
Integrating Boruta, LASSO, and SHAP for Clinically Interpretable Glioma Classification Using Machine Learning


Abstract
1. Introduction
- Identify key genetic and clinical markers for glioma classification through a systematic integration of Boruta, LASSO, and SHAP feature selection methods.
- Optimize and compare multiple ML models (RF, SVM, XGBoost, and Logistic Regression) to assess classification performance using the selected biomarkers.
- Enhance the interpretability of glioma prediction models by analyzing the contributions of selected biomarkers.
- Develop a reproducible framework for glioma classification that can extend to other genomic-based cancer studies.
2. Related Work
2.1. Glioma Classification: The Role of Genetic and Patient-Specific Factors
2.2. Machine Learning for Glioma Classification
2.3. Feature Selection for Biomarker Discovery in Gliomas
3. Methodology
3.1. Data Collection and Preprocessing
3.2. Feature Selection Techniques
3.2.1. Boruta: All-Relevant Feature Selection
- is the Gini importance of the feature in the t-th tree of the Random Forest model.
- T is the total number of trees in the forest.
3.2.2. LASSO: L1 Regularization-Based Feature Reduction
- is the target variable (glioma grade).
- represents the input feature values.
- are the regression coefficients.
- is the regularization parameter controlling feature selection.
3.2.3. SHAP: Explainability-Based Feature Ranking
- is a full set of features.
- is a subset of features excluding
- is the model prediction using only subset S.
- is the model prediction after adding a feature
- The fraction represents the weighting factor based on SHapley values from cooperative game theory.
3.2.4. Intersection of Selected Features and Rationale for Selection
3.3. Machine Learning Models for Classification
3.3.1. Random Forest (RF)
3.3.2. Support Vector Machine (SVM)
3.3.3. XGBoost
3.3.4. Logistic Regression
3.4. Model Training and Evaluation
3.4.1. Training Procedure and Cross-Validation Strategy
3.4.2. Performance Metrics for Model Assessment
- Accuracy: Measures the overall correctness of predictions, calculated as [87]:
- Precision: Evaluates the proportion of correctly identified positive cases among predicted positives [87]:
- Recall (Sensitivity): Measures the model’s ability to correctly identify positive cases [87]:
- F1-score: Provides a balance between precision and recall [87]:
3.4.3. Comparative Analysis of Model Performance
3.5. Sample Size Justification and Overfitting Control
4. Results
4.1. Descriptive Statistics
4.2. Feature Selection Results
4.2.1. Individual Feature Selection Outcomes
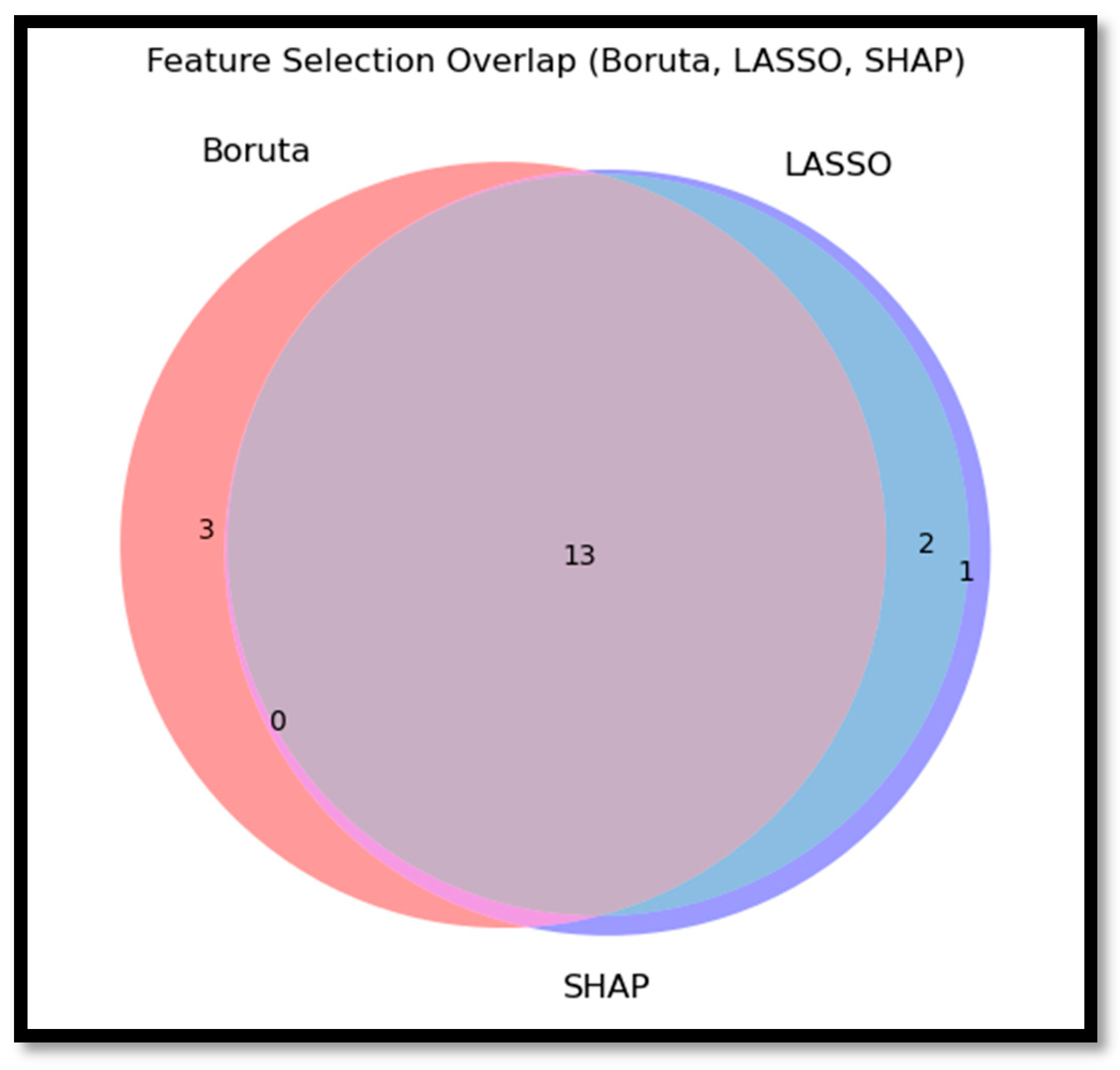
- Boruta Selection: This all-relevant feature selection method identified 16 features as significant, confirming their importance through an iterative Random Forest-based process. Features retained included Age at Diagnosis, race, IDH1, TP53, ATRX, PTEN, EGFR, CIC, MUC16, NF1, PIK3R1, FUBP1, RB1, NOTCH1, SMARCA4, and IDH2.
- LASSO Selection: LASSO identified a set of 15 predictive features, encompassing both genetic and clinical attributes. Notably, gender was selected in this method, alongside Age at Diagnosis, race, IDH1, TP53, ATRX, PTEN, EGFR, CIC, MUC16, NF1, PIK3R1, NOTCH1, GRIN2A, and IDH2.
- SHAP Selection: The SHAP-selected feature set largely overlapped with LASSO, with 16 features identified: gender, Age at Diagnosis, race, IDH1, TP53, ATRX, PTEN, EGFR, CIC, MUC16, PIK3CA, NF1, PIK3R1, NOTCH1, GRIN2A, and IDH2.
4.2.2. Intersection of Selected Features
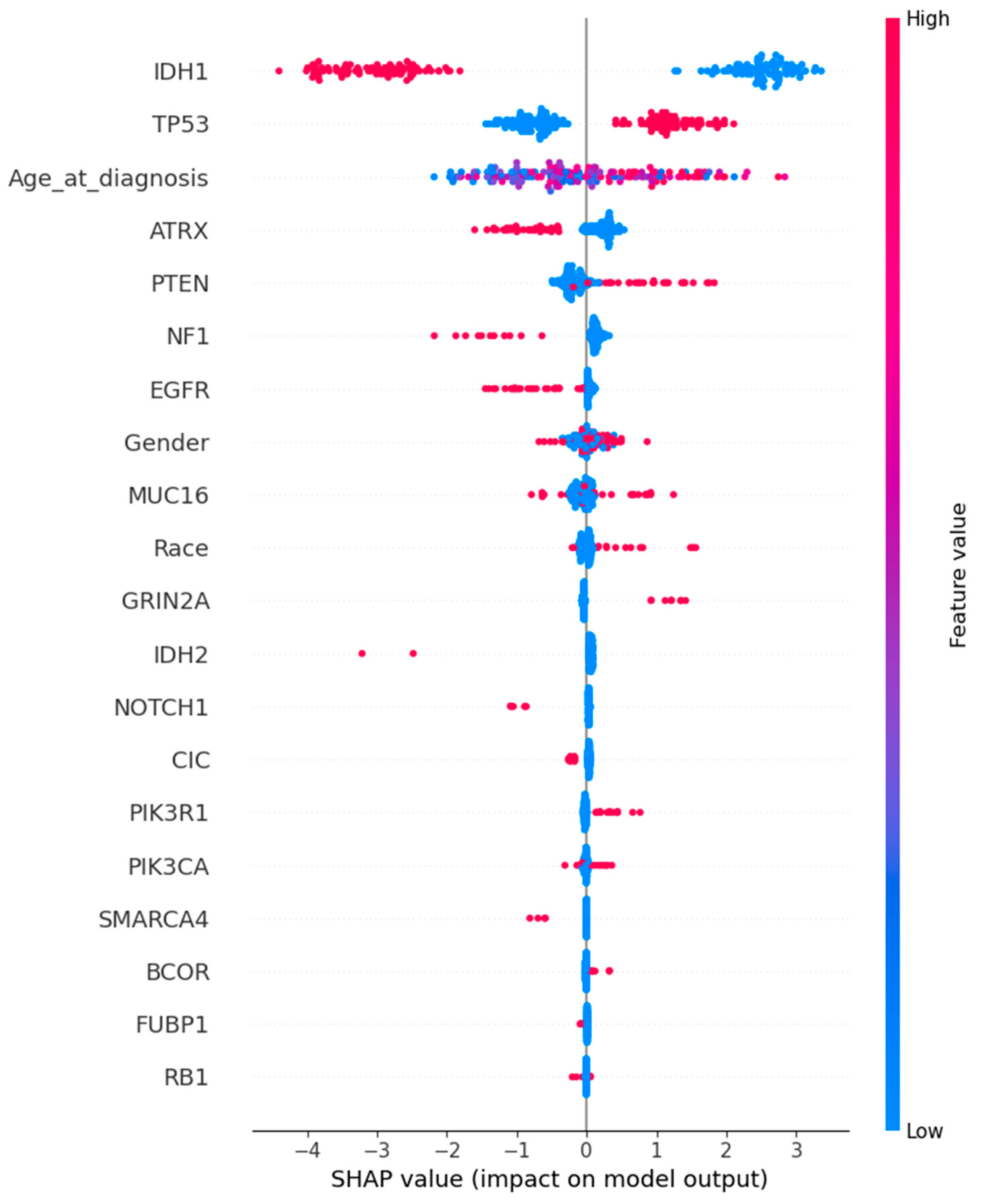
4.2.3. SHAP Feature Importance Analysis
4.3. ML Model Performance
4.3.1. Hyperparameter Tuning Results
4.3.2. Training and Testing Performance
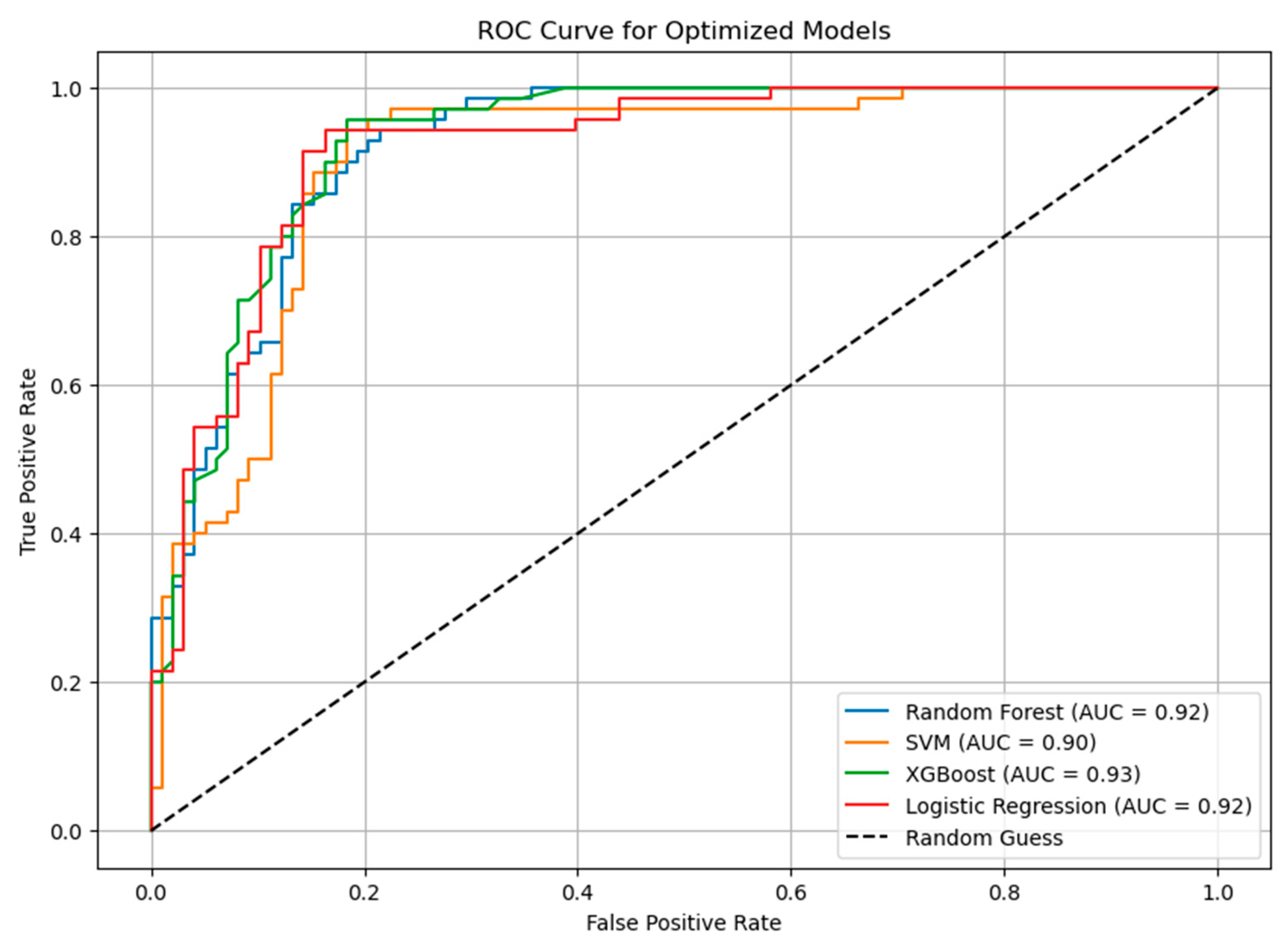
4.3.3. Model Comparison
- Random Forest achieved the highest accuracy on the training set (90.16%) and maintained an acceptable generalization to the test set with an accuracy of 84.53%. It demonstrated a balanced trade-off between precision and recall.
- SVM obtained an accuracy of 88.22% during training but showed a slight decline on the test set (85.71%), suggesting potential overfitting.
- XGBoost exhibited stable performance, with an accuracy of 87.48% in training and 86.30% in testing. This model effectively balanced recall and precision, making it well-suited for identifying glioma subtypes.
- Logistic Regression served as a baseline model, achieving an accuracy of 87.92% in training and 88.09% in testing, demonstrating consistent performance.
4.3.4. ROC Curve Analysis
4.4. Class Distribution and Minority Class Performance Analysis
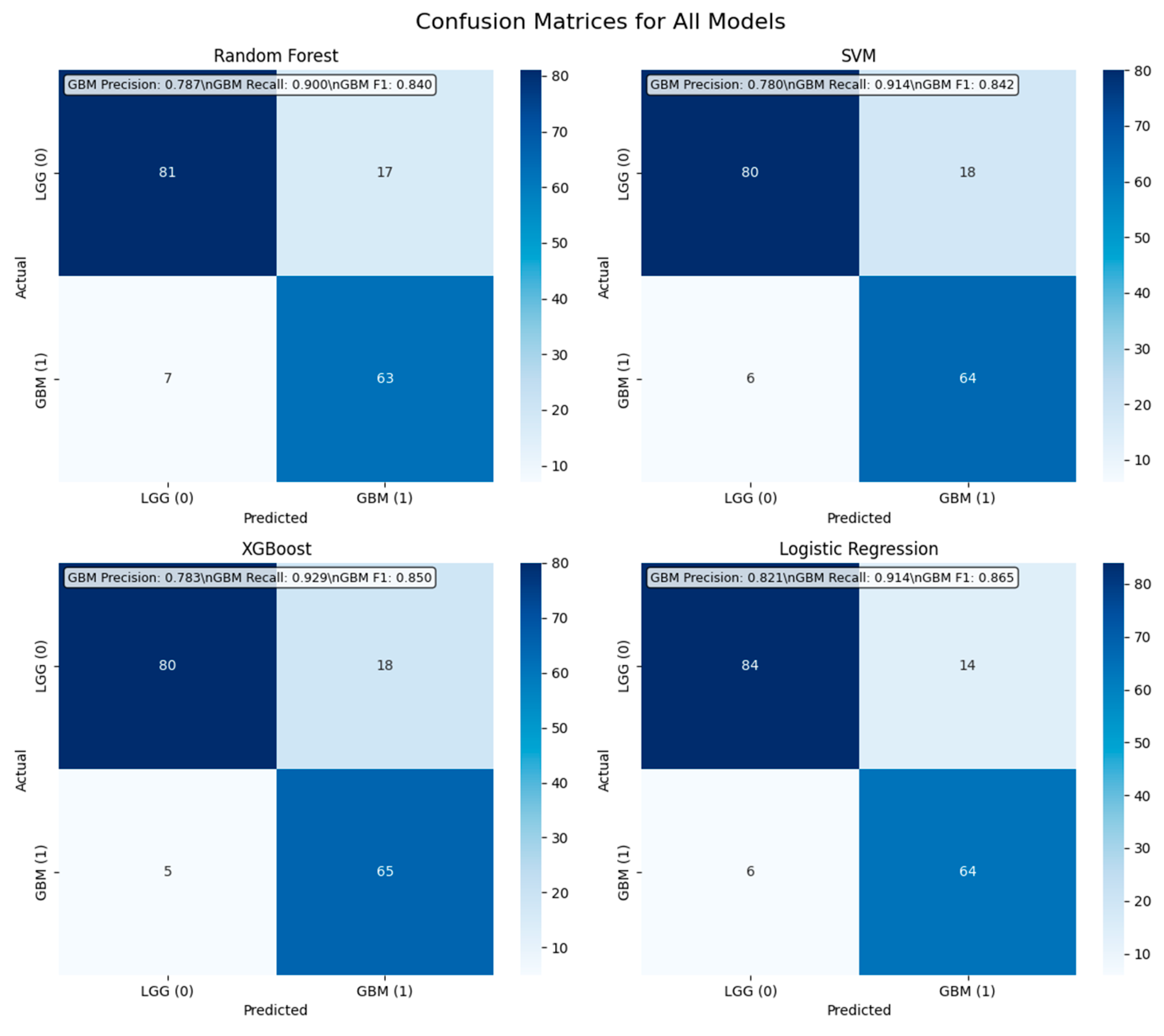
4.4.1. Confusion Matrix Analysis
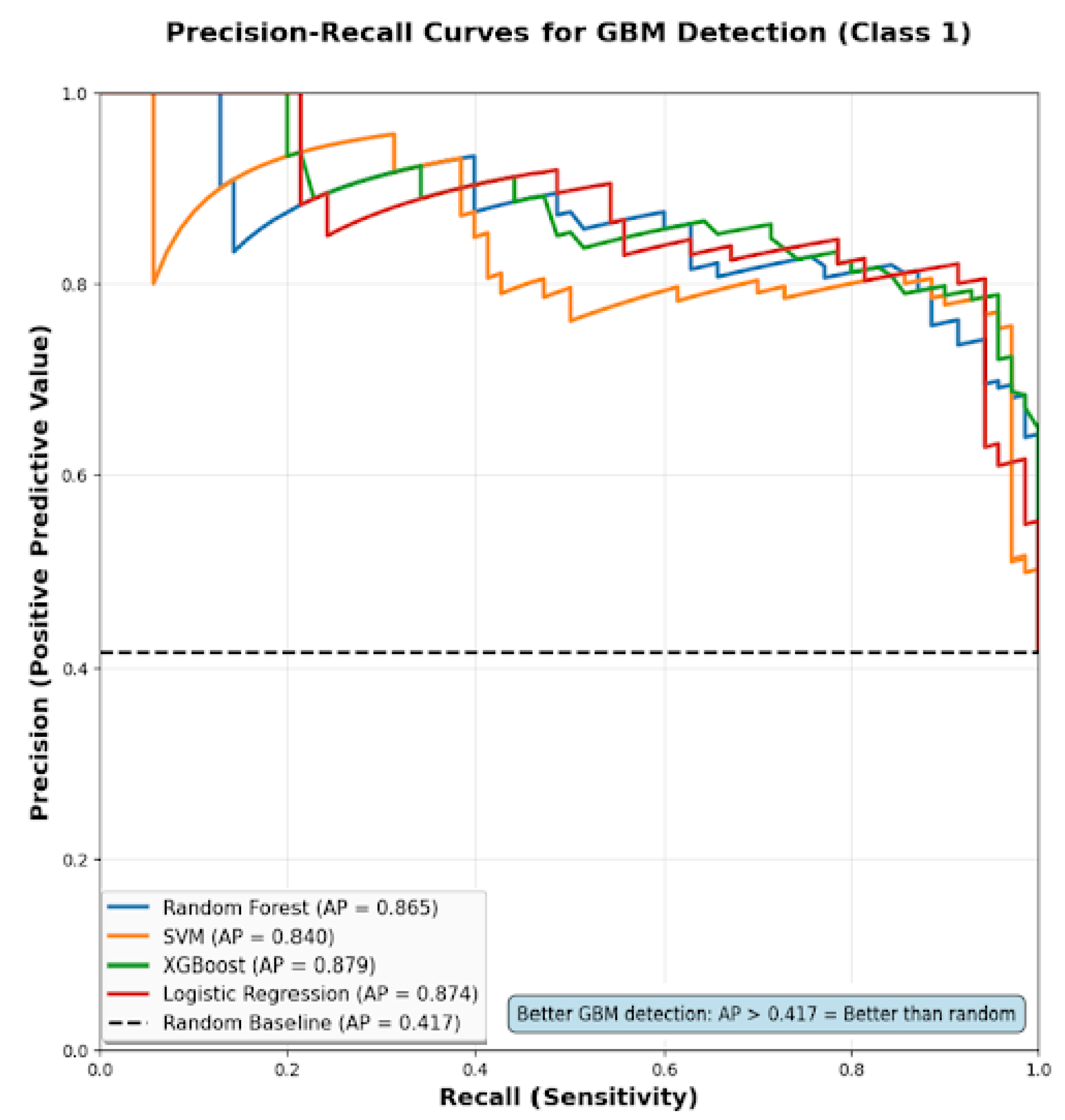
4.4.2. Precision–Recall Analysis for GBM Detection
4.4.3. Class Balancing Techniques Evaluation
4.5. Clinical Utility Assessment
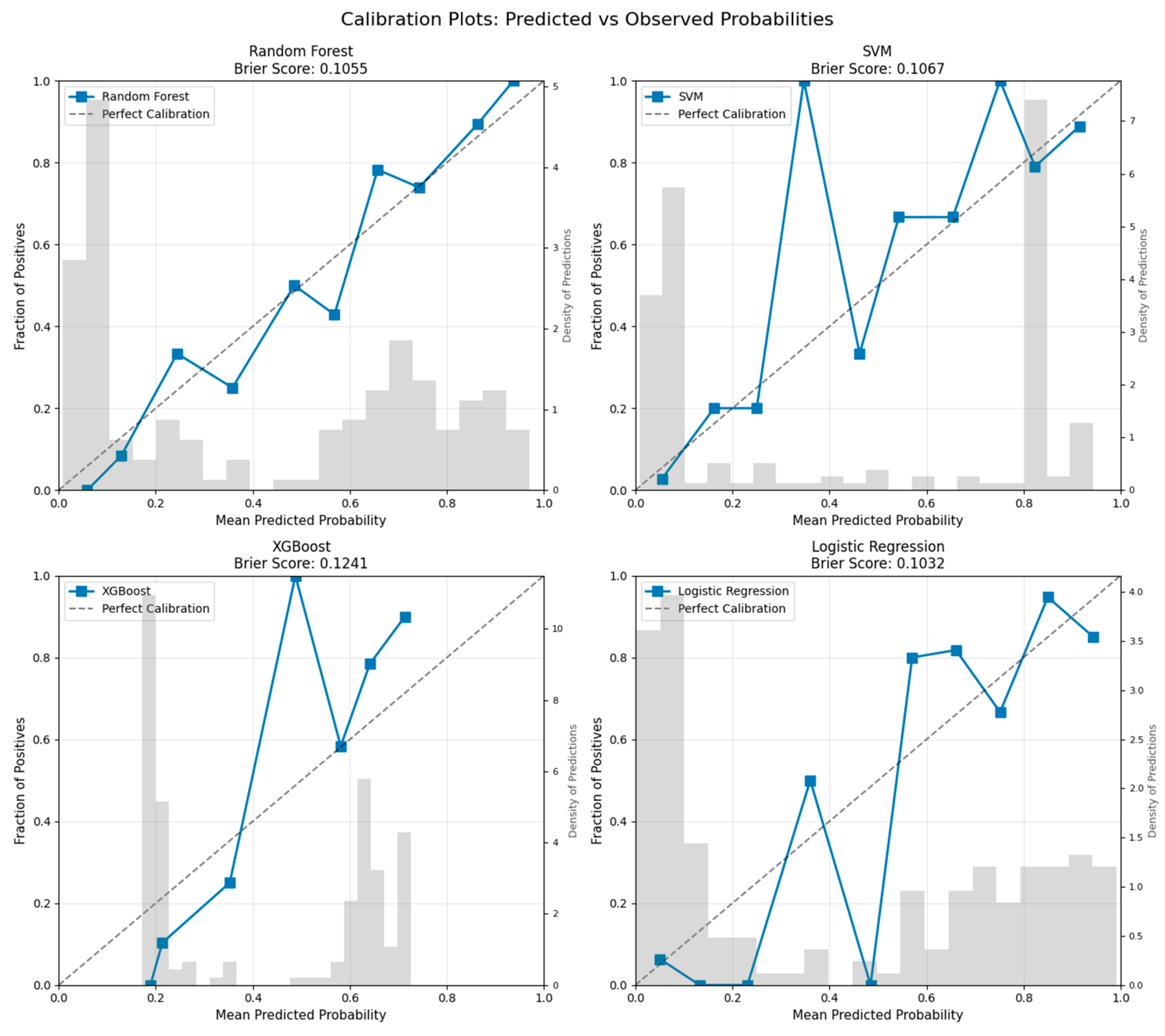
4.5.1. Model Calibration Analysis
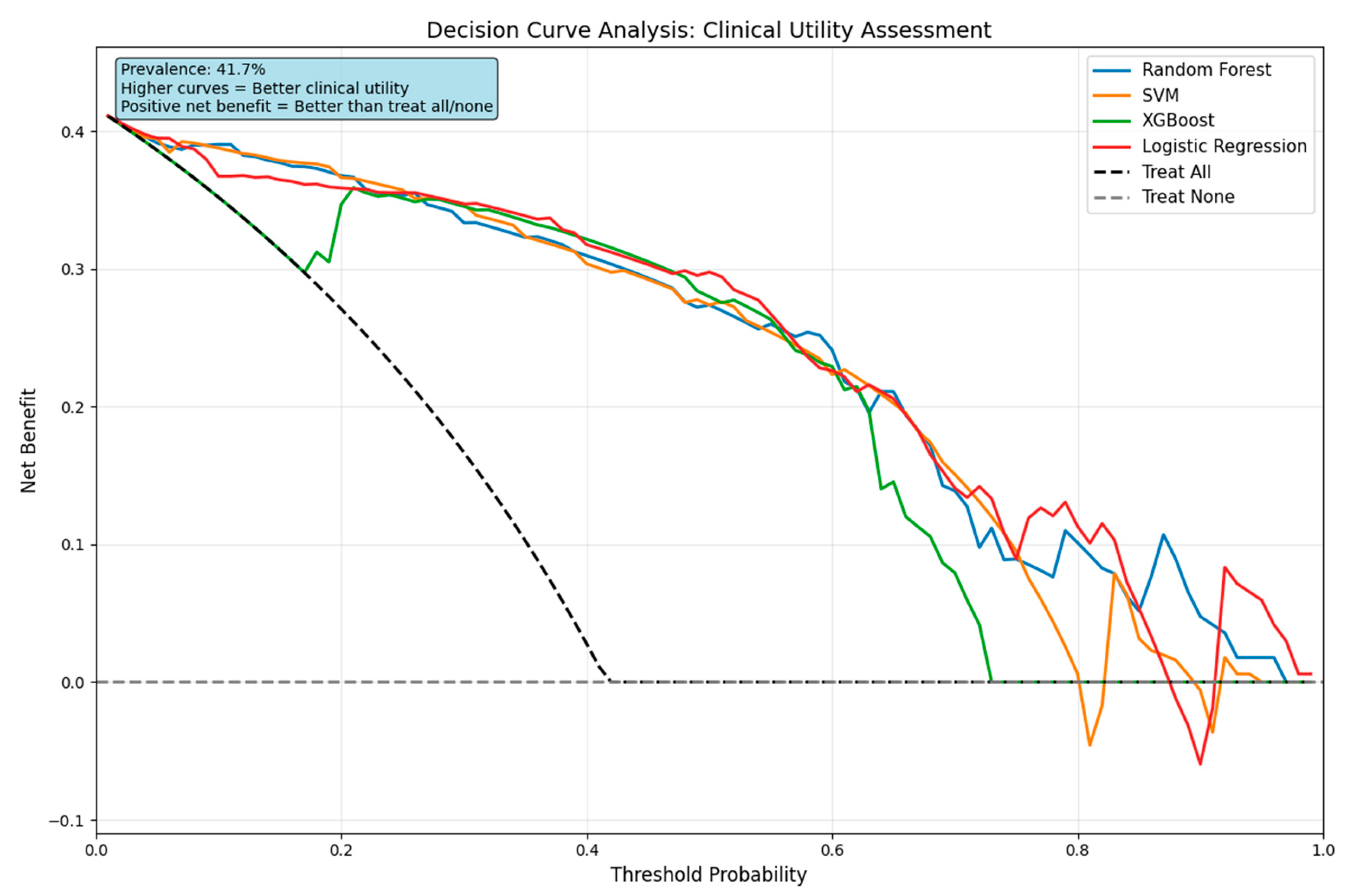
4.5.2. Decision Curve Analysis
4.5.3. Clinical Decision Support Metrics
5. Discussion
5.1. Machine Learning Model Performance Analysis
5.2. Clinical Applications and Implementation
5.3. Study Limitations and Methodological Considerations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Finch, A.; Solomou, G.; Wykes, V.; Pohl, U.; Bardella, C.; Watts, C. Advances in Research of Adult Gliomas. Int. J. Mol. Sci. 2021, 22, 924. [Google Scholar] [CrossRef]
- Claus, E.B.; Walsh, K.M.; Wiencke, J.K.; Molinaro, A.M.; Wiemels, J.L.; Schildkraut, J.M.; Bondy, M.L.; Berger, M.; Jenkins, R.; Wrensch, M. Survival and Low-Grade Glioma: The Emergence of Genetic Information. Neurosurg. Focus 2015, 38, E6. [Google Scholar] [CrossRef]
- Louis, D.N.; Holland, E.C.; Cairncross, J.G. Glioma Classification: A Molecular Reappraisal. Am. J. Pathol. 2001, 159, 779. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Smith-Cohn, M.; Cohen, A.L.; Colman, H. Glioma Subclassifications and Their Clinical Significance. Neurotherapeutics 2017, 14, 284. [Google Scholar] [CrossRef] [PubMed]
- Trinh, D.L.; Kim, S.H.; Yang, H.J.; Lee, G.S. The Efficacy of Shape Radiomics and Deep Features for Glioblastoma Survival Prediction by Deep Learning. Electronics 2022, 11, 1038. [Google Scholar] [CrossRef]
- Wankhede, D.S.; Selvarani, R. Dynamic Architecture Based Deep Learning Approach for Glioblastoma Brain Tumor Survival Prediction. Neurosci. Inform. 2022, 2, 100062. [Google Scholar] [CrossRef]
- Poursaeed, R.; Mohammadzadeh, M.; Safaei, A.A. Survival Prediction of Glioblastoma Patients Using Machine Learning and Deep Learning: A Systematic Review. BMC Cancer 2024, 24, 1581. [Google Scholar] [CrossRef] [PubMed]
- Leanne McDonald, K.; Australia, U.; Giles, K.; Palanichamy, K.; Zong, X.; Liu, A.; Hou, C.; Chen, H.; Zong, P. Genetics and Epigenetics of Glioblastoma: Applications and Overall Incidence of IDH1 Mutation. Front. Oncol. 2016, 6, 16. [Google Scholar] [CrossRef]
- Xie, Y.; Tan, Y.; Yang, C.; Zhang, X.; Xu, C.; Qiao, X.; Xu, J.; Tian, S.; Fang, C.; Kang, C. Omics-Based Integrated Analysis Identified ATRX as a Biomarker Associated with Glioma Diagnosis and Prognosis. Cancer Biol. Med. 2019, 16, 784. [Google Scholar] [CrossRef]
- Liu, H.Q.; Li, W.X.; An, Y.W.; Wu, T.; Jiang, G.Y.; Dong, Y.; Chen, W.X.; Wang, J.C.; Wang, C.; Song, S. Integrated Analysis of the Genomic and Transcriptional Profile of Gliomas with Isocitrate Dehydrogenase-1 and Tumor Protein 53 Mutations. Int. J. Immunopathol. Pharmacol. 2022, 36, 03946320221139262. [Google Scholar] [CrossRef]
- Takano, S.; Ishikawa, E.; Sakamoto, N.; Matsuda, M.; Akutsu, H.; Noguchi, M.; Kato, Y.; Yamamoto, T.; Matsumura, A. Immunohistochemistry on IDH 1/2, ATRX, P53 and Ki-67 Substitute Molecular Genetic Testing and Predict Patient Prognosis in Grade III Adult Diffuse Gliomas. Brain Tumor Pathol. 2016, 33, 107–116. [Google Scholar] [CrossRef]
- Squalli Houssaini, A.; Lamrabet, S.; Senhaji, N.; Sekal, M.; Nshizirungu, J.P.; Mahfoudi, H.; Elfakir, S.; Karkouri, M.; Bennis, S. Prognostic Value of ATRX and P53 Status in High-Grade Glioma Patients in Morocco. Cureus 2024, 16. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Fathi Kazerooni, A.; Toorens, E.; Akbari, H.; Yu, F.; Sako, C.; Mamourian, E.; Shinohara, R.T.; Koumenis, C.; Bagley, S.J.; et al. Integrating Imaging and Genomic Data for the Discovery of Distinct Glioblastoma Subtypes: A Joint Learning Approach. Sci. Rep. 2024, 14, 4922. [Google Scholar] [CrossRef]
- Sánchez-Marqués, R.; García, V.; Sánchez, J.S. A Data-Centric Machine Learning Approach to Improve Prediction of Glioma Grades Using Low-Imbalance TCGA Data. Sci. Rep. 2024, 14, 17195. [Google Scholar] [CrossRef] [PubMed]
- Abusamra, H. A Comparative Study of Feature Selection and Classification Methods for Gene Expression Data of Glioma. Procedia Comput. Sci. 2013, 23, 5–14. [Google Scholar] [CrossRef]
- Whitfield, B.T.; Huse, J.T. Classification of Adult-Type Diffuse Gliomas: Impact of the World Health Organization 2021 Update. Brain Pathol. 2022, 32, e13062. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro Oncol. 2021, 23, 1231. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.L.; Holmen, S.L.; Colman, H. IDH1 and IDH2 Mutations in Gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345. [Google Scholar] [CrossRef]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the P53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466. [Google Scholar] [CrossRef]
- Amorim, J.P.; Santos, G.; Vinagre, J.; Soares, P. The Role of ATRX in the Alternative Lengthening of Telomeres (ALT) Phenotype. Genes 2016, 7, 66. [Google Scholar] [CrossRef]
- Jacome, M.A.; Wu, Q.; Piña, Y.; Etame, A.B. Evolution of Molecular Biomarkers and Precision Molecular Therapeutic Strategies in Glioblastoma. Cancers 2024, 16, 3635. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Liu, Y.; Sun, Y.; Wu, M. Insight into Deep Learning for Glioma IDH Medical Image Analysis: A Systematic Review. Medicine 2024, 103, e37150. [Google Scholar] [CrossRef] [PubMed]
- Brandmaier, A.; Hou, S.Q.; Shen, W.H. Cell Cycle Control by PTEN. J. Mol. Biol. 2017, 429, 2265. [Google Scholar] [CrossRef] [PubMed]
- Makino, R.; Higa, N.; Akahane, T.; Yonezawa, H.; Uchida, H.; Takajo, T.; Fujio, S.; Kirishima, M.; Hamada, T.; Yamahata, H.; et al. Alterations in EGFR and PDGFRA Are Associated with the Localization of Contrast-Enhancing Lesions in Glioblastoma. Neurooncol. Adv. 2023, 5, vdad110. [Google Scholar] [CrossRef]
- Darabi, S.; Xiu, J.; Samec, T.; Kesari, S.; Carrillo, J.; Aulakh, S.; Walsh, K.M.; Sengupta, S.; Sumrall, A.; Spetzler, D.; et al. Capicua (CIC) Mutations in Gliomas in Association with MAPK Activation for Exposing a Potential Therapeutic Target. Med. Oncol. 2023, 40, 197. [Google Scholar] [CrossRef]
- Fernando, T.M.; Piskol, R.; Bainer, R.; Sokol, E.S.; Trabucco, S.E.; Zhang, Q.; Trinh, H.; Maund, S.; Kschonsak, M.; Chaudhuri, S.; et al. Functional Characterization of SMARCA4 Variants Identified by Targeted Exome-Sequencing of 131,668 Cancer Patients. Nat. Commun. 2020, 11, 5551. [Google Scholar] [CrossRef]
- Noviandy, T.R.; Idroes, G.M.; Hardi, I. Integrating Explainable Artificial Intelligence and Light Gradient Boosting Machine for Glioma Grading. Inform. Health 2025, 2, 1–8. [Google Scholar] [CrossRef]
- Karakas, B.; Bachman, K.E.; Park, B.H. Mutation of the PIK3CA Oncogene in Human Cancers. Br. J. Cancer 2006, 94, 455–459. [Google Scholar] [CrossRef]
- Felder, M.; Kapur, A.; Gonzalez-Bosquet, J.; Horibata, S.; Heintz, J.; Albrecht, R.; Fass, L.; Kaur, J.; Hu, K.; Shojaei, H.; et al. MUC16 (CA125): Tumor Biomarker to Cancer Therapy, a Work in Progress. Mol. Cancer 2014, 13, 129. [Google Scholar] [CrossRef]
- Carrano, A.; Juarez, J.J.; Incontri, D.; Ibarra, A.; Cazares, H.G. Sex-Specific Differences in Glioblastoma. Cells 2021, 10, 1783. [Google Scholar] [CrossRef]
- Wang, G.M.; Cioffi, G.; Patil, N.; Waite, K.A.; Lanese, R.; Ostrom, Q.T.; Kruchko, C.; Berens, M.E.; Connor, J.R.; Lathia, J.D.; et al. Importance of the Intersection of Age and Sex to Understand Variation in Incidence and Survival for Primary Malignant Gliomas. Neuro Oncol. 2021, 24, 302. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cote, D.J.; Ascha, M.; Kruchko, C.; Barnholtz-Sloan, J.S. Adult Glioma Incidence and Survival by Race or Ethnicity in the United States From 2000 to 2014. JAMA Oncol. 2018, 4, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Rabin, E.E.; Huang, J.; Kim, M.; Mozny, A.; Lauing, K.L.; Penco-Campillo, M.; Zhai, L.; Bommi, P.; Mi, X.; Power, E.A.; et al. Age-Stratified Comorbid and Pharmacologic Analysis of Patients with Glioblastoma. Brain Behav. Immun. Health 2024, 38, 100753. [Google Scholar] [CrossRef] [PubMed]
- Nizamutdinov, D.; Stock, E.M.; Dandashi, J.A.; Vasquez, E.A.; Mao, Y.; Dayawansa, S.; Zhang, J.; Wu, E.; Fonkem, E.; Huang, J.H. Survival Outcomes Prognostication in Glioblastoma Diagnosed Patients. World Neurosurg. 2017, 109, e67. [Google Scholar] [CrossRef]
- Stabellini, N.; Krebs, H.; Patil, N.; Waite, K.; Barnholtz-Sloan, J.S. Sex Differences in Time to Treat and Outcomes for Gliomas. Front. Oncol. 2021, 11, 630597. [Google Scholar] [CrossRef]
- Colopi, A.; Fuda, S.; Santi, S.; Onorato, A.; Cesarini, V.; Salvati, M.; Balistrieri, C.R.; Dolci, S.; Guida, E. Impact of Age and Gender on Glioblastoma Onset, Progression, and Management. Mech. Ageing Dev. 2023, 211, 111801. [Google Scholar] [CrossRef]
- Wanis, H.A.; Møller, H.; Ashkan, K.; Davies, E.A. The Influence of Ethnicity on Survival from Malignant Primary Brain Tumours in England: A Population-Based Cohort Study. Cancers 2023, 15, 1464. [Google Scholar] [CrossRef]
- Jiang, W.; Rixiati, Y.; Kuerban, Z.; Simayi, A.; Huang, C.; Jiao, B. Racial/Ethnic Disparities and Survival in Pediatrics with Gliomas Based on the Surveillance, Epidemiology, and End Results Database in the United States. World Neurosurg. 2020, 141, e524–e529. [Google Scholar] [CrossRef]
- Zöllner, F.G.; Emblem, K.E.; Schad, L.R. SVM-Based Glioma Grading: Optimization by Feature Reduction Analysis. Z. Med. Phys. 2012, 22, 205–214. [Google Scholar] [CrossRef]
- Basthikodi, M.; Chaithrashree, M.; Ahamed Shafeeq, B.M.; Gurpur, A.P. Enhancing Multiclass Brain Tumor Diagnosis Using SVM and Innovative Feature Extraction Techniques. Sci. Rep. 2024, 14, 26023. [Google Scholar] [CrossRef]
- Kumar, A.; Jha, A.K.; Agarwal, J.P.; Yadav, M.; Badhe, S.; Sahay, A.; Epari, S.; Sahu, A.; Bhattacharya, K.; Chatterjee, A.; et al. Machine-Learning-Based Radiomics for Classifying Glioma Grade from Magnetic Resonance Images of the Brain. J. Pers. Med. 2023, 13, 920. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.F.; Al-Zurfi, A.N.; Abed, M.H.; Ahmed, K. An Effective Ensemble Learning Approach for Classification of Glioma Grades Based on Novel MRI Features. Sci. Rep. 2024, 14, 11977. [Google Scholar] [CrossRef] [PubMed]
- Bhatele, K.R.; Bhadauria, S.S. Machine Learning Application in Glioma Classification: Review and Comparison Analysis. Arch. Comput. Methods Eng. 2022, 29, 247–274. [Google Scholar] [CrossRef]
- Joo, B.; Ahn, S.S.; An, C.; Han, K.; Choi, D.; Kim, H.; Park, J.E.; Kim, H.S.; Lee, S.K. Fully Automated Radiomics-Based Machine Learning Models for Multiclass Classification of Single Brain Tumors: Glioblastoma, Lymphoma, and Metastasis. J. Neuroradiol. 2023, 50, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Vidyadharan, S.; Rao, B.V.V.S.N.P.; Yogeeswari, P.; Kesavadas, C.; Rajagopalan, V. Accurate Low and High Grade Glioma Classification Using Free Water Eliminated Diffusion Tensor Metrics and Ensemble Machine Learning. Sci. Rep. 2024, 14, 19844. [Google Scholar] [CrossRef]
- Dorfner, F.J.; Patel, J.B.; Kalpathy-Cramer, J.; Gerstner, E.R.; Bridge, C.P. A Review of Deep Learning for Brain Tumor Analysis in MRI. NPJ Precis. Oncol. 2025, 9, 2. [Google Scholar] [CrossRef]
- Mohamed Musthafa, M.; Mahesh, T.R.; Vinoth Kumar, V.; Guluwadi, S. Enhancing Brain Tumor Detection in MRI Images through Explainable AI Using Grad-CAM with Resnet 50. BMC Med. Imaging 2024, 24, 107. [Google Scholar] [CrossRef]
- Alshuhail, A.; Thakur, A.; Chandramma, R.; Mahesh, T.R.; Almusharraf, A.; Vinoth Kumar, V.; Khan, S.B. Refining Neural Network Algorithms for Accurate Brain Tumor Classification in MRI Imagery. BMC Med. Imaging 2024, 24, 118. [Google Scholar] [CrossRef]
- Hegazy, R.T.; Khalifa, S.K.; Mortada, R.A.; Amin, B.A.; Elfattah, A.A. Brain Tumor Classification: Leveraging Transfer Learning via EfficientNet-B0 Pretrained Model. Int. Integr. Intell. Syst. 2025, 2. [Google Scholar] [CrossRef]
- Sudha, G.; Saranya, S.; Manikandan, S.; Abdul Arshath, M.M.; Bharathan, S. Automated Glioma Detection Using Machine Learning Techniques. In Proceedings of the 4th International Conference on Power, Energy, Control and Transmission Systems: Harnessing Power and Energy for an Affordable Electrification of India, ICPECTS 2024, Chennai, India, 10–11 December 2020. [Google Scholar] [CrossRef]
- Fountzilas, E.; Pearce, T.; Baysal, M.A.; Chakraborty, A.; Tsimberidou, A.M. Convergence of Evolving Artificial Intelligence and Machine Learning Techniques in Precision Oncology. NPJ Digit. Med. 2025, 8, 75. [Google Scholar] [CrossRef]
- Lin, H.; Liu, C.; Hu, A.; Zhang, D.; Yang, H.; Mao, Y. Understanding the Immunosuppressive Microenvironment of Glioma: Mechanistic Insights and Clinical Perspectives. J. Hematol. Oncol. 2024, 17, 31. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Wang, Y.; Yuan, L.; Ye, L.; Hu, Y.; Cheng, H.; Li, Y. Machine Learning-Based New Classification for Immune Infiltration of Gliomas. PLoS ONE 2024, 19, e0312071. [Google Scholar] [CrossRef]
- Azeez, O.; Azeez, O.A.; Abdulazeez, A.M. Classification of Brain Tumor Based on Machine Learning Algorithms: A Review. J. Appl. Sci. Technol. Trends 2025, 6, 1–15. [Google Scholar] [CrossRef]
- Tasci, E.; Popa, M.; Zhuge, Y.; Chappidi, S.; Zhang, L.; Cooley Zgela, T.; Sproull, M.; Mackey, M.; Kates, H.R.; Garrett, T.J.; et al. MetaWise: Combined Feature Selection and Weighting Method to Link the Serum Metabolome to Treatment Response and Survival in Glioblastoma. Int. J. Mol. Sci. 2024, 25, 10965. [Google Scholar] [CrossRef]
- Labory, J.; Njomgue-Fotso, E.; Bottini, S. Benchmarking Feature Selection and Feature Extraction Methods to Improve the Performances of Machine-Learning Algorithms for Patient Classification Using Metabolomics Biomedical Data. Comput. Struct. Biotechnol. J. 2024, 23, 1274–1287. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Z.; Wang, Y. Utilizing Feature Selection Techniques for AI-Driven Tumor Subtype Classification: Enhancing Precision in Cancer Diagnostics. Biomolecules 2025, 15, 81. [Google Scholar] [CrossRef] [PubMed]
- Ting-Yu, C.; Yang, L.; Liang, C.; Jie, L.; Chao, Z.; Xian-Feng, S. Identification of the Potential Biomarkers in Patients with Glioma: A Weighted Gene Co-Expression Network Analysis. Carcinogenesis 2019, 41, 743. [Google Scholar] [CrossRef]
- Li, Y.; Sun, H. Multi-Omics Analysis Identifies Novels Genes Involved in Glioma Prognosis. Sci. Rep. 2025, 15, 5806. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Cheng, J.; Xia, J.; Yang, Z.; Xu, L. Identification of Critical Biomarkers and Immune Landscape Patterns in Glioma Based on Multi-Database. Discov. Oncol. 2025, 16, 35. [Google Scholar] [CrossRef]
- Liu, Y.; Kannan, K.; Huse, J.; Hickman, R.; Miller, A.M.; Holle, B.M.; Jee, J.; Liu, S.-Y.; Ross, D.; Yu, H.; et al. BIOM-49. Patient-centric integrated graph database reveals critical biomarkers in the recurrence of idh wild-type glioma. Neuro Oncol. 2024, 26, viii30. [Google Scholar] [CrossRef]
- Carrilho, J.F.; Coletti, R.; Costa, B.M.; Lopes, M.B. Multi-Omics Biomarker Selection and Outlier Detection across WHO Glioma Classifications via Robust Sparse Multinomial Regression. medRxiv 2024. medRxiv:2024.08.26.24312601. [Google Scholar] [CrossRef]
- Vieira, F.G.; Bispo, R.; Lopes, M.B. Integration of Multi-Omics Data for the Classification of Glioma Types and Identification of Novel Biomarkers. Bioinform. Biol. Insights 2024, 18, 11779322241249564. [Google Scholar] [CrossRef] [PubMed]
- Paplomatas, P.; Douroumi, I.E.; Vlamos, P.; Vrahatis, A. Genetic Optimization in Uncovering Biologically Meaningful Gene Biomarkers for Glioblastoma Subtypes. BioMedInformatics 2024, 4, 811–822. [Google Scholar] [CrossRef]
- Cattelani, L.; Ghosh, A.; Rintala, T.; Fortino, V. A Comprehensive Evaluation Framework for Benchmarking Multi-Objective Feature Selection in Omics-Based Biomarker Discovery. IEEE/ACM Trans. Comput. Biol. Bioinform. 2024, 21, 2432–2446. [Google Scholar] [CrossRef]
- Tasci, E.; Zhuge, Y.; Kaur, H.; Camphausen, K.; Krauze, A.V. Hierarchical Voting-Based Feature Selection and Ensemble Learning Model Scheme for Glioma Grading with Clinical and Molecular Characteristics. Int. J. Mol. Sci. 2022, 23, 14155. [Google Scholar] [CrossRef] [PubMed]
- Harding-Larsen, D.; Funk, J.; Madsen, N.G.; Gharabli, H.; Acevedo-Rocha, C.G.; Mazurenko, S.; Welner, D.H. Protein Representations: Encoding Biological Information for Machine Learning in Biocatalysis. Biotechnol. Adv. 2024, 77, 108459. [Google Scholar] [CrossRef] [PubMed]
- Data Standardization: How to Do It and Why It Matters|Built In. Available online: https://builtin.com/data-science/when-and-why-standardize-your-data (accessed on 24 February 2025).
- Train-Test Split for Evaluating Machine Learning Algorithms—MachineLearningMastery.Com. Available online: https://machinelearningmastery.com/train-test-split-for-evaluating-machine-learning-algorithms/ (accessed on 24 February 2025).
- Kursa, M.B.; Jankowski, A.; Rudnicki, W.R. Boruta—A System for Feature Selection. Fundam Inf. 2010, 101, 271–285. [Google Scholar] [CrossRef]
- Habibi, A.; Delavar, M.R.; Sadeghian, M.S.; Nazari, B.; Pirasteh, S. A Hybrid of Ensemble Machine Learning Models with RFE and Boruta Wrapper-Based Algorithms for Flash Flood Susceptibility Assessment. Int. J. Appl. Earth Obs. Geoinf. 2023, 122, 103401. [Google Scholar] [CrossRef]
- Sarkar, D.; Bali, R.; Sharma, T. Feature Engineering and Selection. In Practical Machine Learning with Python: A Problem-Solver’s Guide to Building Real-World Intelligent Systems; Sarkar, D., Bali, R., Sharma, T., Eds.; Apress: Berkeley, CA, USA, 2018; pp. 177–253. ISBN 978-1-4842-3207-1. [Google Scholar]
- Chatterjee, T.; Chowdhury, R. Improved Sparse Approximation Models for Stochastic Computations. In Handbook of Neural Computation; Elsevier Inc.: Amsterdam, The Netherlands, 2017; pp. 201–223. ISBN 9780128113196. [Google Scholar]
- Hastie, T.; Tibshirani, R.; Friedman, J. Linear Methods for Regression. In The Elements of Statistical Learning: Data Mining, Inference, and Prediction; Hastie, T., Tibshirani, R., Friedman, J., Eds.; Springer: New York, NY, USA, 2009; pp. 43–99. ISBN 978-0-387-84858-7. [Google Scholar]
- Santos, M.R.; Guedes, A.; Sanchez-Gendriz, I. SHapley Additive ExPlanations (SHAP) for Efficient Feature Selection in Rolling Bearing Fault Diagnosis. Mach. Learn. Knowl. Extr. 2024, 6, 316–341. [Google Scholar] [CrossRef]
- Lundberg, S.M.; Lee, S.I. A Unified Approach to Interpreting Model Predictions. In Advances in Neural Information Processing Systems; MIT Press: Cambridge, MA, USA, 2017; pp. 4766–4775. [Google Scholar]
- Adnan, M.; Alarood, A.A.S.; Uddin, M.I.; Rehman, I. ur Utilizing Grid Search Cross-Validation with Adaptive Boosting for Augmenting Performance of Machine Learning Models. PeerJ Comput. Sci. 2022, 8, e803. [Google Scholar] [CrossRef]
- Bulagang, A.F.; Weng, N.G.; Mountstephens, J.; Teo, J. A Review of Recent Approaches for Emotion Classification Using Electrocardiography and Electrodermography Signals. Inf. Med. Unlocked 2020, 20, 100363. [Google Scholar] [CrossRef]
- Thomas, N.S.; Kaliraj, S. An Improved and Optimized Random Forest Based Approach to Predict the Software Faults. SN Comput. Sci. 2024, 5, 530. [Google Scholar] [CrossRef]
- Cervantes, J.; Garcia-Lamont, F.; Rodríguez-Mazahua, L.; Lopez, A. A Comprehensive Survey on Support Vector Machine Classification: Applications, Challenges and Trends. Neurocomputing 2020, 408, 189–215. [Google Scholar] [CrossRef]
- Xia, Y. Chapter Eleven—Correlation and Association Analyses in Microbiome Study Integrating Multiomics in Health and Disease. In Progress in Molecular Biology and Translational Science; Sun, J., Ed.; Academic Press: Cambridge, MA, USA, 2020; Volume 171, pp. 309–491. ISBN 1877–1173. [Google Scholar]
- Cao, Y.; Forssten, M.P.; Sarani, B.; Montgomery, S.; Mohseni, S. Development and Validation of an XGBoost-Algorithm-Powered Survival Model for Predicting In-Hospital Mortality Based on 545,388 Isolated Severe Traumatic Brain Injury Patients from the TQIP Database. J. Pers. Med. 2023, 13, 1401. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar] [CrossRef]
- Pavlou, M.; Ambler, G.; Seaman, S.; De iorio, M.; Omar, R.Z. Review and Evaluation of Penalised Regression Methods for Risk Prediction in Low-dimensional Data with Few Events. Stat. Med. 2015, 35, 1159. [Google Scholar] [CrossRef] [PubMed]
- Zabor, E.C.; Reddy, C.A.; Tendulkar, R.D.; Patil, S. Logistic Regression in Clinical Studies. Int. J. Radiat. Oncol. Biol. Phys. 2022, 112, 271–277. [Google Scholar] [CrossRef]
- Montesinos López, O.A.; Montesinos López, A.; Crossa, J. Overfitting, Model Tuning, and Evaluation of Prediction Performance. In Multivariate Statistical Machine Learning Methods for Genomic Prediction; Montesinos López, O.A., Montesinos López, A., Crossa, J., Eds.; Springer International Publishing: Cham, Switzerland, 2022; pp. 109–139. ISBN 978-3-030-89010-0. [Google Scholar]
- Dalianis, H. Evaluation Metrics and Evaluation. In Clinical Text Mining: Secondary Use of Electronic Patient Records; Dalianis, H., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 45–53. [Google Scholar]
- Nahm, F.S. Receiver Operating Characteristic Curve: Overview and Practical Use for Clinicians. Korean J. Anesth. 2022, 75, 25. [Google Scholar] [CrossRef]
- Bahar, R.C.; Merkaj, S.; Cassinelli Petersen, G.I.; Tillmanns, N.; Subramanian, H.; Brim, W.R.; Zeevi, T.; Staib, L.; Kazarian, E.; Lin, M.D.; et al. Machine Learning Models for Classifying High- and Low-Grade Gliomas: A Systematic Review and Quality of Reporting Analysis. Front. Oncol. 2022, 12, 856231. [Google Scholar] [CrossRef]
- Hashido, T.; Saito, S.; Ishida, T. Radiomics-Based Machine Learning Classification for Glioma Grading Using Diffusion- And Perfusion-Weighted Magnetic Resonance Imaging. J. Comput. Assist Tomogr. 2021, 45, 606–613. [Google Scholar] [CrossRef]
- Wang, Z.; Xiao, X.; He, K.; Wu, D.; Pang, P.; Wu, T. A Study of MRI-Based Machine-Learning Methods for Glioma Grading. Int. J. Radiat. Res. 2022, 20, 115–120. [Google Scholar] [CrossRef]
- Rosenblatt, M.; Tejavibulya, L.; Jiang, R.; Noble, S.; Scheinost, D. Data Leakage Inflates Prediction Performance in Connectome-Based Machine Learning Models. Nat. Commun. 2024, 15, 1829. [Google Scholar] [CrossRef]
- Mallampati, S.B.; Hari, S. A Comparative Study on the Impacts of Data Leakage During Feature Selection Using the CIC-IoT 2023 Intrusion Detection Dataset. In Proceedings of the 10th International Conference on Electrical Energy Systems, ICEES 2024, Chennai, India, 22–24 August 2024. [Google Scholar]
- Piovani, D.; Sokou, R.; Tsantes, A.G.; Vitello, A.S.; Bonovas, S. Optimizing Clinical Decision Making with Decision Curve Analysis: Insights for Clinical Investigators. Healthcare 2023, 11, 2244. [Google Scholar] [CrossRef]
- Gerds, T.A.; Andersen, P.K.; Kattan, M.W. Calibration Plots for Risk Prediction Models in the Presence of Competing Risks. Stat. Med. 2014, 33, 3191–3203. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Rousson, V.; Lee, W.-C.; Ferdynus, C.; Chen, M.; Qian, X.; Guo, Y.; written on behalf of AME Big-Data Clinical Trial Collaborative Group. Decision Curve Analysis: A Technical Note. Ann. Transl. Med. 2018, 6, 308. [Google Scholar] [CrossRef]
- Tian, Y.; Chen, L.; Jiang, Y. LASSO-Based Screening for Potential Prognostic Biomarkers Associated with Glioblastoma. Front. Oncol. 2023, 12, 1057383. [Google Scholar] [CrossRef]
- Mirchia, K.; Pan, S.; Payne, E.; Liu, J.; Peeran, Z.; Shukla, P.; Young, J.; Gupta, R.; Wu, J.; Pak, J.; et al. PATH-53. DNA mutation sequencing and methylation analysis of somaticnf1 mutant idh-wildtype glioblastoma identifies three epigenetic groups andcdkn2a/b loss as a negative prognostic biomarker. Neuro Oncol. 2024, 26, viii191. [Google Scholar] [CrossRef]
- Noor, H.; Briggs, N.E.; McDonald, K.L.; Holst, J.; Vittorio, O. Tp53 Mutation Is a Prognostic Factor in Lower Grade Glioma and May Influence Chemotherapy Efficacy. Cancers 2021, 13, 5362. [Google Scholar] [CrossRef] [PubMed]
- Rathore, F.A.; Khan, H.S.; Ali, H.M.; Obayya, M.; Rasheed, S.; Hussain, L.; Kazmi, Z.H.; Nour, M.K.; Mohamed, A.; Motwakel, A. Survival Prediction of Glioma Patients from Integrated Radiology and Pathology Images Using Machine Learning Ensemble Regression Methods. Appl. Sci. 2022, 12, 10357. [Google Scholar] [CrossRef]
- Zhao, R.; Zhuge, Y.; Camphausen, K.; Krauze, A.V. Machine Learning Based Survival Prediction in Glioma Using Large-Scale Registry Data. Health Inform. J 2022, 28, 14604582221135427. [Google Scholar] [CrossRef]
- Agrawal, A.; Maan, V. Computational Predictions of MGMT Promoter Methylation in Gliomas: A Mathematical Radiogenomics Approach. Commun. Appl. Nonlinear Anal. 2024, 31, 229–252. [Google Scholar] [CrossRef]
- Du, P.; Liu, X.; Wu, X.; Chen, J.; Cao, A.; Geng, D. Predicting Histopathological Grading of Adult Gliomas Based on Preoperative Conventional Multimodal MRI Radiomics: A Machine Learning Model. Brain Sci. 2023, 13, 912. [Google Scholar] [CrossRef]
- Liang, H.X.; Wang, Z.Y.; Li, Y.; Ren, A.N.; Chen, Z.F.; Wang, X.Z.; Wang, X.M.; Yuan, Z.G. The Application Value of Support Vector Machine Model Based on Multimodal MRI in Predicting IDH-1mutation and Ki-67 Expression in Glioma. BMC Med. Imaging 2024, 24, 244. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhang, X.; Wang, Y.; Li, H.; Qi, Z.; Du, Z.; Chu, Y.; Feng, D.; Xie, Q.; Song, J.; et al. Multimodal Data Integration Using Deep Learning Predicts Overall Survival of Patients with Glioma. View 2024, 5, 20240001. [Google Scholar] [CrossRef]
- A Ilemobayo, J.; Durodola, O.; Alade, O.; J Awotunde, O.; T Olanrewaju, A.; Falana, O.; Ogungbire, A.; Osinuga, A.; Ogunbiyi, D.; Ifeanyi, A.; et al. Hyperparameter Tuning in Machine Learning: A Comprehensive Review. J. Eng. Res. Rep. 2024, 26, 388–395. [Google Scholar] [CrossRef]
- Likhitha, G.; Sree, B.R.; Ratan, C.; Karthikeyan, C.; Samkumar, G.V. Advancing Brain Tumor Classification Using CNN and EXtreme Gradient Boosting. In Proceedings of the 2024 International Conference on Expert Clouds and Applications, Bengaluru, India, 18–19 April 2024; pp. 985–991. [Google Scholar] [CrossRef]
- Yan, Z.; Wang, J.; Dong, Q.; Zhu, L.; Lin, W.; Jiang, X. XGBoost Algorithm and Logistic Regression to Predict the Postoperative 5-Year Outcome in Patients with Glioma. Ann. Transl. Med. 2022, 10, 860. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Rue, J.; Mohinta, S.; Rees, J.; Brandner, S.; Nachev, P.; Hyare, H.; Bakhsh, A.; Scott, I.; Jenkinson, M.; et al. Mathematical modelling of survival in low grade gliomas at malignant transformation with xgboost. Neuro Oncol. 2024, 26, vii12–vii13. [Google Scholar] [CrossRef]
- Mitchell, R.; Frank, E. Accelerating the XGBoost Algorithm Using GPU Computing. PeerJ Comput. Sci. 2017, 3, e127. [Google Scholar] [CrossRef]
- Chen, D.W.; Miao, R.; Deng, Z.Y.; Lu, Y.Y.; Liang, Y.; Huang, L. Sparse Logistic Regression with L1/2 Penalty for Emotion Recognition in Electroencephalography Classification. Front. Neuroinform. 2020, 14, 29. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Song, S.; Li, X.; Wu, F.; Li, B.; Wu, Q. Prediction of Stroke Hematoma Expansion Using a Machine Learning Model with Stacked Generalization. In Proceedings of the 2024 IEEE/ACIS 24th International Conference on Computer and Information Science, ICIS 2024—Proceedings, Shanghai, China, 20–22 September 2024; pp. 90–94. [Google Scholar]
- Singh, P.; Hasija, T.; Ramkumar, K.R. Optimizing Phishing Detection Systems with Ensemble Learning: Insights from a Multi-Model Voting Classifier. In Proceedings of the 5th International Conference on Smart Electronics and Communication, ICOSEC 2024, Kongunadu, India, 18–20 September 2024; pp. 1336–1341. [Google Scholar]
- Akhy, S.A.; Mia, M.B.; Mustafa, S.; Chakraborti, N.R.; Krishnachalitha, K.C.; Rabbany, G. A Comprehensive Study on Ensemble Feature Selection Techniques for Classification. In Proceedings of the 2024 11th International Conference on Computing for Sustainable Global Development, INDIACom 2024, New Delhi, India, 28 February–1 March 2024; pp. 1319–1324. [Google Scholar]
- Ballard, J.L.; Wang, Z.; Li, W.; Shen, L.; Long, Q. Deep Learning-Based Approaches for Multi-Omics Data Integration and Analysis. BioData Min. 2024, 17, 38. [Google Scholar] [CrossRef]
- Munquad, S.; Das, A.B. DeepAutoGlioma: A Deep Learning Autoencoder-Based Multi-Omics Data Integration and Classification Tools for Glioma Subtyping. BioData Min. 2023, 16, 32. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Best Parameters |
|---|---|
| Random Forest | {‘n_estimators’: 300, ‘min_samples_split’: 5, ‘min_samples_leaf’: 2, ‘max_depth’: 30} |
| SVM | {‘kernel’: ‘rbf’, ‘gamma’: ‘auto’, ‘C’: 10} |
| XGBoost | {‘subsample’: 0.7, ‘n_estimators’: 100, ‘max_depth’: 3, ‘learning_rate’: 0.01} |
| Logistic Regression | {‘solver’: ‘liblinear’, ‘penalty’: ‘l1’, ‘C’: 1} |
| Dataset | Model | Precision | Recall | F1-Score | Accuracy |
|---|---|---|---|---|---|
| Training set | Random Forest | 0.9044 | 0.9016 | 0.9020 | 0.9016 |
| SVM | 0.8962 | 0.8822 | 0.8890 | 0.8822 | |
| XGBoost | 0.8819 | 0.8748 | 0.8755 | 0.8748 | |
| Logistic Regression | 0.8863 | 0.8809 | 0.8816 | 0.8792 | |
| Testing set | Random Forest | 0.8523 | 0.8453 | 0.8488 | 0.8453 |
| SVM | 0.8604 | 0.8571 | 0.8588 | 0.8571 | |
| XGBoost | 0.8711 | 0.8630 | 0.8670 | 0.8630 | |
| Logistic Regression | 0.8924 | 0.8809 | 0.8866 | 0.8809 |
| Model | Threshold | Sensitivity | Specificity | PPV | NPV | NNS |
|---|---|---|---|---|---|---|
| Random Forest | 0.3 | 0.929 | 0.786 | 0.756 | 0.939 | 1.323 |
| 0.5 | 0.900 | 0.827 | 0.788 | 0.920 | 1.270 | |
| 0.7 | 0.600 | 0.918 | 0.840 | 0.763 | 1.190 | |
| SVM | 0.3 | 0.943 | 0.816 | 0.786 | 0.952 | 1.273 |
| 0.5 | 0.886 | 0.837 | 0.795 | 0.911 | 1.258 | |
| 0.7 | 0.829 | 0.857 | 0.806 | 0.875 | 1.241 | |
| XGBoost | 0.3 | 0.957 | 0.786 | 0.761 | 0.962 | 1.313 |
| 0.5 | 0.929 | 0.816 | 0.783 | 0.941 | 1.277 | |
| 0.7 | 0.257 | 0.980 | 0.900 | 0.649 | 1.111 | |
| Logistic Regression | 0.3 | 0.943 | 0.816 | 0.786 | 0.952 | 1.273 |
| 0.5 | 0.914 | 0.857 | 0.821 | 0.933 | 1.219 | |
| 0.7 | 0.671 | 0.898 | 0.825 | 0.793 | 1.213 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samara, M.N.; Harry, K.D. Integrating Boruta, LASSO, and SHAP for Clinically Interpretable Glioma Classification Using Machine Learning. BioMedInformatics 2025, 5, 34. https://doi.org/10.3390/biomedinformatics5030034
Samara MN, Harry KD. Integrating Boruta, LASSO, and SHAP for Clinically Interpretable Glioma Classification Using Machine Learning. BioMedInformatics. 2025; 5(3):34. https://doi.org/10.3390/biomedinformatics5030034
Chicago/Turabian StyleSamara, Mohammad Najeh, and Kimberly D. Harry. 2025. "Integrating Boruta, LASSO, and SHAP for Clinically Interpretable Glioma Classification Using Machine Learning" BioMedInformatics 5, no. 3: 34. https://doi.org/10.3390/biomedinformatics5030034
APA StyleSamara, M. N., & Harry, K. D. (2025). Integrating Boruta, LASSO, and SHAP for Clinically Interpretable Glioma Classification Using Machine Learning. BioMedInformatics, 5(3), 34. https://doi.org/10.3390/biomedinformatics5030034