connected to a vapor condenser (SNC-Lavalin, Brussels, Belgium and Bioprocess Technologies, Madrid, Spain) in order to avoid any liquid loss. The vapor condenser was connected to a chiller water machine where the set-point was kept at 4 °C and condensate was fed back to the bioreactor.
2.3. Bioreactor Disassembly
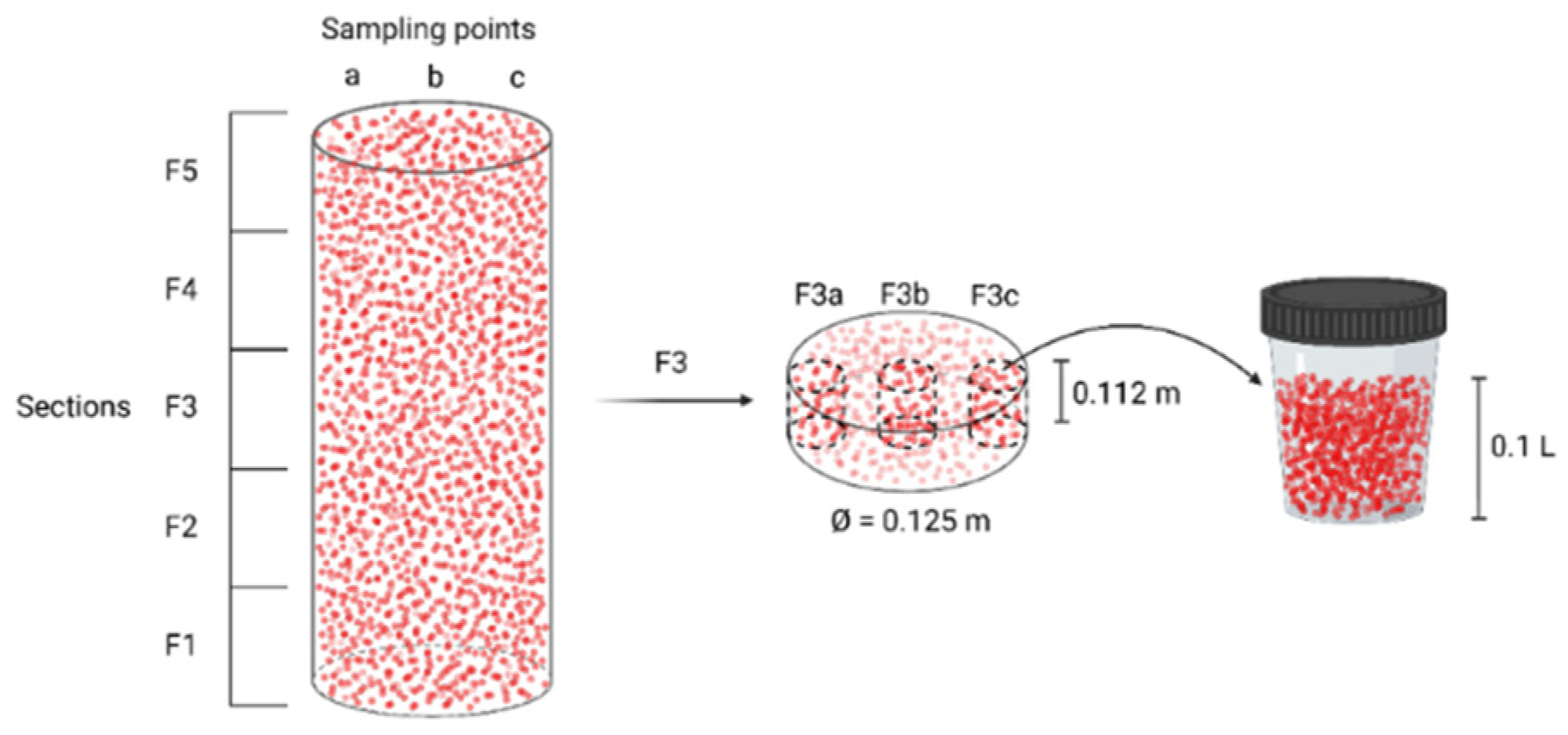
Collection of the samples was performed using sterile material, but the bioreactor was opened to the air, when stopping it after 840 days of continuous operation. The packed-bed was divided vertically in five different sections (F1-F2-F3-F4-F5), each divided in three radial sampling points (a, b, c) (Figure 2). The height of each vertical section was set at 0.112 m. From each sampling point, a total amount of 0.1 L beads were collected. In addition, one 0.1 L sample (F0) below the packed-bed was taken. After collection, each 0.1 L sample was split into two. One part was flash-frozen in liquid N2 and stored at −80 °C, and the second was submerged in phosphate-buffered saline (PBS) and stored at 4 °C.
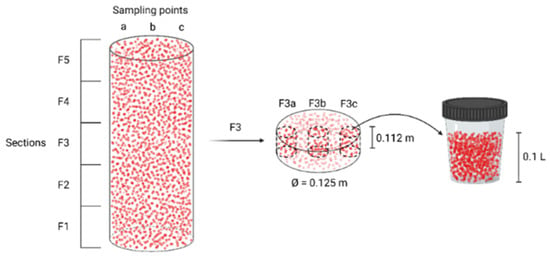
Figure 2.
Division of the packed-bed into different sections for sampling. The example of fraction F3c is detailed.
2.4. Processing of Bioreactor Samples
A volume of 3 to 5 mL packed-beads was taken out of the frozen stock at −80°C and transferred to a 15 mL tube. All tubes were weighed before and after the collection of the beads. In total, 8 ml of cold PBS was added to the beads and vortexed. In order to remove the Biostyr® beads (Veolia Water Technologies, Saint-Maurice, France), which are less dense than water, the tubes were centrifuged for 5 min at 4 °C and 5000× g and the beads on top of the liquid suspension were removed with a sterile spatula. The pellet was resuspended and fractionated in 2 mL collection tubes. Finally, the 2 mL suspensions were centrifuged for 2 min at 13,000× g, the supernatants were discarded and the pellets flash-frozen in liquid nitrogen before it was stored in −80° for further analysis.
2.4.1. ATP Measurements
Samples stored at 4 °C were used to measure ATP. Two mL of sample of the packed-beads and biofilm was taken and 2 mL of PBS was added and vortexed. In total, 50 µL of this cell suspension was used as a sample for ATP measurement (BioThema Intracellular ATP Kit, Handen, Sweden). Equivalent active cells (EAC) were calculated from the ATP concentrations, assuming 10−18 mol ATP equals 1 EAC, following the manufacturer’s instructions.
2.4.2. qPCR and 16S rRNA Gene Amplicon Sequencing
DNA was extracted from the frozen 2 mL pellets with the QiaAmp DNA mini kit (Qiagen, Antwerp, Belgium). A final concentration of DNA was measured with QuantiFluor® ONE ds DNA System (Promega, Leiden, The Netherlands) in a multiwell using the Clariostar (BMG Labtech, De Meern, The Netherlands). Quantitative PCR (qPCR) with 16S rRNA gene-targeting primers (910F/1141R, Neu1265F/Neu1422R, and Nwi70F/Nwi165R, see [25]) was performed on a Rotorgene Q (Qiagen, Antwerp, Belgium). A reaction mixture of 20 µL was prepared for each sample, containing 10 µL 2× reaction buffer (QuantiNova Sybr Green RT-PCR, Qiagen, Antwerp, Belgium), 1 µL primer forward (10 µM), 1 µL primer reverse (10 µM), 7 µL RNase free water, and 1 µL of template (<100 ng/µL). The genomic DNA standards, the samples, and non-template controls were treated similarly and all performed in duplicate. The amplification program was 5 min at 95 °C, followed by 35 cycles of 1 min at 95 °C, 1 min at 56 °C and 1 min at 72 °C. The 2−ΔΔCT method was used to relatively quantify N. europaea and N. winogradskyi.
Next to qPCR, 16S rRNA gene amplicon sequencing was performed. High-throughput amplicon sequencing of the V3–V4 hypervariable region was performed with the Illumina MiSeq platform (V3 chemistry paired-end), according to the manufacturer’s guidelines at BaseClear (Leiden, The Netherlands). Data were processed using the OCToPUS pipeline (V1.0, SCK CEN, Mol, Belgium) [34]. For each operational taxonomic unit (OTU), the relative abundance was calculated based on 3% dissimilatory cut-off.
2.4.3. Metaproteomic Analysis
A frozen 2 mL pellet, wet mass of ca. 125 mg, was dissolved in 1 mL 6M guanidine HCl (in K2HPO4 buffer 20 mM (pH 7.5)). Samples were lysed by ultrasonication (3 × 20 s, amplitude 40%, 1 cycle, in between 1 min on ice) (Imlab, Boutersem, Belgium). The concentration of the samples was measured using the Bradford method (Bio-Rad Protein Assay, Bio-Rad, Temse, Belgium) following precipitation with acetone (1:4 v/v cold acetone and incubation at −20 °C for 1 h).
LC-MS/MS analysis was performed using a nanoElute UHPLC (Bruker Daltonics, Bremen, Germany) connected to a QTOF-MS instrument (Impact II, Bruker Daltonics, Germany) via a CaptiveSpray nanoflow electrospray source (Bruker Daltonics, Bremen, Germany). Samples were run in a random order in order to identify a potential batch effect. In total, 2 µg of tryptic digest (in 4% acetonitrile/solvent A) was injected onto a trapping column setup (300 µm × 5 mm, C18 PepMap 300, 5 µm, 300 Å; Bruker Daltonics, Bremen, Germany) using a flow rate of 200 µL/min. Subsequently, peptides were separated using a C18 Reprosil AQ, 1.9 µm, 120 Å, 0.075 × 400 mm column operated at 40 °C (Bruker Daltonics, Bremen, Germany) at a flow rate of 0.2 µL/min. Gradient conditions were: 2–35% solvent B for 100 min; 35–95% solvent B for 10 min; and 95% solvent B held for 10 min (solvent A, 0.1% formic acid in water; solvent B, 0.1% formic acid in acetonitrile). Drying gas flow and temperature of the CaptiveSpray were set to 4 L/min and 180 °C, respectively, and nebulizer gas pressure was set to 0.4 bar. MS acquisition rate was set to 2 Hz and data have been acquired over a 150–2200 m/z mass range. In all the full-scan measurements, a lock-mass (m/z 1221.9906, Hexakis (1H, 1H, 4H-hexafluorobutyloxy)phosphazine) (Bruker Daltonics, Bremen, Germany) was used as internal calibrator.
Instant Expertise method (Compass otofSeries 4.1, Bruker Daltonics, Bremen, Germany) was used to select as many as possible of the most intense ions per cycle of 3 s MS/MS accumulation depended on the MS1 level. Threshold (per 1000 summation) absolute was 2500 cts (spectral rate of 2 Hz). Peptide fragmentation was performed with nitrogen gas on the most abundant and at least doubly charged to five charged ions detected in the initial MS scan. Active exclusion was performed after 1 spectrum for 0.50 min unless the intensity of the precursor ions was more than 3 times higher than in the previous scan. The mass spectrometry proteomic data have been deposited with the ProteomeXchange Consortium [35] via the PRIDE [36] partner repository with the dataset identifier PXD030632.
All MS/MS spectra were searched against the All entries database in SwissProt using the Mascot v2.6.0 algorithm (http://www.matrixscience.com/, accessed on 15 November 2021) with the following settings: database: swissprot_2019_03; enzyme: trypsin; variable modifications: oxidation (M); fixed modifications: carbamidomethyl (C); missed cleavages: 1; taxonomy: Bacteria (Eubacteria) or an in-house combined Nitrosomonas.ATCC 19718/Nitrobacter.ATCC 25391 database when specified; instrument type CID: ESI-QUAD-TOF; peptide tolerance: 15 ppm; MS/MS tolerance: 0.02 Da; peptide charge: 1+, 2+ and 3+; minimum peptide length: 5; peptide decoy: ON; adjust FDR (%): 1; ion score cut-off: 15; ion score threshold for significant peptide IDs: 35; percolator: ON. Proteins identified with only one peptide were rejected for data interpretation.
Species-specific protein abundance was derived from the exponentially modified Protein Abundance Index (emPAI), which represents the number of identified peptides divided by the number of theoretical tryptic peptides [37,38]. This index was directly calculated by the Mascot research server, but had to be normalized from one experiment to another to avoid bias inherent to data-based acquired spectra (i.e., sample preparation, peptides ionization). This was achieved by calculating the following percentage [38]: Normalized emPAI = (emPAIprotein/emPAItotal) × 100. Where emPAIprotein and emPAItotal are the emPAI for a given protein and the sum of the emPAI of all the identified protein within a given experiment, respectively.
3. Results and Discussion
3.1. Nitrifying Packed-Bed Operation
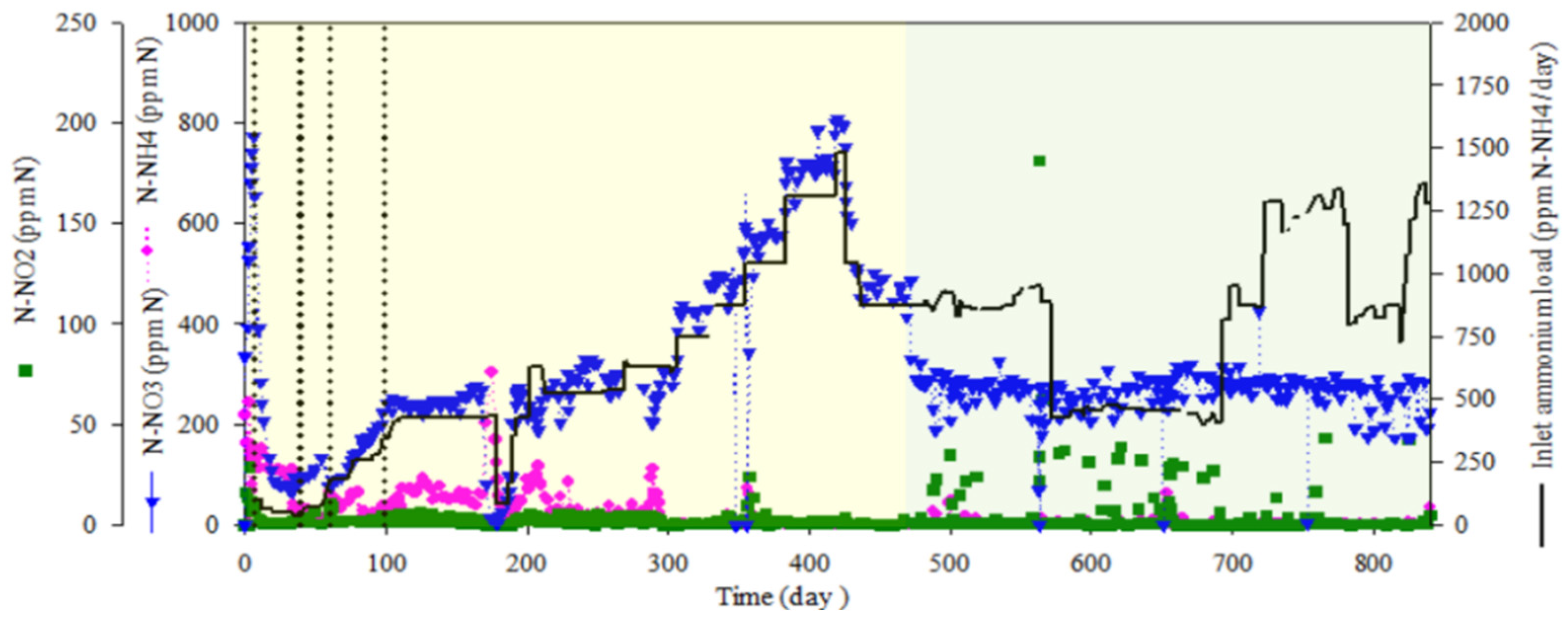
The complete bioreactor operation, including nitrogen species profiles and ammonium load fed into the bioreactor, is presented in Figure 3. After the inoculation of the packed-bed bioreactor, a batch mode phase was maintained for five days. Pulses of concentrated ammonium (N-NH4+; 100 g × L−1) up to a final target concentration of 100 mg N-NH4+·L−1 in the bioreactor were added when depleted. After five days in batch phase, a continuous culture mode was established in the packed-bed reactor at a hydraulic residence time (HRT) of 80 h for 30 days. HRT was stepwise decreased from 80 to 60, 40 and, finally, to 14 h on day 60. From day 60 to day 435, ammonium was progressively increased until a maximum of 1478 mg N-NH4+·L−1·day−1 by increasing the ammonium concentration in the inlet medium. From day 435 to 475, the ammonium load was progressively decreased to 860 mg N-NH4+·L−1·day−1 by decreasing the ammonium concentration in the inlet medium in order to prepare the next experimental step. From day 475 until the end of the test, the system was fed with different ammonium loads by means of reducing HRT from 14 h to 6 h, reaching phases where the ammonium load was at 1285 mg N-NH4·L−1·day−1 (Table 1). Complete nitrification was obtained from day 300, when an inlet ammonium load of 750 mg N-NH4+·L−1·day−1 was fed into the bioreactor. From day 300 to the end of the test, 99% ammonium removal was obtained with no nitrite accumulation, indicating a full nitrification result. The major achievement of the test was maintaining continuous full nitrification over 1.5 years, where the nitrification rate was recovered very fast after occasional NH4+/NO2− accumulation events due to operational changes. Additionally, the bioreactor was exposed to different ammonium loads, by changing ammonium concentration in the inlet media or HRT, and the system was able to achieve 99% ammonium removal with a rapid adaptation to the new conditions whatever the load was tested, indicating robust biofilm formation. The temperature was maintained at 30 °C. The pH was initially set to 7.5 and increased to 8.1 when the packed-bed was fully colonized. Recirculation liquid flow from the top to the bottom section of the bioreactor was maintained at 108 L·day−1 to improve the homogeneity of the liquid phase.
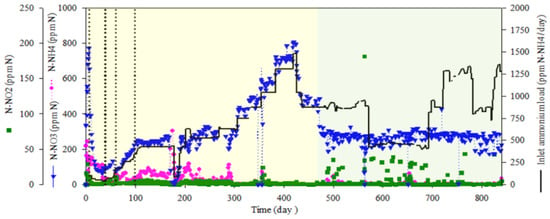

Table 1.
Hydraulic residence time (HRT) and ammonium load during the different operation periods of the nitrifying bioreactor.
3.2. Cell Abundance in the Biofilm Biomass
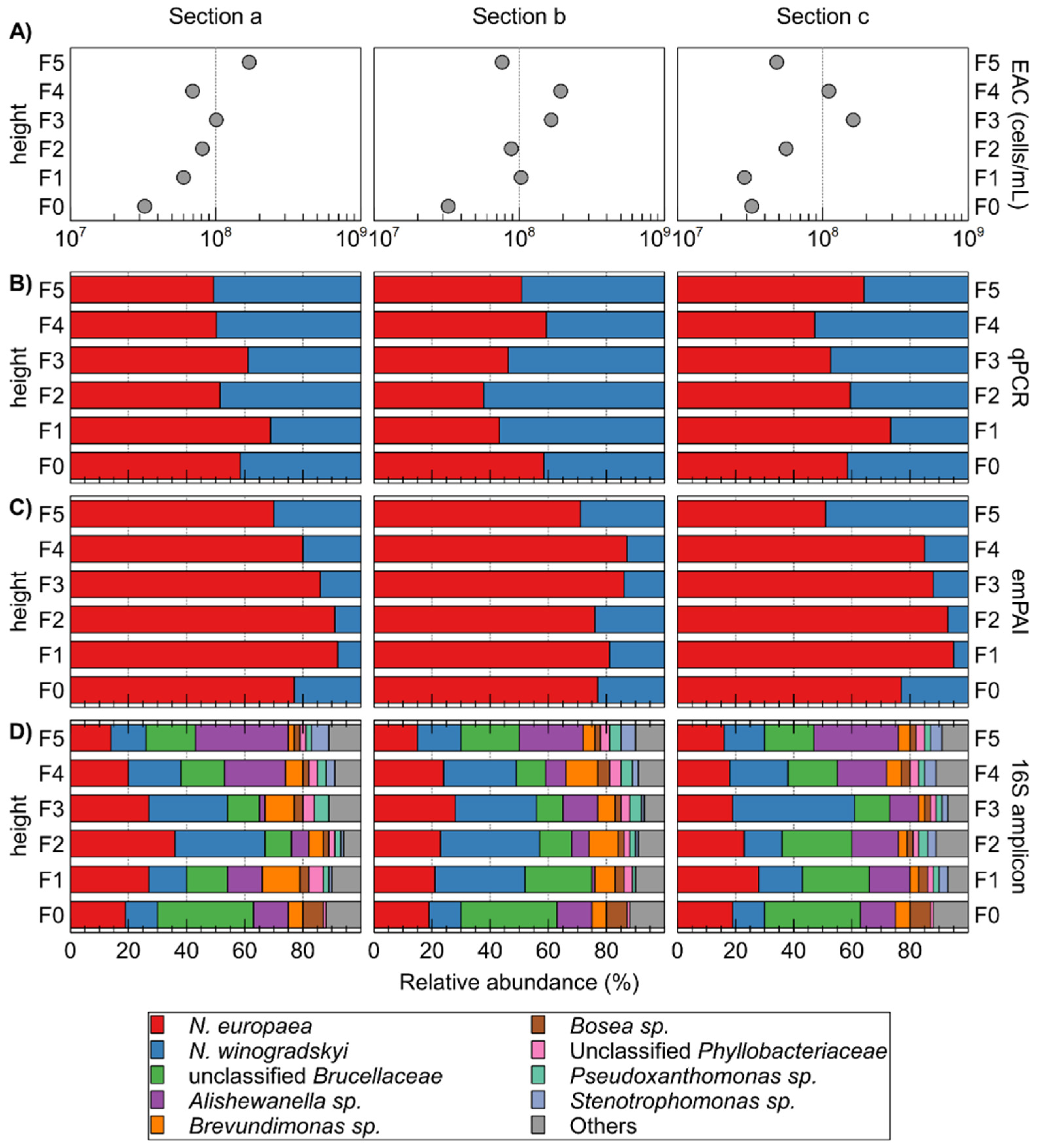
The total number of cells within the different sections, based on ATP measurements, fluctuated around 8.95 ± 5.10 × 107 cells/mL beads, with generally less biomass in the lower sections (Figure 4). Although these results are not directly comparable with the weight-based biomass determination by Montras et al. [25], it indicates that the cell numbers/biomass profile along the vertical axis varies from operation to operation.
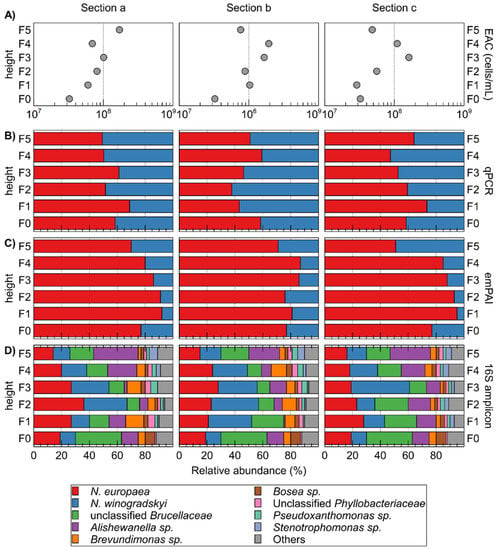
Figure 4.
Characterization of different sections (five vertical sections, each divided into three horizontal sections a, b, and c) of the packed-bed based on ATP-measured equivalent active cells (EAC) counts (A); qPCR- (B); and emPAI-based (C) determination of the relative abundance of N. europaea versus N. winogradskyi; and relative abundance of OTUs in 16S rRNA gene amplicons (D). Relative abundance N. europaea versus N. winogradskyi based on 16S rRNA gene amplicons is also plotted (cross) on qPCR panels (B).
3.3. Relative Abundance of N. europaea and N. winogradskyi
The relative abundance of autotrophic bacteria N. europaea and N. winogradskyi was calculated using qPCR (Figure 4). It is noteworthy that the extracted amount of DNA was roughly similar for each sample with 18.3 ± 3.7 ng µL−1. N. europaea was almost always more abundant than N. winogradskyi, except for fractions F1b, F2b, F3b, F4c, and F5a. However, with an average relative abundance of 55% for N. europaea and 45% for N. winogradskyi, this difference was not pronounced and with standard deviations ranging from to 7.5 to 16.3% in the different horizontal sections, heterogeneity within a horizontal section was observed. Nevertheless, as N. europaea converts ammonium to nitrite, a more abundant population would provide the necessary growth substrate for N. winogradskyi. Furthermore, the gradual increase of N. winogradskyi with increasing height was much less pronounced than observed by Montras et al. [25]. Differences in bioreactor operation conditions could possibly be the cause for these observations. In the last 5 months of its operation, the Montras bioreactor ammonium load at the inlet was 0.22 g N-NH4+·L−1·day−1 [25] while in our experiment, the ammonium load fluctuated between 0.3–1.3 g N-NH4+·L−1·day−1. Hence, a higher ammonium concentration will be available higher in the reactor, causing N. europaea to be able to proliferate more and increasing its relative abundancy in comparison to N. winogradskyi at increased height along the bioreactor.
3.4. Composition of the Microbial Community
Based on 16S amplicons, 28 OTUs were identified next to N. europaea and N. winogradskyi. The latter constituted overall 44.07 ± 11.75% of the reads (Figure 4D; Supplementary Table S1). The N. europaea and N. winogradskyi ratio correlated well (except for section F3c) with qPCR results (Figure 4B). Next to N. europaea and N. winogradskyi, the most abundant OTUs belonged to unclassified Brucellaceae, Alishewanella sp., Brevundimonas sp., Bosea sp., unclassified Phyllobacteriaceae, Pseudoxanthomonas sp., and Stenotrophomonas sp. (Figure 3). Montras et al. [25] showed a limited presence of γ-Proteobacteria (less than 3%) in their nitrifying bioreactor, whereas, in our case, the γ-proteobacterial Alishewanella sp. OTU was more abundant (15.10 ± 8.13%).
3.5. Metaproteomics
3.5.1. Database Search Restricted to Nitrosomonas and Nitrobacter
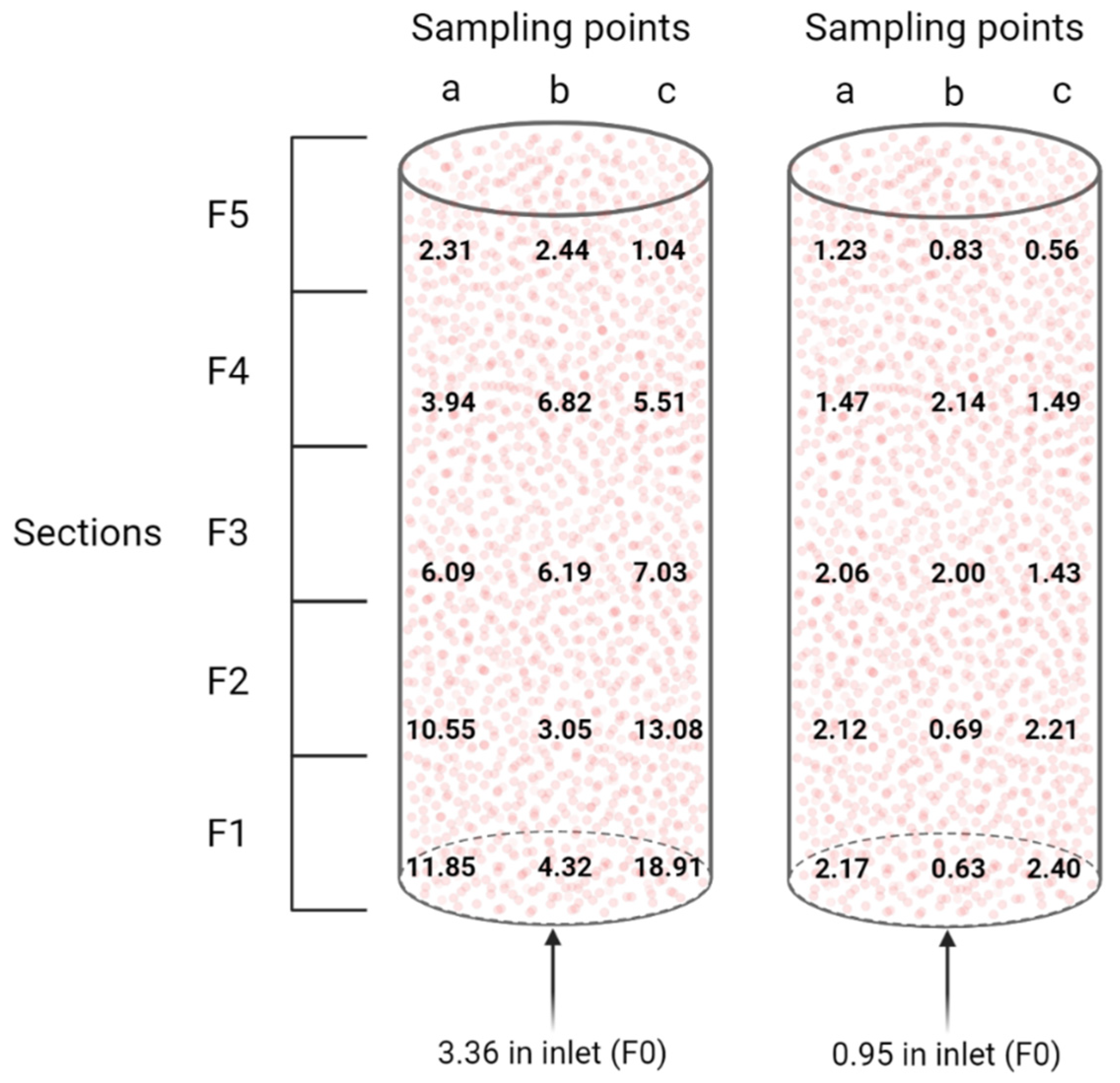
The total number of proteins identified with at least two peptides when using an in-house combined N. europaea (2524 CDS)/N. winogradskyi (3609 CDS) database, as well as the percentage of N. europaea and N. winogradskyi proteins in terms of specific protein abundance, are reported in Table 2. The relative abundance based on normalized emPAI showed the same trend as for the qPCR approach, i.e., N. europaea mostly outnumbering N. winogradskyi. More specifically, N. europaea constituted on average 82 ± 11% based on protein levels. However, looking at the samples from the periphery, namely “a” and “c”, N. europaea was less abundant compared to N. winogradskyi when moving from the bottom to the top of the nitrifying bioreactor, whereas the center of the packed-bed appeared less prone to large variations (Figure 5). In other words, in the bottom of the bioreactor, where ammonium is the main nitrogen compound, N. europaea proteins were more abundant. Then, nitrite is produced, mixed within the bioreactor, and transported towards the middle and the upper parts of the packed-bed. The later zones included the highest abundance of N. winogradskyi proteins. We do not have a clear explanation for this phenomenon since the liquid phase recirculation provided adequate mixing and, therefore, no local gradients would be expected favoring differences in relative biomass abundance. It is noteworthy that the discrepancy between N. europaea and N. winogradskyi abundances estimated by a DNA-based (55:45) and protein-based (82:18) approach urges a cautious interpretation of bioreactor functional characterization based on a single method.
Table 2.
The total number of proteins identified in each section of the nitrifying reactor with at least two peptides using an in-house combined N. europaea ATCC 19718 (Ns)/N. winogradskyi ATCC 25391 (Nb) database. For each section, the Ns and Nb protein percentages based on normalized emPAI are indicated.
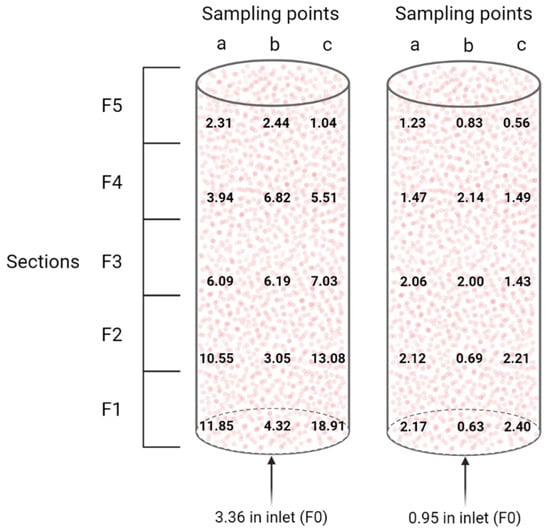
Figure 5.
Visualization of the N. europaea to N. winogradskyi ratio (left) and the N. europaea and N. winogradskyi to other bacteria ratio (right) distribution inside the packed-bed nitrifying bioreactor, based on normalized emPAI proteomic data.
More recently, Perez et al. [27] determined the relative cell densities of N. europaea and N. winogradskyi grown in a suspension co-culture in mineral medium in a continuous chemostat. By means of a multi-level approach, including ammonium- and nitrite-dependent oxygen uptake, as well as qPCR and fluorescence in situ hybridization, they reported a ratio of approximately 4:1, i.e., 80% N. europaea and 20% N. winogradskyi in suspension at steady-state. This is consistent with the ratios that we reported, based on proteomic data, for the major part of the nitrifying reactor. While for sections F1a, F1c, F2a, and F2c, there is a clear “overpopulation” of N. europaea, and in all sections F5, an “overpopulation” of N. winogradskyi. This could possibly be correlated to localized accumulation of ammonium and nitrite, respectively.
Regarding the nitrifying process, AmoB of N. europaea (NE0943/NE2062), beta subunit of the enzyme catalyzing the first and rate-limiting step in nitrification, i.e. the oxidation of ammonia to hydroxylamine, was detected in all sections and it was the most abundant in section F0, where the fresh NH4+ solution is fed to the nitrifying bioreactor (Figure 1B). N. europaea HAO enzyme (NE20444/NE09624/NE23394), catalyzing the second step in nitrification, i.e., oxidation of hydroxylamine to nitrite, was also detected in all sections of the bioreactor with a very similar abundance index (Table 3). During the final step, nitrite is converted to nitrate by nitrite oxidoreductase. Performing manual curation using the MaGe platform [39], Q3SUK0 and Q3SU14 annotation in N. winogradskyi could be identified as nitrite oxidoreductase beta subunit (encoded by the nxrB gene). The highest abundance of the latter protein is mostly associated with the highest general abundance of N. winogradskyi along the different sections of the bioreactor (i.e., F1b, F2b, F3a, F4c, and F5c in Figure 5 and Table 3).
Table 3.
Most abundant proteins in regard to the sample section (F0–F5) and position (a, b, c) in the nitrifying bioreactor expressed in terms of normalized emPAI proteomic analysis. In bold, the highest abundance detected for the protein of interest was used as ranking parameter. ND: not detected. Protein database search restricted to N. europaea ATCC 19718 (Ns) and N. winogradskyi ATCC 25391 (Nb).
In addition to proteins involved in the nitrification process, NorQ protein involved in nitrate reduction could be identified in 14 sections out of 16 of the packed-bed (Q82TG8, Table 3). Furthermore, a certain level of nitrite reductase, aniA/nirK, was also found in both N. europaea (Q82VX5, Table 3) and, to a lower extent, (emPAI mostly below 0.1) in N. winogradskyi (Q3SP9, Supplementary Table S2). Taking into account that oxygen is injected into the lower section of the bioreactor and that the pO2 set-point at the top section is 80%, the aeration cannot be considered suboptimal, but anaerobic sections may exist along the packed-bed because of biofilm density that may be associated with localized denitrification activity. However, NirK in N. europaea has also been reported to reduce sensitivity of N. europaea to accumulation of NO2− and, more importantly, could be acting as an electron sink during ammonium oxidation and preventing auto-oxidation of NH2OH, allowing for a more efficient nitrification activity. Hence, it plays a significant role in the central nitrogen metabolism of N. europaea [41,42]. Moreover, it is not essential in its denitrification pathway [43]. As a consequence, the presence of N. europaea NirK protein in the bioreactor does not necessarily indicate denitrification activity. This is supported by the fact that no denitrification was observed in the bioreactor overall behavior.
Notably, the most abundant protein from N. winogradskyi was Nwi_1037, a very small protein (55 amino acids) that was detected for the first time using mass spectrometry, but for which no function could be identified so far (Table 3). For N. europaea, the protein encoded by NE1907 was also identified for the first time by mass spectrometry. Further sequence analysis using the MaGe platform [39] allowed us to assign a putative role in membrane permeability.
3.5.2. Database Search Broadened to Bacteria (Eubacteria)
Broadening the protein database search to bacteria (eubacteria) allowed us to detect proteins from other bacteria next to N. europaea and N. winogradskyi. Proteins from this pair represented 57.53 ± 12.04% of the total identified proteins (Supplementary Table S3). Therefore, the relative protein abundance was significantly higher than the relative species abundance, indicating a higher metabolic activity. Although in some apical (F5b, F5c) and basal (F0, F1b, F2b) fractions, non-Nitrosomonas/Nitrobacter proteins were more abundant (Figure 5). Interestingly, several proteins related to denitrification processes taking place in anaerobic conditions were identified, i.e., pseudoazurin from Alcaligenes faecalis, respiratory nitrate reductase beta chain from Bradyrhizobium sp., the CbbQ protein from Hydrogenophilus thermoluteolus that has a high similarity to NirQ/NorQ in denitrifying bacteria, nitrous-oxide reductase from Brucella melitensis and Paracoccus denitrificans, and copper-containing nitrite reductase, also from A. faecalis. Pseudoazurin was the second most abundant non-Nitrosomonas/Nitrobacter protein. It is induced in the presence of nitrite and serves as a direct electron donor to nitrite reductase, under anaerobic growth conditions [44]. Moreover, the related UniProt Cluster ID (UniRef100_P04377) reports pseudoazurin from Ochrobactrum sp. And, more specifically, O. anthropi as a 100% identical protein. Most likely this abundant protein belongs to the unclassified_Brucellaceae group (OTU4) reported above in the 16S amplicon sequencing section, because Ochrobactrum and Brucella cannot be discriminated based on 16S (i.e., 100% sequence identity with O. anthropi ATCC 49188).
However, these proteins have normalized emPAI values below 1 (or below 0.5 when considering only the packed-bed) (Table 4), while the AmoB protein from N. europaea in section F0 has a normalized emPAI value of 7.18 (and 6.34 in section F1b) (Supplementary Table S3). Therefore, the possible denitrification activity originating from the contaminants appeared to be much lower than the nitrification process, and, in any case, no denitrification was detected at the macroscopic level during the 840 days operation data (see Section 3.5.1). Next to these proteins, the major carboxysome shell protein CsoS1A was the most abundant non-Nitrobacter/Nitrosomonas protein and was found in section F0. It belongs to the sulfur bacterium Halothiobacillus neapolitanus, a chemolithoautotroph just like nitrifiers, so they may enter in competition for inorganic carbon.
Table 4.
Most abundant proteins not belonging to N. europaea or N. winogradskyi in regard to the sample section (F0–F5) and position (a, b, c) in the nitrifying bioreactor and expressed in terms of normalized emPAI proteomic analysis. In bold, the highest abundance detected for the protein of interest was used as ranking parameter. ND: not detected. Protein database search opened to eubacteria.
Overall, the most abundant proteins identified after 840 days of continuous operation indicated that nitrification is the major metabolic process taking place inside the nitrifying bioreactor, in agreement with the operational data showing full nitrification over the course of the experiment. While the bioreactor was originally inoculated with an axenic consortium, the heterotrophic community could reach around 50% of the total population after such a long operation period. However, this did not prevent the bioreactor to reach full nitrification, which is in accordance with Sedlacek et al. (2016) who reported heterotrophic bacteria (Alphaproteobacteria, Betaproteobacteria, Gammaprotebacteria and Bacteriodetes) to increase the abundancy of proteins related to the ammonia oxidation pathway and reduce stress response protein content in batch co-cultures with Nitrosomonas sp. Is79 [45].
4. Conclusions
Our experimental packed-bed bioreactor was exposed to different ammonium loads and hydraulic residence times over a prolonged period and could achieved full nitrification over a period of about 1.5 years, indicating robust and steady nitrifying activity. This is crucial from the BLSS point of view for resource recovery, when considering long-term space missions without possible resupply from Earth. Surprisingly, dismantling after 840 days of autotrophic continuous cultivation showed that N. europaea and N. winogradskyi constituted roughly only half of the total microbial population. Nevertheless, the heterotrophic population did not appear to affect the bioreactor’s nitrification function. Interestingly, although DNA-based abundance estimates showed that N. europaea was slightly more abundant than N. winogradskyi, protein-based abundance estimates indicated a much higher abundance of N. europaea. The latter indicates that the relationships between gene abundances and metabolic processes need to be further studied to better link genomic data with metabolic processes in the nitrifying bioreactor. Lastly, our proteomic approach also revealed that the central part of the bioreactor has the most stable nitrifier composition. This will be further investigated in order to improve the design of future nitrifying packed-bed bioreactor to avoid potential detrimental localized nutrient accumulation in the periphery, especially in the bottom and top sections of the bioreactor.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/applmicrobiol2010020/s1: Table S1: Identification and relative abundance of OTUs derived from 16S rRNA amplicon sequencing; Table S2: Protein identification and emPAI-based quantification derived from metaproteome analysis with protein database search restricted to N. europaea ATCC 19718 and N. winogradskyi ATCC 25391; Table S3: Protein identification and emPAI-based quantification derived from metaproteome analysis with protein database search opened to Eubacteria.
Author Contributions
Conceptualization, F.M., C.A. and R.V.H.; methodology, F.M., C.A. and R.V.H.; software, F.M. and M.M.; validation, F.M., C.A., T.V. and R.V.H.; formal analysis, F.M., C.A., T.V. and R.V.H.; data curation, F.M., C.A., T.V. and R.V.H.; writing—original draft preparation, F.M., C.A., T.V. and R.V.H.; writing—review and editing, F.M., C.A., T.V. and R.V.H.; visualization, F.M., C.A., T.V. and R.V.H.; supervision, F.G. and N.L.; project administration, F.G. and N.L.; funding acquisition, F.G. All authors have read and agreed to the published version of the manuscript.
Funding
The MELiSSA Pilot Plant is funded from ESA contributions from Spain (main contributor), Belgium, France, Italy, and Norway, under Frame Contract C4000109802/13/NL/CP. Co-funding from Ministerio de Ciencia e Innovación (RD 788/2020).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The 16S rRNA amplicon dataset generated and analyzed during the current study is available in the NCBI Sequence Read Archive (SRA) repository (PRJNA788718), https://www.ncbi.nlm.nih.gov/sra/ (accessed on 15 November 2021). The mass spectrometry proteomics data have been deposited with the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD030632.
Acknowledgments
MELiSSA is an international consortium of 15 partners led by the European Space Agency (for information: https://www.melissafoundation.org, accessed on 15 November 2021). Its activities are governed by a Memorandum of Understanding (ESA 4000100293/10/NL/PA). Generalitat de Catalunya and Universitat Autònoma de Barcelona are also acknowledged. We are also thankful to Ann Janssen and Ilse Coninx for providing technical support. Figure 1B, Figure 2, and Figure 5 were created using BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Anderson, M.S.; Ewert, M.K.; Keener, J.F. Life Support Baseline Values and Assumptions Document; NASA/TP-2015-218570; NASA: Houston, TX, USA, 2018.
- Pickett, M.T.; Roberson, L.B.; Calabria, J.L.; Bullard, T.J.; Turner, G.; Yeh, D.H. Regenerative water purification for space applications: Needs, challenges, and technologies towards ‘closing the loop’. Life Sci. Space Res. 2020, 24, 64–82. [Google Scholar] [CrossRef] [PubMed]
- Volpin, F.; Badeti, U.; Wang, C.; Jiang, J.; Vogel, J.; Freguia, S.; Fam, D.; Cho, J.; Phuntsho, S.; Shon, H.K. Urine Treatment on the International Space Station: Current Practice and Novel Approaches. Membranes 2020, 10, 327. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, Z.; Abney, M.; Brown, B.; Fox, E.; Stanley, C. State of NASA Oxygen Recovery. In Proceedings of the 48th International Conference on Environmental Systems, Albuquerque, NM, USA, 8–12 July 2018. [Google Scholar]
- Verbeelen, T.; Leys, N.; Ganigue, R.; Mastroleo, F. Development of Nitrogen Recycling Strategies for Bioregenerative Life Support Systems in Space. Front. Microbiol. 2021, 12, 700810. [Google Scholar] [CrossRef] [PubMed]
- Lasseur, C.; Brunet, J.; De Weever, H.; Dixon, M.; Dussap, G.; Godia, F.; Leys, N.; Mergeay, M.; Van Der Straeten, D. MELiSSA: The European Project of a Closed Life Support System. Gravit. Space Biol. 2010, 23, 3–12. [Google Scholar]
- Hendrickx, L.; De Wever, H.; Hermans, V.; Mastroleo, F.; Morin, N.; Wilmotte, A.; Janssen, P.; Mergeay, M. Microbial ecology of the closed artificial ecosystem MELiSSA (Micro-Ecological Life Support System Alternative): Reinventing and compartmentalizing the Earth’s food and oxygen regeneration system for long-haul space exploration missions. Res. Microbiol. 2006, 157, 77–86. [Google Scholar] [CrossRef]
- Hendrickx, L.; Mergeay, M. From the deep sea to the stars: Human life support through minimal communities. Curr. Opin. Microbiol. 2007, 10, 231–237. [Google Scholar] [CrossRef]
- Mastroleo, F.; Van Houdt, R.; Leroy, B.; Benotmane, M.A.; Janssen, A.; Mergeay, M.; Vanhavere, F.; Hendrickx, L.; Wattiez, R.; Leys, N. Experimental design and environmental parameters affect Rhodospirillum rubrum S1H response to space flight. ISME J. 2009, 3, 1402–1419. [Google Scholar] [CrossRef] [Green Version]
- Paradiso, R.; De Micco, V.; Buonomo, R.; Aronne, G.; Barbieri, G.; De Pascale, S. Soilless cultivation of soybean for Bioregenerative Life-Support Systems: A literature review and the experience of the MELiSSA Project—Food characterisation Phase I. Plant Biol. 2014, 16 (Suppl. 1), 69–78. [Google Scholar] [CrossRef]
- Alemany, L.; Peiro, E.; Arnau, C.; Garcia, D.; Poughon, L.; Cornet, J.F.; Dussap, C.G.; Gerbi, O.; Lamaze, B.; Lasseur, C.; et al. Continuous controlled long-term operation and modeling of a closed loop connecting an air-lift photobioreactor and an animal compartment for the development of a life support system. Biochem. Eng. J. 2019, 151, 107323. [Google Scholar] [CrossRef]
- Poughon, L.; Laroche, C.; Creuly, C.; Dussap, C.G.; Paille, C.; Lasseur, C.; Monsieurs, P.; Heylen, W.; Coninx, I.; Mastroleo, F.; et al. Limnospira indica PCC8005 growth in photobioreactor: Model and simulation of the ISS and ground experiments. Life Sci. Space Res. 2020, 25, 53–65. [Google Scholar] [CrossRef]
- Albiol, J.; Gòdia, F.; Luis Montesinos, J.; Pérez, J.; Vernerey, A.; Cabello, F.; Creus, N.; Morist, A.; Mengual, X.; Lasseur, C. Biological Life Support System Demostration Facility: The Melissa Pilot Plant; SAE Technical Paper 2000-01-2379; SAE: Warrendale, PA, USA, 2000. [Google Scholar]
- Gòdia, F.; Albiol, J.; Perez, J.; Creus, N.; Cabello, F.; Montras, A.; Masot, A.; Lasseur, C. The MELISSA pilot plant facility as an integration test-bed for advanced life support systems. Adv. Space Res.-Ser. 2004, 34, 1483–1493. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gragera, D.; Arnau, C.; Peiro, E.; Dussap, C.G.; Poughon, L.; Gerbi, O.; Lamaze, B.; Lasseur, C.; Godia, F. Integration of Nitrifying, Photosynthetic and Animal Compartments at the MELiSSA Pilot Plant. Front. Astron. Space 2021, 8, 750616. [Google Scholar] [CrossRef]
- Clauwaert, P.; Muys, M.; Alloul, A.; De Paepe, J.; Luther, A.; Sun, X.; Ilgrande, C.; Christiaens, M.E.R.; Hu, X.; Zhang, D.; et al. Nitrogen cycling in Bioregenerative Life Support Systems: Challenges for waste refinery and food production processes. Prog. Aerosp. Sci. 2017, 91, 87–98. [Google Scholar] [CrossRef]
- Heinonen-Tanski, H.; van Wijk-Sijbesma, C. Human excreta for plant production. Bioresour. Technol. 2005, 96, 403–411. [Google Scholar] [CrossRef]
- Chain, P.; Lamerdin, J.; Larimer, F.; Regala, W.; Lao, V.; Land, M.; Hauser, L.; Hooper, A.; Klotz, M.; Norton, J.; et al. Complete genome sequence of the ammonia-oxidizing bacterium and obligate chemolithoautotroph Nitrosomonas europaea. J. Bacteriol. 2003, 185, 2759–2773. [Google Scholar] [CrossRef] [Green Version]
- Caranto, J.D.; Lancaster, K.M. Nitric oxide is an obligate bacterial nitrification intermediate produced by hydroxylamine oxidoreductase. Proc. Natl. Acad. Sci. USA 2017, 114, 8217–8222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starkenburg, S.R.; Chain, P.S.G.; Sayavedra-Soto, L.A.; Hauser, L.; Land, M.L.; Larimer, F.W.; Malfatti, S.A.; Klotz, M.G.; Bottomley, P.J.; Arp, D.J.; et al. Genome sequence of the chemolithoautotrophic nitrite-oxidizing bacterium Nitrobacter winogradskyi Nb-255. Appl. Environ. Microbiol. 2006, 72, 2050–2063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, I.; van Spanning, R.J.M.; Jetten, M.S.M. Denitrification and ammonia oxidation by Nitrosomonas europaea wild-type, and NirK- and NorB-deficient mutants. Microbiology 2004, 150, 4107–4114. [Google Scholar] [CrossRef] [Green Version]
- Freitag, A.; Rudert, M.; Bock, E. Growth of Nitrobacter by Dissimilatoric Nitrate Reduction. FEMS Microbiol. Lett. 1987, 48, 105–109. [Google Scholar] [CrossRef]
- Starkenburg, S.R.; Arp, D.J.; Bottomley, P.J. Expression of a putative nitrite reductase and the reversible inhibition of nitrite-dependent respiration by nitric oxide in Nitrobacter winogradskyi Nb-255. Environ. Microbiol. 2008, 10, 3036–3042. [Google Scholar] [CrossRef]
- Perez, J.; Montesinos, J.L.; Albiol, J.; Godia, F. Nitrification by immobilized cells in a micro-ecological life support system using packed-bed bioreactors: An engineering study. J. Chem. Technol. Biotechnol. 2004, 79, 742–754. [Google Scholar] [CrossRef]
- Montras, A.; Pycke, B.; Boon, N.; Godia, F.; Mergeay, M.; Hendrickx, L.; Perez, J. Distribution of Nitrosomonas europaea and Nitrobacter winogradskyi in an autotrophic nitrifying biofilm reactor as depicted by molecular analyses and mathematical modelling. Water Res. 2008, 42, 1700–1714. [Google Scholar] [CrossRef] [PubMed]
- Cruvellier, N.; Poughon, L.; Creuly, C.; Dussap, C.G.; Lasseur, C. High ammonium loading and nitrification modelling in a fixed-bed bioreactor. J. Water Process. Eng. 2017, 20, 90–96. [Google Scholar] [CrossRef]
- Perez, J.; Buchanan, A.; Mellbye, B.; Ferrell, R.; Chang, J.H.; Chaplen, F.; Bottomley, P.J.; Arp, D.J.; Sayavedra-Soto, L.A. Interactions of Nitrosomonas europaea and Nitrobacter winogradskyi grown in co-culture. Arch. Microbiol. 2015, 197, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Lorenzo, C.; Rodriguez-Diaz, M.; Sipkema, D.; Juarez-Jimenez, B.; Rodelas, B.; Smidt, H.; Gonzalez-Lopez, J. Effect of salinity on nitrification efficiency and structure of ammonia-oxidizing bacterial communities in a submerged fixed bed bioreactor. Chem. Eng. J. 2015, 266, 233–240. [Google Scholar] [CrossRef]
- Jeong, D.; Cho, K.; Lee, C.H.; Lee, S.; Bae, H. Effects of salinity on nitrification efficiency and bacterial community structure in a nitrifying osmotic membrane bioreactor. Process Biochem. 2018, 73, 132–141. [Google Scholar] [CrossRef]
- Wang, Y.L.; Niu, Q.G.; Zhang, X.; Liu, L.; Wang, Y.B.; Chen, Y.Q.; Negi, M.; Figeys, D.; Li, Y.Y.; Zhang, T. Exploring the effects of operational mode and microbial interactions on bacterial community assembly in a one-stage partial-nitritation anammox reactor using integrated multi-omics. Microbiome 2019, 7, 122. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Wang, L. Multi-omics analysis reveals structure and function of biofilm microbial communities in a pre-denitrification biofilter. Sci. Total Environ. 2021, 757, 143908. [Google Scholar] [CrossRef]
- Jose, D.; Preena, P.G.; Kumar, V.J.R.; Philip, R.; Singh, I.S.B. Metaproteomic insights into ammonia oxidising bacterial consortium developed for bioaugmenting nitrification in aquaculture systems. Biologia 2020, 75, 1751–1757. [Google Scholar] [CrossRef]
- Salerno, C.; Berardi, G.; Laera, G.; Pollice, A. Functional Response of MBR Microbial Consortia to Substrate Stress as Revealed by Metaproteomics. Microb. Ecol. 2019, 78, 873–884. [Google Scholar] [CrossRef]
- Mysara, M.; Njima, M.; Leys, N.; Raes, J.; Monsieurs, P. From reads to operational taxonomic units: An ensemble processing pipeline for MiSeq amplicon sequencing data. GigaScience 2017, 6, giw017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deutsch, E.W.; Csordas, A.; Sun, Z.; Jarnuczak, A.; Perez-Riverol, Y.; Ternent, T.; Campbell, D.S.; Bernal-Llinares, M.; Okuda, S.; Kawano, S.; et al. The ProteomeXchange consortium in 2017: Supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017, 45, D1100–D1106. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.W.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Mastroleo, F.; Leroy, B.; van Houdt, R.; s’Heeren, C.; Mergeay, M.; Hendrickx, L.; Wattiez, R. Shotgun Proteome Analysis of Rhodospirillum rubrum S1H: Integrating Data from Gel-Free and Gel-Based Peptides Fractionation Methods. J. Proteome Res. 2009, 8, 2530–2541. [Google Scholar] [CrossRef]
- Ishihama, Y.; Oda, Y.; Tabata, T.; Sato, T.; Nagasu, T.; Rappsilber, J.; Mann, M. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell Proteom. 2005, 4, 1265–1272. [Google Scholar] [CrossRef] [Green Version]
- Vallenet, D.; Labarre, L.; Rouy, Z.; Barbe, V.; Bocs, S.; Cruveiller, S.; Lajus, A.; Pascal, G.; Scarpelli, C.; Médigue, C. MaGe: A microbial genome annotation system supported by synteny results. Nucleic Acids Res. 2006, 34, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Meincke, M.; Bock, E.; Kastrau, D.; Kroneck, P.M.H. Nitrite Oxidoreductase from Nitrobacter-Hamburgensis—Redox Centers and Their Catalytic Role. Arch. Microbiol. 1992, 158, 127–131. [Google Scholar] [CrossRef]
- Beaumont, H.J.E.; Lens, S.I.; Westerhoff, H.V.; van Spanning, R.J.A. Novel nirK cluster genes in Nitrosomonas europaea are required for NirK-dependent tolerance to nitrite. J. Bacteriol. 2005, 187, 6849–6851. [Google Scholar] [CrossRef] [Green Version]
- Jason, J.; Cantera, L.; Stein, L.Y. Role of nitrite reductase in the ammonia-oxidizing pathway of Nitrosomonas europaea. Arch. Microbiol. 2007, 188, 349–354. [Google Scholar]
- Kozlowski, J.A.; Price, J.; Stein, L.Y. Revision of N2O-Producing Pathways in the Ammonia-Oxidizing Bacterium Nitrosomonas europaea ATCC. Appl. Environ. Microbol. 2014, 80, 4930–4935. [Google Scholar] [CrossRef] [Green Version]
- Pearson, I.V.; Page, M.D.; van Spanning, R.J.; Ferguson, S.J. A mutant of Paracoccus denitrificans with disrupted genes coding for cytochrome c550 and pseudoazurin establishes these two proteins as the in vivo electron donors to cytochrome cd1 nitrite reductase. J. Bacteriol. 2003, 185, 6308–6315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedlacek, C.J.; Nielsen, S.; Greis, K.D.; Haffey, W.D.; Revsbech, N.P.; Ticak, T.; Laanbroek, H.J.; Bollmann, A. Effects of Bacterial Community Members on the Proteome of the Ammonia-Oxidizing Bacterium Nitrosomonas sp Strain Is79. Appl. Environ. Microbiol. 2016, 82, 4776–4788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).