
An Examination of Fungal and Bacterial Assemblages in Bulk and Rhizosphere Soils under Solanum tuberosum in Southeastern Wyoming, USA

Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Extraction and Library Preparation
2.2. Sequence Data Analysis
2.3. Statistical Analysis of Sequence Data
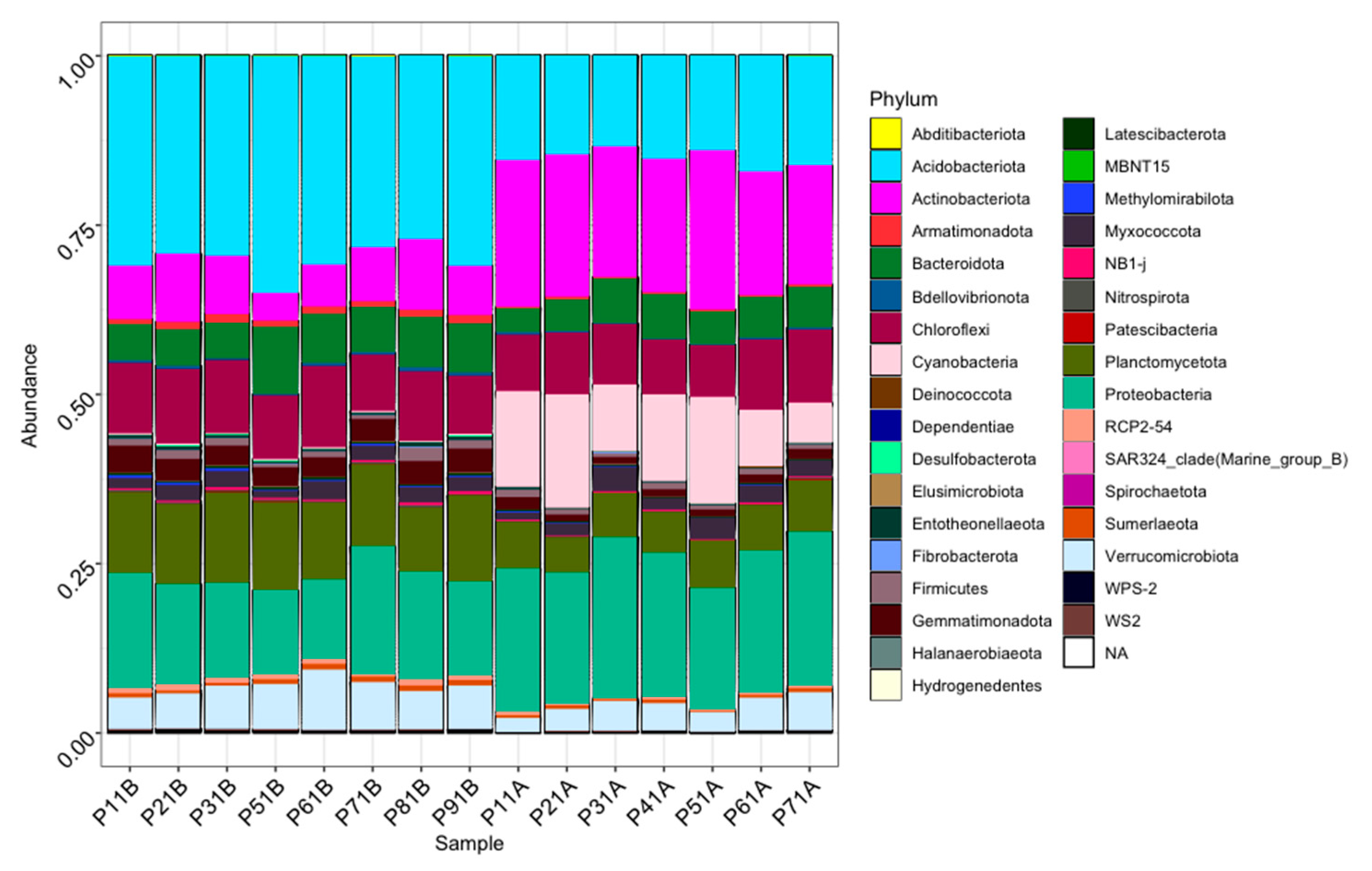
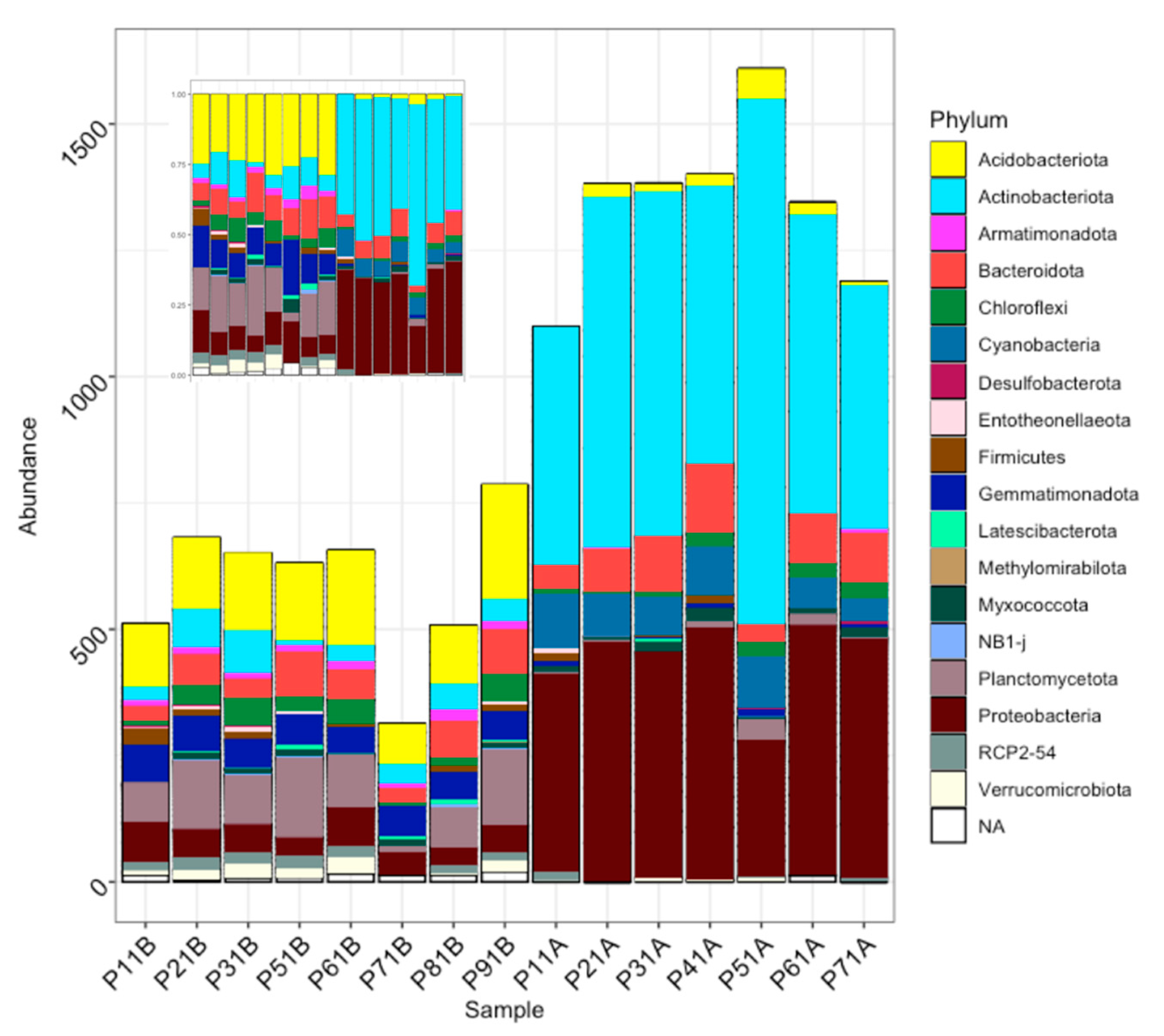
3. Results
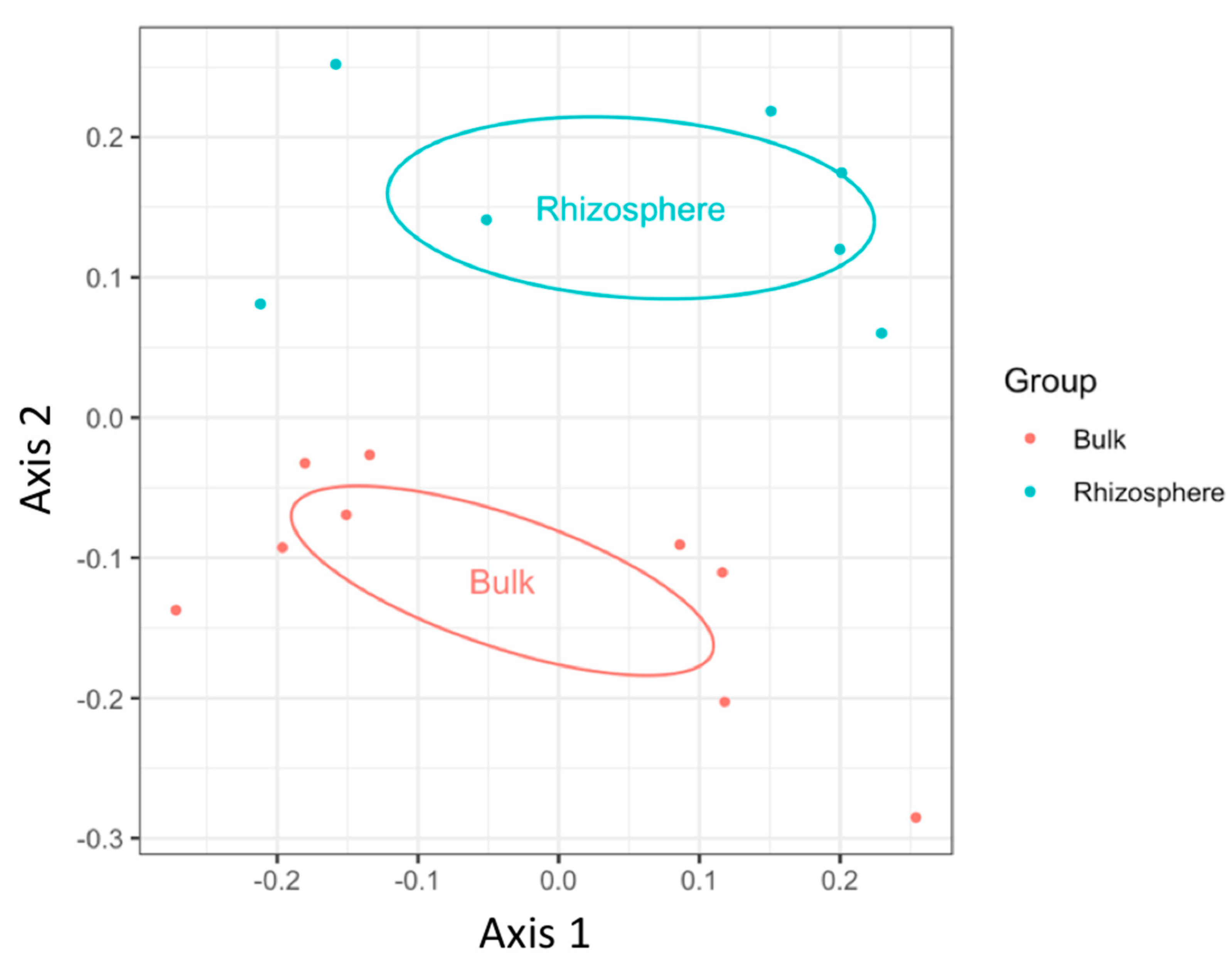
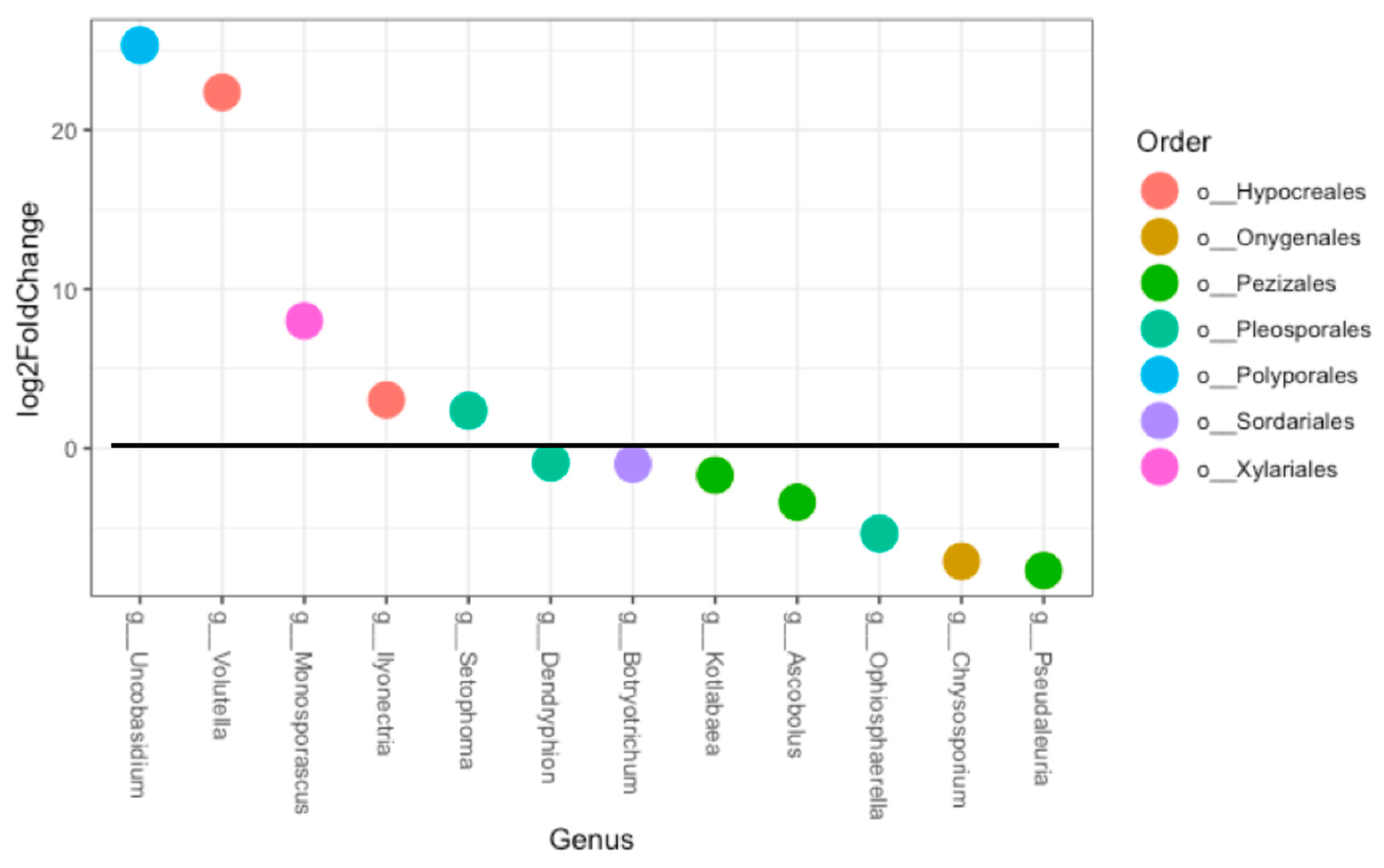
3.1. Fungal Diversity
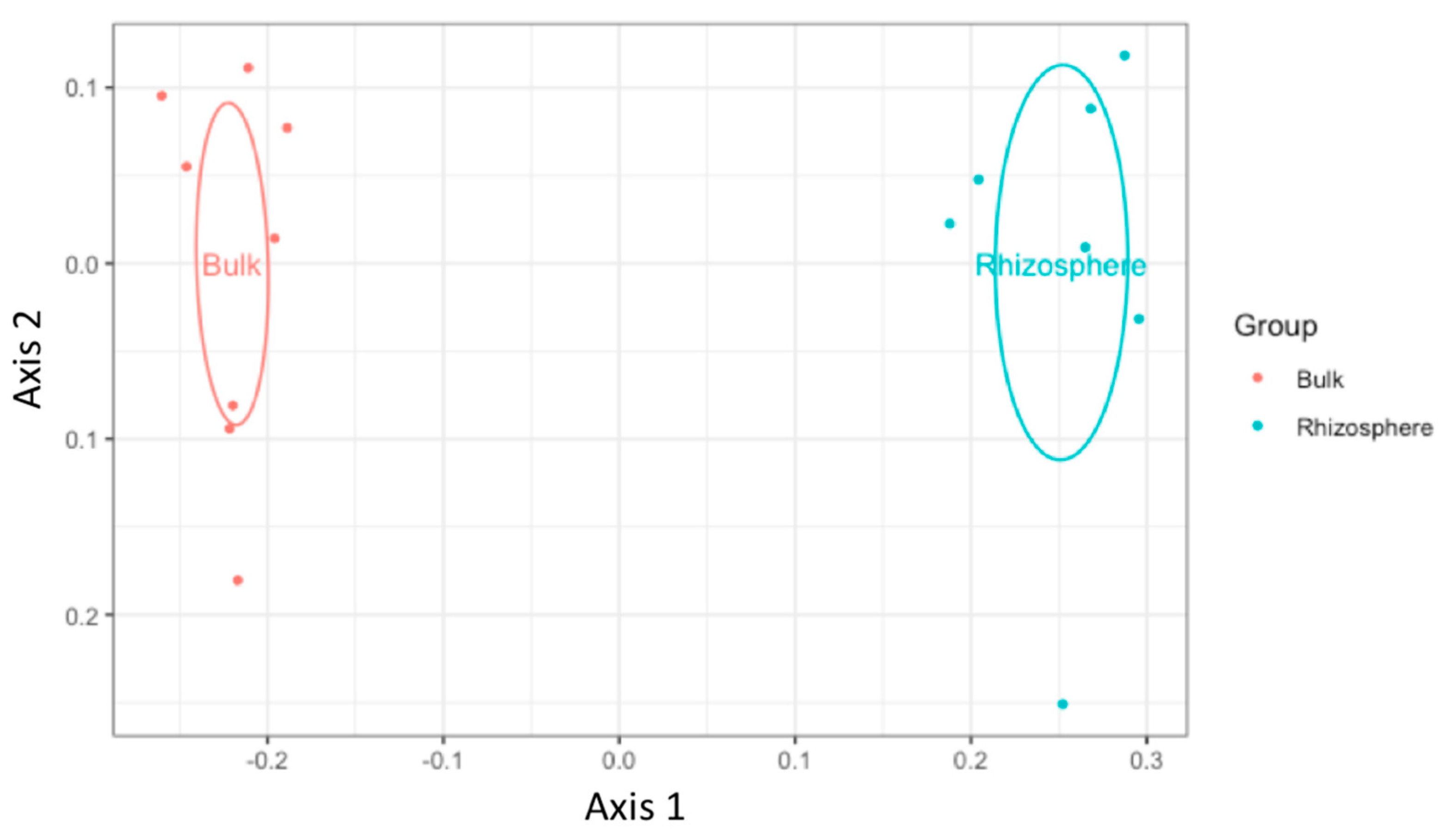
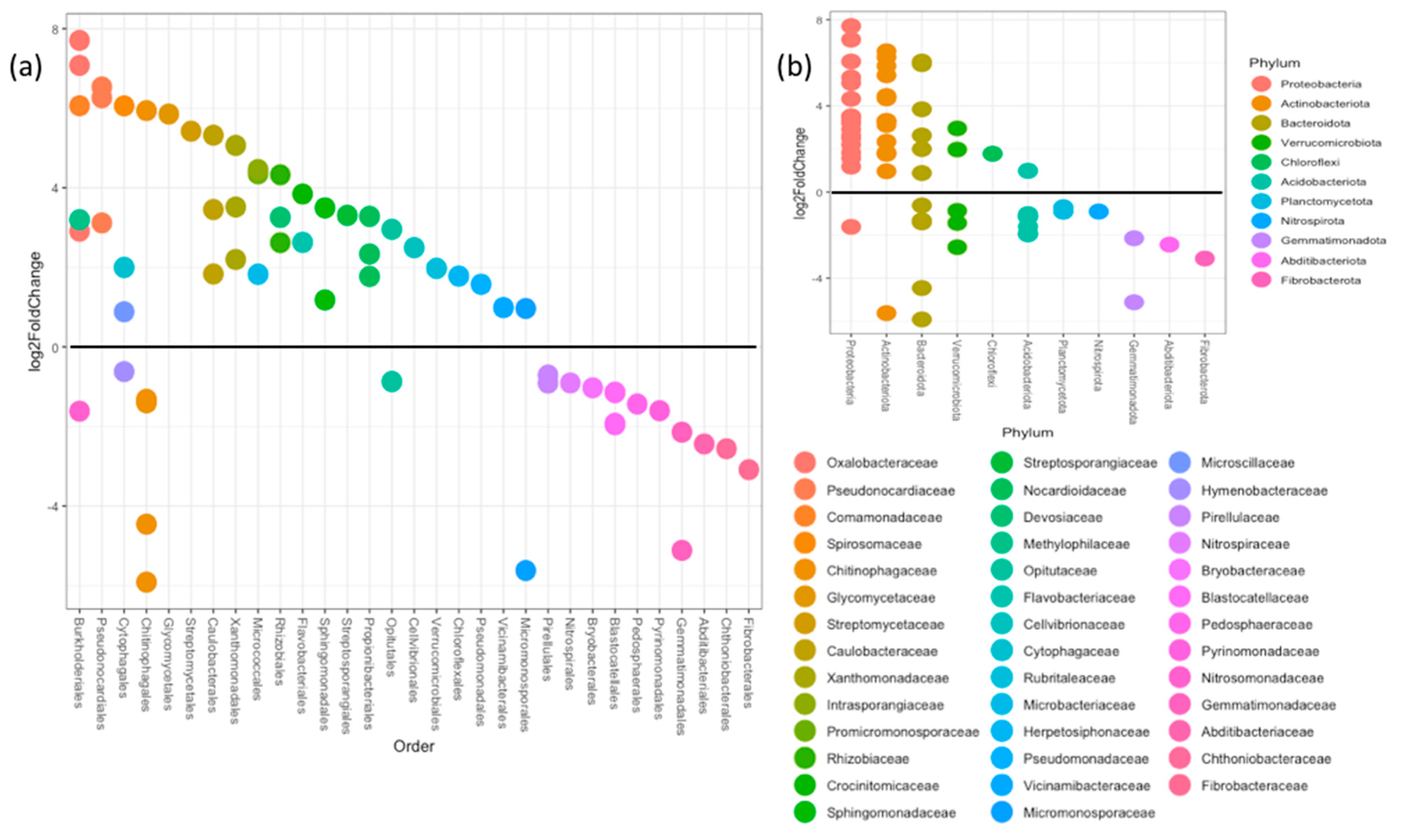
3.2. Bacterial Diversity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ding, Q.; Chen, X.; Hilborn, R.; Chen, Y. Vulnerability to impacts of climate change on marine fisheries and food security. Mar. Policy 2017, 83, 55–61. [Google Scholar] [CrossRef]
- Garcia, J.; Kao-Kniffin, J. Microbial group dynamics in plant rhizospheres and their implications on nutrient cycling. Front. Microbiol. 2018, 9, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Read, D.J.; Perez-Moreno, J. Mycorrhizas and nutrient cycling in ecosystems—A journey towards relevance? New Phytol. 2003, 157, 475–492. [Google Scholar] [CrossRef] [PubMed]
- Van der Heijden, M.G.A.; Klironomos, J.N.; Ursic, M.; Moutoglis, P.; Streitwolf-Engel, R.; Boller, T.; Wiemken, A.; Sanders, I.R. Mycorrhizal fungal diversity determines plant biodiversity, ecosystem variability and productivity. Nature 1998, 396, 69–72. [Google Scholar] [CrossRef]
- Timmusk, S.; Abd El-Daim, I.A.; Copolovici, L.; Tanilas, T.; Kännaste, A.; Behers, L.; Nevo, E.; Seisenbaeva, G.; Stenström, E.; Niinemets, Ü. Drought-tolerance of wheat improved by rhizosphere bacteria from harsh environments: Enhanced biomass production and reduced emissions of stress volatiles. PLoS ONE 2014, 9, e96086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zolla, G.; Badri, D.V.; Bakker, M.G.; Manter, D.K.; Vivanco, J.M. Soil microbiomes vary in their ability to confer drought tolerance to Arabidopsis. Appl. Soil Ecol. 2013, 68, 1–9. [Google Scholar] [CrossRef]
- Chen, C.; Condron, L.; Davis, M.; Sherlock, R. Phosphorus dynamics in the rhizosphere of perennial ryegrass (Lolium perenne L.) and radiata pine (Pinus radiata D. Don.). Soil Biol. Biochem. 2002, 34, 487–499. [Google Scholar] [CrossRef]
- Richardson, A.E.; Barea, J.-M.; McNeill, A.M.; Prigent-Combaret, C. Acquisition of phosphorus and nitrogen in the rhizosphere and plant growth promotion by microorganisms. Plant Soil 2009, 321, 305–339. [Google Scholar] [CrossRef]
- Richardson, A.E.; Simpson, R.J. Soil microorganisms mediating phosphorus availability update on microbial phosphorus. Plant Physiol. 2011, 156, 989–996. [Google Scholar] [CrossRef] [Green Version]
- Pineda, A.; Zheng, S.-J.; Van Loon, J.J.A.; Pieterse, C.M.J.; Dicke, M. Helping plants to deal with insects: The role of beneficial soil-borne microbes. Trends Plant Sci. 2010, 15, 507–514. [Google Scholar] [CrossRef]
- Hubbard, C.J.; Li, B.; McMinn, R.; Brock, M.T.; Maignien, L.; Ewers, B.E.; Kliebenstein, D.; Weinig, C. The effect of rhizosphere microbes outweighs host plant genetics in reducing insect herbivory. Mol. Ecol. 2019, 28, 1801–1811. [Google Scholar] [CrossRef]
- Durán, P.; Thiergart, T.; Garrido-Oter, R.; Agler, M.; Kemen, E.; Schulze-Lefert, P.; Hacquard, S. Microbial interkingdom interactions in roots promote Arabidopsis survival. Cell 2018, 175, 973–983. [Google Scholar] [CrossRef]
- Oberholster, T.; Vikram, S.; Cowan, D.; Valverde, A. Key microbial taxa in the rhizosphere of sorghum and sunflower grown in crop rotation. Sci. Total Environ. 2018, 624, 530–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravanbakhsh, M.; Sasidharan, R.; Voesenek, L.A.C.J.; Kowalchuk, G.A.; Jousset, A. Microbial modulation of plant ethylene signaling: Ecological and evolutionary consequences. Microbiome 2018, 6, 1–10. [Google Scholar] [CrossRef]
- Lugtenberg, B.; Kamilova, F. Plant-growth-promoting rhizobacteria. Annu. Rev. Microbiol. 2009, 63, 541–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantó, C.D.L.F.; Simonin, M.; King, E.; Moulin, L.; Bennett, M.J.; Castrillo, G.; Laplaze, L. An extended root phenotype: The rhizosphere, its formation and impacts on plant fitness. Plant J. 2020, 103, 951–964. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick, C.R.; Salas-González, I.; Conway, J.M.; Finkel, O.M.; Gilbert, S.; Russ, D.; Teixeira, P.J.P.L.; Dangl, J.L. The plant microbiome: From ecology to reductionism and beyond. Annu. Rev. Microbiol. 2020, 74, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, F.; Wu, Y.; Hu, H.; Dai, X. Progress of potato staple food research and industry development in China. J. Integr. Agric. 2017, 16, 2924–2932. [Google Scholar] [CrossRef]
- Van der Wolf, J.M.; Boer, S.H.D. Bacterial Pathogens of Potato. In Potato Biology and Biotechnology; Elsevier: Amsterdam, The Netherlands, 2007; pp. 595–617. [Google Scholar]
- Haas, B.J.; Kamoun, S.; Zody, M.C.; Jiang, R.H.Y.; Handsaker, R.E.; Cano, L.M.; Grabherr, M.; Kodira, C.D.; Raffaele, S.; Torto-Alalibo, T.; et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 2009, 461, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, N.; Tiedje, J.M.; Quensen, J.F.; Meng, Q.; Hao, J.J. Microbial communities associated with potato common scab-suppressive soil determined by pyrosequencing analyses. Plant Dis. 2012, 96, 718–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlatter, D.; Kinkel, L.; Thomashow, L.; Weller, D.; Paulitz, T. Disease suppressive soils: New insights from the soil microbiome. Phytopathology 2017, 107, 1284–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Meng, Z.; Liu, X.; Zhang, X.-H. Microbial assembly, interaction, functioning, activity and diversification: A review derived from community compositional data. Mar. Life Sci. Technol. 2019, 1, 112–128. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, S.; Mitter, B.; Oswald, A.; Schloter-Hai, B.; Schloter, M.; Declerck, S.; Sessitsch, A. Rhizosphere microbiomes of potato cultivated in the High Andes show stable and dynamic core microbiomes with different responses to plant development. FEMS Microbiol. Ecol. 2017, 93, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Yeboah, S.; Xu, X.; Liu, Y.; Yu, B. Analysis on fungal diversity in rhizosphere soil of continuous cropping potato subjected to different furrow-ridge mulching managements. Front. Microbiol. 2017, 8, 845–855. [Google Scholar] [CrossRef]
- Brempong, M.B.; Norton, U.; Norton, J.B. Compost and soil moisture effects on seasonal carbon and nitrogen dynamics, greenhouse gas fluxes and global warming potential of semi-arid soils. Int. J. Recycl. Org. Waste Agric. 2019, 8, 367–376. [Google Scholar] [CrossRef] [Green Version]
- Luster, J.; Finlay, R. Handbook of Methods Used in Rhizosphere Research; Swiss Federal Research Institute WSL: Birmensdorf, Switzerland, 2006; ISBN 3-905621-35-5. [Google Scholar]
- Custer, G.F.; VanDiepen, L.T.A.; Stump, W.L. Structural and functional dynamics of soil microbes following spruce beetle infestation. Appl. Environ. Microbiol. 2020, 86, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef]
- Apprill, A.; Mcnally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Ihrmark, K.; Bödeker, I.T.M.; Cruz-Martinez, K.; Friberg, H.; Kubartova, A.; Schenck, J.; Strid, Y.; Stenlid, J.; Brandström-Durling, M.; Clemmensen, K.E.; et al. New primers to amplify the fungal ITS2 region—Evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol. Ecol. 2012, 82, 666–677. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and Direct Sequencing of Fungal Ribosomal RNA Genes for Phylogenetics. In PCR Protocols; Elsevier: Amsterdam, The Netherlands, 1990; pp. 315–322. ISBN 008088671X. [Google Scholar]
- R Development Core Team. A Language and Environment for Statistical Computing; R Foundation Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Abarenkov, K.; Nilsson, R.H.; Larsson, K.H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Høiland, K.; Kjøller, R.; Larsson, E.; Pennanen, T.; et al. The UNITE database for molecular identification of fungi—Recent updates and future perspectives. New Phytol. 2010, 186, 281–285. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, P.; Solymos, P.; et al. Vegan: Community Ecology Package; R Development Core Team: Vienna, Austria, 2018. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Domagalski, R.; Neal, Z.; Sagan, B. Backbone: An R Package for extracting the backbone of bipartite projections. PLoS ONE 2019, 16, e0244363. [Google Scholar]
- Neal, Z. Identifying statistically significant edges in one-mode projections. Soc. Netw. Anal. Min. 2013, 3, 915–924. [Google Scholar] [CrossRef]
- Inceoǧlu, Ö.; Al-Soud, W.A.; Salles, J.F.; Semenov, A.V.; Van Elsas, J.D. Comparative analysis of bacterial communities in a potato field as determined by pyrosequencing. PLoS ONE 2011, 6, e23321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loit, K.; Soonvald, L.; Astover, A.; Runno-Paurson, E.; Öpik, M.; Tedersoo, L. Temporal and cultivar-specific effects on potato root and soil fungal diversity. Agronomy 2020, 10, 1535. [Google Scholar] [CrossRef]
- Stefańczyk, E.; Sobkowiak, S.; Brylińska, M.; Śliwka, J. Diversity of Fusarium spp. associated with dry rot of potato tubers in Poland. Eur. J. Plant Pathol. 2016, 145, 871–884. [Google Scholar] [CrossRef] [Green Version]
- Leplat, J.; Friberg, H.; Abid, M.; Steinberg, C. Survival of Fusarium graminearum, the causal agent of Fusarium head blight. A review. Agron. Sustain. Dev. 2013, 33, 97–111. [Google Scholar] [CrossRef]
- Loria, R.; Bukhalid, R.A.; Fry, B.A.; King, R.R. Plant pathogenicity in the genus Streptomyces. Plant Dis. 1997, 81, 836–846. [Google Scholar] [CrossRef] [Green Version]
- Amaresan, N.; Kumar, K.; Naik, J.H.; Bapatla, K.G.; Mishra, R.K. Streptomyces in Plant Growth Promotion. In New and Future Developments in Microbial Biotechnology and Bioengineering; Elsevier: Amsterdam, The Netherlands, 2018; pp. 125–135. [Google Scholar]
- Morrissey, E.M.; Mau, R.L.; Schwartz, E.; Koch, B.J.; Hayer, M.; Hungate, B.A. Taxonomic patterns in the nitrogen assimilation of soil prokaryotes. Environ. Microbiol. 2018, 20, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Meier, M.A.; Lopez-Guerrero, M.G.; Guo, M.; Schmer, M.R.; Herr, J.R.; Schnable, J.C.; Alfano, J.R.; Yang, J. Rhizosphere microbiomes in a historical maize/soybean rotation system respond to host species and nitrogen fertilization at genus and sub-genus levels. bioRxiv 2020, 1–43. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, D.; Yang, Y.; Pan, Y.; Zhao, D.; Zhu, J.; Zhang, L.; Zhihui, Y. Dissecting the effect of continuous cropping of potato on soil bacterial communities as revealed by high-throughput sequencing. PLoS ONE 2020, 15, e0233356. [Google Scholar] [CrossRef]
- Banerjee, S.; Schlaeppi, K.; Van der Heijden, M.G.A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.; Delgado-Baquerizo, M.; Trivedi, C.; Hamonts, K.; Anderson, I.C.; Singh, B.K. Keystone microbial taxa regulate the invasion of a fungal pathogen in agro-ecosystems. Soil Biol. Biochem. 2017, 111, 10–14. [Google Scholar] [CrossRef]
- Zhao, J.; Li, W.; Shi, L.; Wang, H.; Wang, Y.; Zhao, Y.; Xiang, W.; Wang, X. Lechevalieria rhizosphaerae sp. nov., a novel actinomycete isolated from rhizosphere soil of wheat (Triticum aestivum L.) and emended description of the genus Lechevalieria. Int. J. Syst. Evol. Microbiol. 2017, 67, 4655–4659. [Google Scholar] [CrossRef] [PubMed]
- Labeda, D.P. The Family Actinosynnemataceae. In The Prokaryotes; Springer: New York, NY, USA, 2006; pp. 654–668. [Google Scholar]
- Ofek, M.; Hadar, Y.; Minz, D. Ecology of root colonizing Massilia (Oxalobacteraceae). PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, A.L.; Liu, Y.; Lazarovits, G. Isolation of bacteria from the rhizosphere and rhizoplane of potato (Solanum tuberosum) grown in two distinct soils using semi selective media and characterization of their biological properties. Am. J. Potato Res. 2012, 89, 294–305. [Google Scholar] [CrossRef]
- Morris, S.J.; Blackwood, C.B. The Ecology of the Soil Biota and their Function, 4th ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2015; ISBN 9780124159556. [Google Scholar]
- Goldford, J.E.; Lu, N.; Bajić, D.; Estrela, S.; Tikhonov, M.; Sanchez-Gorostiaga, A.; Segrè, D.; Mehta, P.; Sanchez, A. Emergent simplicity in microbial community assembly. Science 2018, 361, 469–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konopka, A. What is microbial community ecology? ISME J. 2009, 3, 1223–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martiny, J.B.H.; Jones, S.E.; Lennon, J.T.; Martiny, A.C. Microbiomes in light of traits: A phylogenetic perspective. Science 2015, 350, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fungi | ||||||
| Bulk | Rhizosphere | |||||
| Mean | Standard Deviation | Mean | Standard Deviation | |||
| Shannon Diversity (H’) | p-value | p = 0.36 | 3.54 | 0.28 | 3.67 | 0.09 |
| Test Statistic | X21,14 = 0.891 | |||||
| Richness | p-value | p < 0.05 | 153.33 | 21.99 | 130.29 | 19.99 |
| Test Statistic | F1,14 = 4.672 | |||||
| Bacteria | ||||||
| Bulk | Rhizosphere | |||||
| Mean | Standard Deviation | Mean | Standard Deviation | |||
| Shannon Diversity (H’) | p-value | p < 0.0001 | 6.42 | 0.26 | 5.75 | 0.28 |
| Test Statistic | F1,14 = 22.29 | |||||
| Richness | p-value | p = 0.118 | 1227 | 384 | 952 | 218 |
| Test Statistic | F1,14 = 2.796 | |||||
| Fungal Trophic Modes | ||||||
| Bulk | Rhizosphere | |||||
| Mean | Standard Deviation | Mean | Standard Deviation | |||
| Saprotroph | p-value | p < 0.01 | 0.1861% | 0.0337% | 0.1170% | 0.0283% |
| Test Statistic | Χ21,14 = 8.371 | |||||
| Pathotroph-Saprotroph | p-value | p < 0.01 | 0.0770% | 0.0170% | 0.1159% | 0.0187% |
| Test Statistic | Χ21,14 = 7.085 | |||||
| Pathotroph-Saprotroph-Symbiotroph | p-value | p < 0.01 | 0.0097% | 0.0086% | 0.0024% | 0.0030% |
| Test Statistic | Χ21,14 = 4.371 | |||||
| Fungal Guilds | ||||||
| Bulk | Rhizosphere | |||||
| Mean | Standard Deviation | Mean | Standard Deviation | |||
| Arbuscular-Mycorrhizal | p-value | p < 0.01 | 0.0021% | 0.0037% | 0.0087% | 0.0075% |
| Test Statistic | Χ21,14 = 4.389 | |||||
| Dung-Saprotroph-Soil-Saprotroph-Wood-Saprotroph | p-value | p < 0.05 | 0.0154% | 0.0170% | 0.0169% | 0.0448% |
| Test Statistic | Χ21,14 = 5.980 | |||||
| Dung-Saprotroph-Undefined-Saprotroph | p-value | p < 0.05 | 0.1314% | 0.1572% | 0.0389% | 0.0188% |
| Test Statistic | Χ21,14 = 6.1875 | |||||
| Fungal-Parasite-Plant-Pathogen-Plant-Saprotroph | p-value | p < 0.001 | 0.0227% | 0.0064% | 0.0882% | 0.0611% |
| Test Statistic | Χ21,14 = 11.117 | |||||
| Plant-Pathogen-Plant-Saprotroph | p-value | p < 0.05 | 0.0010% | 0.0030% | 0.0076% | 0.0077% |
| Test Statistic | Χ21,14 = 4.509 | |||||
| Undefined-Saprotroph | p-value | p < 0.01 | 0.1876% | 0.0375% | 0.1156% | 0.0267% |
| Test Statistic | Χ21,14 = 9.100 | |||||
| Mean Relative Abundance | Change (log2 Fold) | Association | Adjusted p-Value | Kingdom | Phylum |
|---|---|---|---|---|---|
| 2780.41682 | 6.905558924 | Rhizosphere | 7.25 × 10−164 | Bacteria | Cyanobacteria |
| 5773.55747 | 2.114174238 | Rhizosphere | 7.78 × 10−28 | Bacteria | Actinobacteriota |
| 3572.14083 | 0.565324082 | Rhizosphere | 1.52 × 10−5 | Bacteria | Chloroflexi |
| 6986.19763 | 1.230783697 | Rhizosphere | 3.50 × 10−19 | Bacteria | Proteobacteria |
| 851.536029 | 0.92383889 | Rhizosphere | 0.0005466 | Bacteria | Myxococcota |
| 764.000925 | −0.727541596 | Bulk | 1.12 × 10−7 | Bacteria | Gemmatimonadota |
| 218.042158 | −1.157547177 | Bulk | 1.23 × 10−8 | Bacteria | Armatimonadota |
| 14.6936551 | −1.880961129 | Bulk | 0.00190362 | Bacteria | Abditibacteriota |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Custer, G.F.; van Diepen, L.T.A.; Stump, W. An Examination of Fungal and Bacterial Assemblages in Bulk and Rhizosphere Soils under Solanum tuberosum in Southeastern Wyoming, USA. Appl. Microbiol. 2021, 1, 162-176. https://doi.org/10.3390/applmicrobiol1020013
Custer GF, van Diepen LTA, Stump W. An Examination of Fungal and Bacterial Assemblages in Bulk and Rhizosphere Soils under Solanum tuberosum in Southeastern Wyoming, USA. Applied Microbiology. 2021; 1(2):162-176. https://doi.org/10.3390/applmicrobiol1020013
Chicago/Turabian StyleCuster, Gordon F., Linda T. A. van Diepen, and William Stump. 2021. "An Examination of Fungal and Bacterial Assemblages in Bulk and Rhizosphere Soils under Solanum tuberosum in Southeastern Wyoming, USA" Applied Microbiology 1, no. 2: 162-176. https://doi.org/10.3390/applmicrobiol1020013
APA StyleCuster, G. F., van Diepen, L. T. A., & Stump, W. (2021). An Examination of Fungal and Bacterial Assemblages in Bulk and Rhizosphere Soils under Solanum tuberosum in Southeastern Wyoming, USA. Applied Microbiology, 1(2), 162-176. https://doi.org/10.3390/applmicrobiol1020013