
Identification of a New Lung Cancer Biomarker Signature Using Data Mining and Preliminary In Vitro Validation


Abstract
1. Introduction
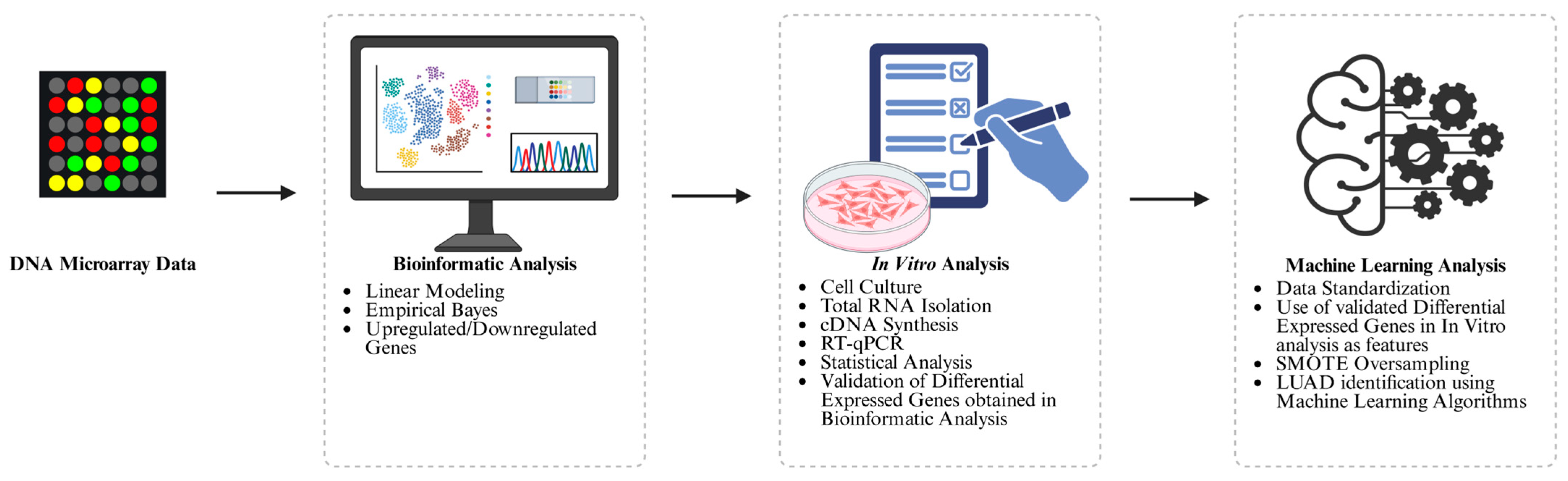
2. Materials and Methods
2.1. Data Description
2.2. Methodology
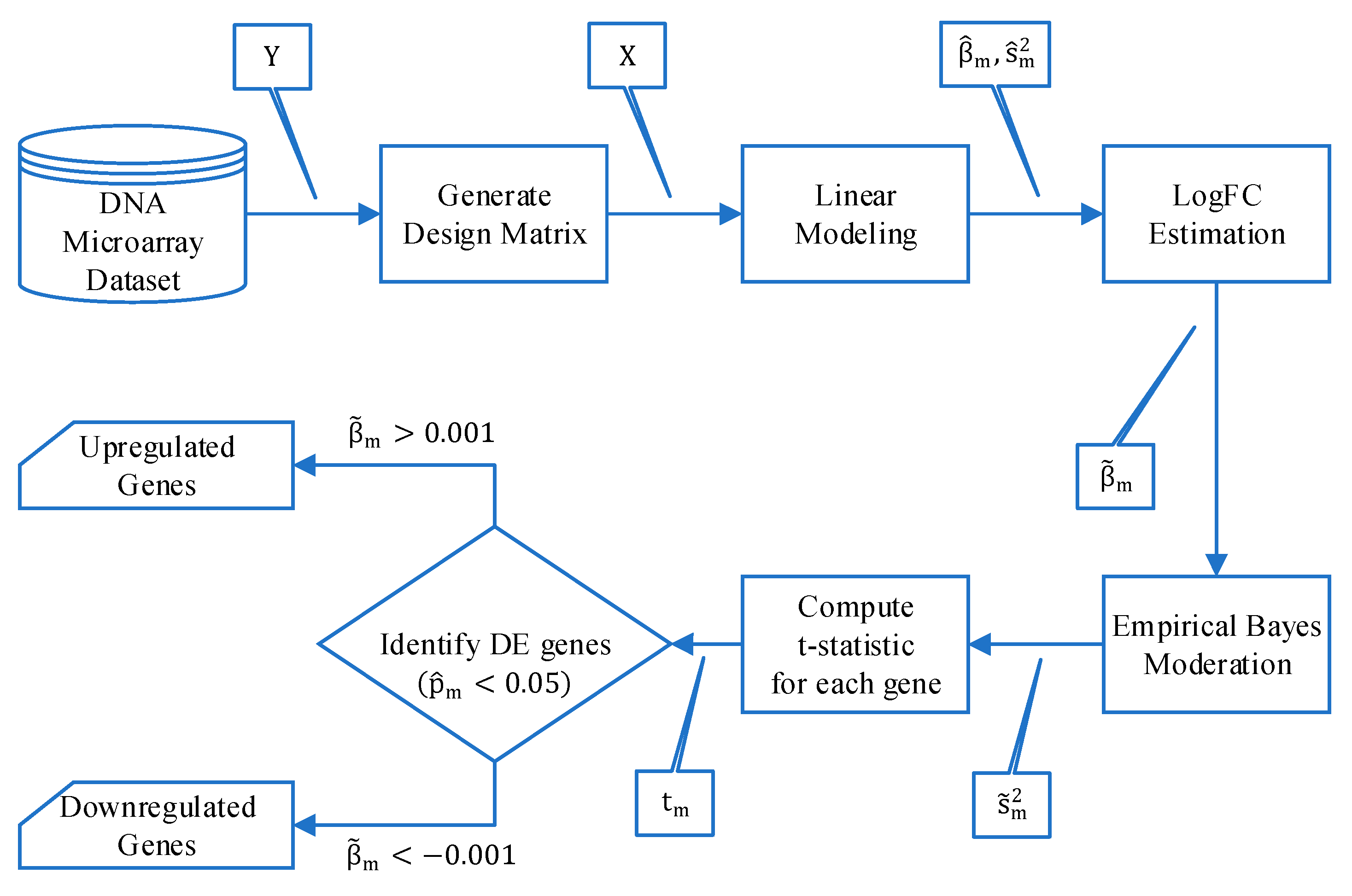
2.2.1. Bioinformatics Analysis
Preprocessing
Biomarker Extraction
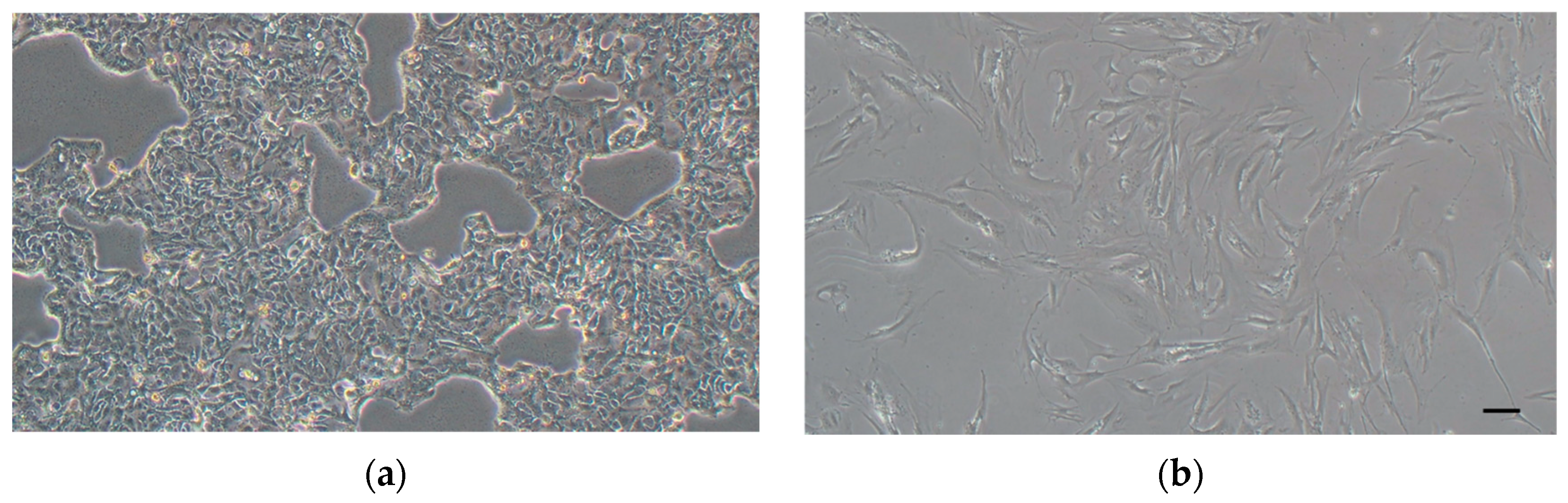
2.2.2. In Vitro Analysis
2.2.3. Machine Learning Analysis
3. Results
3.1. Bioinformatics Analysis
3.2. In Vitro Analysis
3.3. Machine Learning Analysis
4. Discussion and Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| LUAD | Lung adenocarcinoma |
| NSCLC | Non-small cell lung cancer |
| ML | Machine learning |
| SMOTE | Synthetic Minority Oversampling Technique |
| GEO | Gene Expression Omnibus |
| RMA | Robust Multi-array Average |
| DE | Differentially expressed |
| mRMR | minimum Redundancy Maximum Relevance |
| LASSO | Least Absolute Shrinkage and Selection Operator |
| SVM | Support Vector Machine |
| RF | Random Forest |
| LR | Logistic Regression |
| XGBoost | Extreme Gradient Boosting |
| GB | Gradient Boosting |
| AB | AdaBoost |
| ET | Extra Trees |
| kNN | k-Nearest Neighbors |
| LDA | Linear Discriminant Analysis |
References
- Haghjoo, N.; Moeini, A.; Masoudi-Nejad, A. Introducing a Panel for Early Detection of Lung Adenocarcinoma by Using Data Integration of Genomics, Epigenomics, Transcriptomics and Proteomics. Exp. Mol. Pathol. 2020, 112, 104360. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Linear Models and Empirical Bayes Methods for Assessing Differential Expression in Microarray Experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Carlin, B.P.; Louis, T.A. Empirical Bayes: Past, Present and Future. J. Am. Stat. Assoc. 2000, 95, 1286–1289. [Google Scholar] [CrossRef]
- Ding, C.; Peng, H. Minimum Redundancy Feature Selection from Microarray Gene Expression Data. J. Bioinform. Comput. Biol. 2005, 3, 185–205. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression Shrinkage and Selection Via the Lasso. J. R. Stat. Soc. Ser. B Stat. Methodol. 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. SMOTE: Synthetic Minority Over-Sampling Technique. JAIR 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Edgar, R. Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Zhang, Y.; Foreman, O.; Wigle, D.A.; Kosari, F.; Vasmatzis, G.; Salisbury, J.L.; van Deursen, J.; Galardy, P.J. USP44 Regulates Centrosome Positioning to Prevent Aneuploidy and Suppress Tumorigenesis. J. Clin. Invest. 2012, 122, 4362–4374. [Google Scholar] [CrossRef]
- Rousseaux, S.; Debernardi, A.; Jacquiau, B.; Vitte, A.-L.; Vesin, A.; Nagy-Mignotte, H.; Moro-Sibilot, D.; Brichon, P.-Y.; Lantuejoul, S.; Hainaut, P.; et al. Ectopic Activation of Germline and Placental Genes Identifies Aggressive Metastasis-Prone Lung Cancers. Sci. Transl. Med. 2013, 5, 186ra66. [Google Scholar] [CrossRef]
- Tarca, A.L.; Lauria, M.; Unger, M.; Bilal, E.; Boue, S.; Kumar Dey, K.; Hoeng, J.; Koeppl, H.; Martin, F.; Meyer, P.; et al. Strengths and Limitations of Microarray-Based Phenotype Prediction: Lessons Learned from the IMPROVER Diagnostic Signature Challenge. Bioinformatics 2013, 29, 2892–2899. [Google Scholar] [CrossRef]
- Sanchez-Palencia, A.; Gomez-Morales, M.; Gomez-Capilla, J.A.; Pedraza, V.; Boyero, L.; Rosell, R.; Fárez-Vidal, M.E. Gene Expression Profiling Reveals Novel Biomarkers in Nonsmall Cell Lung Cancer. Int. J. Cancer 2011, 129, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Botling, J.; Edlund, K.; Lohr, M.; Hellwig, B.; Holmberg, L.; Lambe, M.; Berglund, A.; Ekman, S.; Bergqvist, M.; Pontén, F.; et al. Biomarker Discovery in Non-Small Cell Lung Cancer: Integrating Gene Expression Profiling, Meta-Analysis, and Tissue Microarray Validation. Clin. Cancer Res. 2013, 19, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A. Exploration, Normalization, and Summaries of High Density Oligonucleotide Array Probe Level Data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.I.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-Vector Networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Nelder, J.A.; Wedderburn, R.W. Generalized Linear Models. J. R. Stat. Soc. Ser. A Stat. Soc. 1972, 135, 370–384. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. Xgboost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM Sigkdd International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- Friedman, J.H. Greedy Function Approximation: A Gradient Boosting Machine. Ann. Stat. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- Freund, Y.; Schapire, R.E. A Decision-Theoretic Generalization of on-Line Learning and an Application to Boosting. J. Comput. Syst. Sci. 1997, 55, 119–139. [Google Scholar] [CrossRef]
- Geurts, P.; Ernst, D.; Wehenkel, L. Extremely Randomized Trees. Mach. Learn. 2006, 63, 3–42. [Google Scholar] [CrossRef]
- Fix, E.; Hodges, J.L. Discriminatory Analysis. Nonparametric Discrimination: Consistency Properties. Int. Stat. Rev./Rev. Int. Stat. 1989, 57, 238. [Google Scholar] [CrossRef]
- Hart, P.E.; Stork, D.G.; Duda, R.O. Pattern Classification; Wiley Hoboken: Hoboken, NJ, USA, 2000; ISBN 0-471-05669-3. [Google Scholar]
- Lyu, Y.; Wang, Q.; Liang, J.; Zhang, L.; Zhang, H. The Ion Channel Gene KCNAB2 Is Associated with Poor Prognosis and Loss of Immune Infiltration in Lung Adenocarcinoma. Cells 2022, 11, 3438. [Google Scholar] [CrossRef]
- Li, Y.; Niu, J.; Sun, Z.; Liu, J. FTO-mediated m6A Methylation of KCNAB2 Inhibits Tumor Property of Non-Small Cell Lung Cancer Cells and M2 Macrophage Polarization by Inactivating the PI3K/AKT Pathway. J. Biochem. Mol. Toxicol. 2025, 39, e70232. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Shen, J.; Fei, J.; Zhu, X.; Yin, M.; Zhou, J. KNDC1 Is a Predictive Marker of Malignant Transformation in Borderline Ovarian Tumors. OncoTargets Ther. 2020, 2020, 709–718. [Google Scholar] [CrossRef]
- Zhang, Y.; Fan, Q.; Guo, Y.; Zhu, K. Eight-Gene Signature Predicts Recurrence in Lung Adenocarcinoma. Cancer Biomark. 2020, 28, 447–457. [Google Scholar] [CrossRef]
- Xia, L.; Ma, H. Identification of a Novel Signature Based on Ferritinophagy-Related Genes to Predict Prognosis in Lung Adenocarcinoma: Focus on AHNAK2. Bioengineering 2024, 11, 1070. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, G.; Cui, T.; Lv, L. Adhesion GPR123 Is an Indicator for Recurrence and Prognosis in Bladder Cancer. Genes. Genom. 2021, 43, 1317–1325. [Google Scholar] [CrossRef]
- Hou, L.; Zhang, S.; Yu, W.; Yang, X.; Shen, M.; Hao, X.; Ren, X.; Sun, Q. Single-Cell Transcriptomics Reveals Tumor-Infiltrating B Cell Function after Neoadjuvant Pembrolizumab and Chemotherapy in Non-Small Cell Lung Cancer. J. Leukoc. Biol. 2024, 116, 555–564. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Training Samples | Test Samples | Total Samples |
|---|---|---|---|
| LUAD | 301 | 75 | 376 |
| Control | 91 | 23 | 114 |
| Total | 392 | 98 | 490 |
| Gene ID | LASSO | mRMR | |
|---|---|---|---|
| KCNAB2 | −0.066271 | −0.001912 | 95.929278 |
| SLC15A1 | 0.075376 | 0.002642 | 39.628941 |
| KNDC1 | −0.061559 | −0.001577 | 36.992438 |
| GPR123 (ADGRA1) | 0.002207 | 0.000941 | 0.575879 |
| GPR183 | −0.00581 | 0.000049 | 0.190521 |
| Gene Features | Oversampling | Best ML Model | Acc. | F1 | Prec | Recall | Spec. | AUC |
|---|---|---|---|---|---|---|---|---|
| SLC15A1, KNDC1 | No | ET | 79.39 | 80.63 | 84.82 | 79.39 | 82.24 | 87.85 |
| SMOTE | SVM linear | 78.16 | 74.89 | 82.45 | 74.18 | 82.45 | 87.94 | |
| KCNAB2, SLC15A1, KNDC1 | No | ET | 82.65 | 83.34 | 85.71 | 82.65 | 80.86 | 90.85 |
| SMOTE | SVM linear | 82.65 | 78.78 | 83.55 | 77.40 | 83.55 | 90.84 | |
| ADGRA1, KCNAB2, SLC15A1, KNDC1 | No | ET | 84.08 | 84.60 | 86.18 | 84.08 | 80.00 | 92.25 |
| SMOTE | ET | 85.10 | 79.92 | 80.86 | 79.55 | 80.86 | 91.72 | |
| ADGRA1, KCNAB2, SLC15A1, GPR183, KNDC1 | No | SVM linear | 83.67 | 84.49 | 87.18 | 83.67 | 84.74 | 91.71 |
| SMOTE | RF | 86.12 | 81.54 | 83.11 | 81.51 | 83.11 | 91.46 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ben Ali, F.; Mustafov, D.; Braoudaki, M.; Adeleke, S.; Mporas, I. Identification of a New Lung Cancer Biomarker Signature Using Data Mining and Preliminary In Vitro Validation. BioMedInformatics 2025, 5, 32. https://doi.org/10.3390/biomedinformatics5020032
Ben Ali F, Mustafov D, Braoudaki M, Adeleke S, Mporas I. Identification of a New Lung Cancer Biomarker Signature Using Data Mining and Preliminary In Vitro Validation. BioMedInformatics. 2025; 5(2):32. https://doi.org/10.3390/biomedinformatics5020032
Chicago/Turabian StyleBen Ali, Ferid, Denis Mustafov, Maria Braoudaki, Sola Adeleke, and Iosif Mporas. 2025. "Identification of a New Lung Cancer Biomarker Signature Using Data Mining and Preliminary In Vitro Validation" BioMedInformatics 5, no. 2: 32. https://doi.org/10.3390/biomedinformatics5020032
APA StyleBen Ali, F., Mustafov, D., Braoudaki, M., Adeleke, S., & Mporas, I. (2025). Identification of a New Lung Cancer Biomarker Signature Using Data Mining and Preliminary In Vitro Validation. BioMedInformatics, 5(2), 32. https://doi.org/10.3390/biomedinformatics5020032