
Cinco de Bio: A Low-Code Platform for Domain-Specific Workflows for Biomedical Imaging Research

 , , , , , ,
, , , , , ,  and
and 
Abstract
1. Introduction
1.1. Use Case: Highly-Plexed Tissue Image Analysis
1.1.1. Image Capture
1.1.2. The Traditional Approach to Computational Analysis of Highly-Plexed Tissue Images
- Handling Raw Data:
- The raw data are uploaded from the experimental machine to the cloud storage system provided by their institution. They then download the data from the cloud storage to their own workstation, where the analysis typically takes place.
- Executing the workflow:
- Initial Data Preparation: In a typical computing environment, these Tag Image File Format (TIFF) files are too large to be loaded into the memory, and most image processing tools are not designed to handle images in the multi-page TIFF format. Therefore, the first step in the workflow is to split the multi-page TIFF into a set of single grey-scale TIFFs, where each grey-scale TIFF is the image of a single protein channel acquired from the experiment. This is typically done in MATLAB or Python, using a TIFF Reader to load a single page from the multi-page TIFF into memory, and then writing that single page to disk as a TIFF file (with the name of the protein marker the page denotes).
- De-Arraying: When handling a TMA, the de-arraying processing step crops each individual tissue sample on the slide. This step can be done manually using a tool to draw the regions of interest, or it can be automated using AI, with a semantic segmentation model drawing bounding boxes for the tissue samples. To manually perform the de-arraying of the TMA, the greyscale image, which contains the nuclear stain (such as 4′,6-diamidino-2-phenylindole (DAPI)), is loaded into MATLAB or Python. The image is resized to fit on the screen, then it is opened in a window where a human manually draws an Region of Interest (ROI). The coordinates of the ROI are then translated back to the full-size image. Every grey-scale image is then cropped using those coordinates, and the cropped images are saved to disk. This process is repeated for every core in the TMA. Automated de-arraying is done using AI tools available as libraries in Python. The steps are the same, but the AI model replaces the need for the windows to manually draw the bounding box.
- Cell Segmentation: As a given tissue sample could comprise tens of thousands to millions of cells, manual cell segmentation is not feasible, and therefore, it is done almost exclusively with AI-based tools. These tools are almost exclusively available as Python libraries. The grey-scale TIFFs containing the protein channels with the strongest nucleus and membrane expressions (in some cases, nucleus only and the membrane region is inferred from the nucleus) are loaded into Python and passed to one of the Python-based AI tools which perform the segmentation. The AI tool outputs a nucleus and a membrane mask in the form of arrays or tensors, which are converted to an image and saved to disk. For a WSI this process is only performed once. However, for a TMA it needs to be performed for each tissue core.
- Extraction: The nucleus mask image, the membrane mask image and each of the protein channel images are loaded into either Python or MATLAB. Then, an image processing library extracts from the masks the indexes for each pixel, which makes up the nucleus and membrane of a cell. The morphological features are extracted solely from the masks (using an image processing library to get the perimeter, etc.), the proteomic features are extracted by using the pixel indexes from the masks as a binary gate on the protein channel images before getting the mean value of the non-zero pixels or similar.
1.2. Background and Related Work
1.2.1. Workflow Management and Execution Tools
1.2.2. Model-Driven LCNC
2. Material and Methods: Cinco de Bio
2.1. Designing a Workflow in CdB
The Application Domain DSL of the Case Study in the CdB
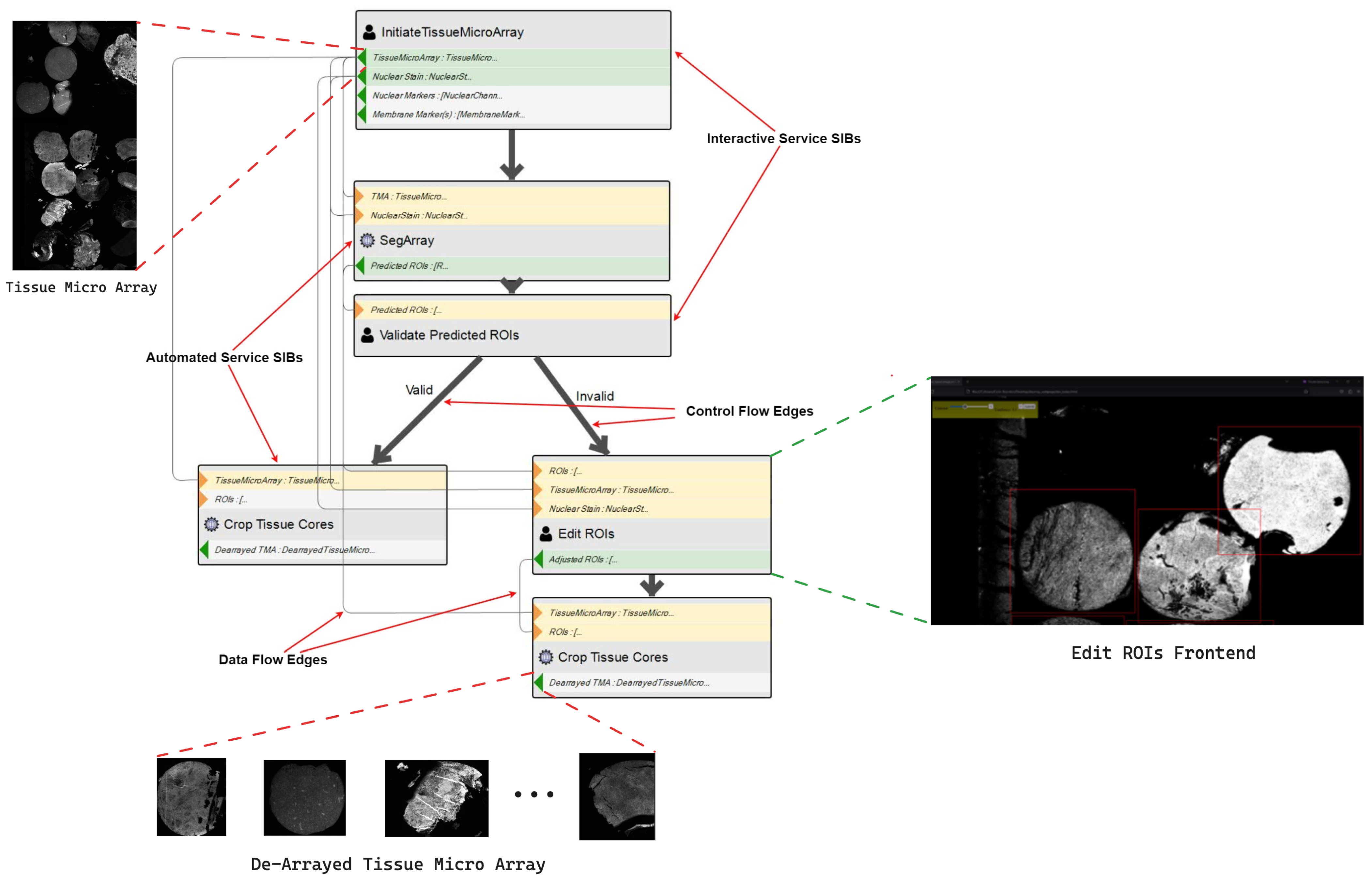
2.2. Interactive vs. Automated SIBs
- An automated SIB pertains to a data processing service which takes some input, follows some set of predefined steps and produces the output without requiring human intervention at any point.
- An interactive SIB instead pertains to a service that takes some input and follows some set of predefined steps. However, at a certain point in the processing journey, the service will require a person to provide some input, blocking the execution. Once that input is available, the service resumes and completes the remaining processing steps, resulting in the output.
- The dotted lines show the definition of WSI and TMA data types outside of CdB: these are both multi-page TIFF files.
- The solid lines denote the type definition of the data within the CdB environment. A TMA is a hash map of TMA Protein Channels, where TMA Protein Channel is a file-pointer to a single page TIFF, whereas a WSI is a hash map WSI Protein Channel and whereby the WSI Protein Channel is also a file-pointer to a single page TIFF.
2.3. Workflow Runtime in CdB
- Transform the workflow model to a program in an imperative programming language which can be executed and orchestrate the workflow.
- The workflow orchestration program submits a data processing job (i.e., one step of a workflow) to be scheduled by the job management system.
- When resources are available to do so, the data processing job is then running, retrieving the data to be processed from the data storage API and subsequently writing the intermediate results to the data storage API for retrieval by subsequent data processing jobs.
- The job management system via the executing API notifies the workflow orchestration program that a data processing job is complete so that it can retrieve the results and proceed to the next step. The execution API simultaneously updates the execution front-end to keep the user informed of the workflow status (or retrieving the final results upon completion of the workflow).
- In the case of interactive services, the data processing job first renders a front-end, which is made available via the data processing API, the URL of which is then presented to the user via execution front-end where they are re-directed to the service front-end to undertake the required task. The results of which are then submitted via the service API, and from that point, the system handles it as if it were a regular automated data processing job.
3. Results and Discussion
3.1. Executing the Analysis Workflow with CdB
- 1.
- Handling Raw Data: The raw data are uploaded from the experimental machine to the CdB data storage via the CdB upload portal.
- 2.
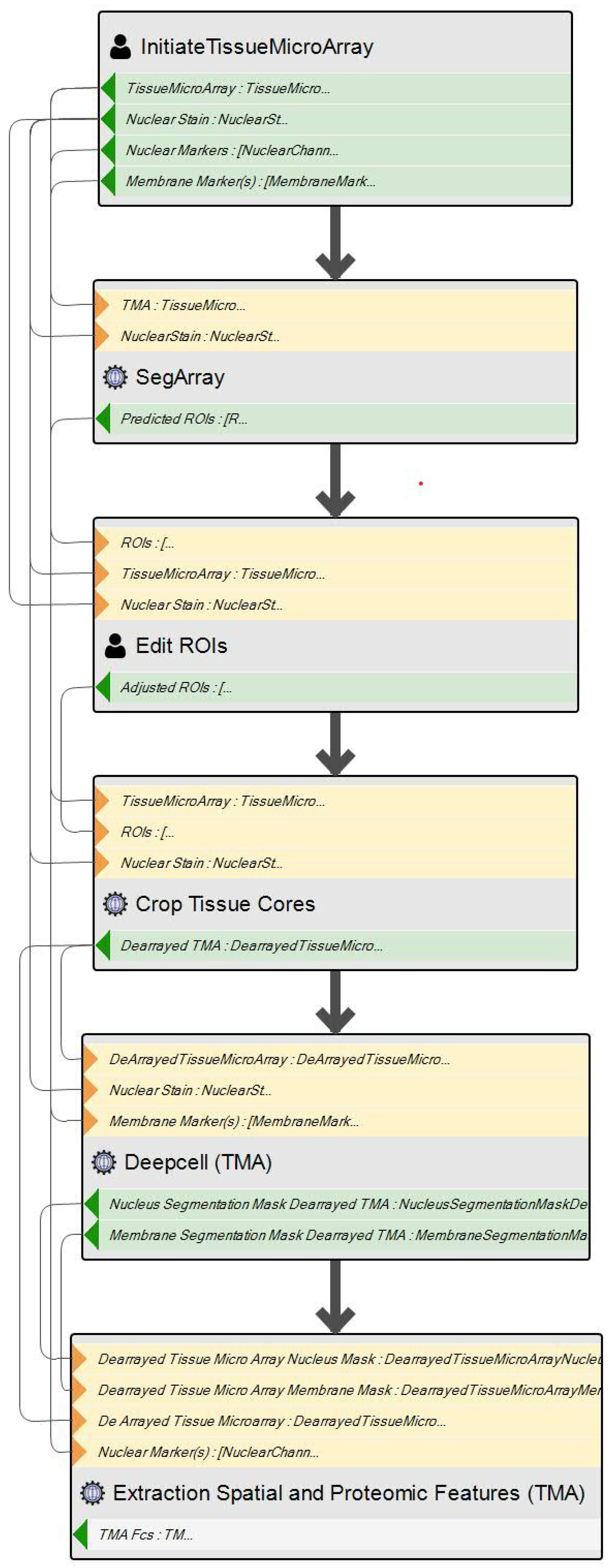
- Modeling: Using CdB’s IME, the researcher models the workflow using the graphical DSL, as seen in the simple sequential workflow in Figure 6. The Initiate TissueMicroArray SIB handles the initial data preparation phase of the workflow. It is an interactive service: the user selects the data on which the workflow will execute and inputs some other parameters. The SegArray, Edit ROIs and Crop Tissue Cores SIBs handle the de-arraying phase. The Deepcell (TMA) SIB conducts the cell segmentation, and finally, the Extraction Spatial and Proteomic Features (TMA) SIB handles the feature extraction.
- 3.
- Executing: Once the modelled workflow is finished and checked as valid, i.e., it passes all the syntactic and static semantic checks of the CdB IME, the user simply hits the execute workflow button. The user is then redirected to the execution front-end to monitor the status of the workflow execution.
- The execution runtime handles the orchestration of the various data processing services which comprise the workflow. Passing the appropriate data to each service to undertake their processing. Updating the status on the execution front-end for the user to monitor the status of their workflow.
- In the case of interactive services such as “Initiate TissueMicroArray” and “Edit ROIs”, the user is presented a redirect URL via the execution front-end, which launches the interaction front-end for the respective services where they can enter the required additional input.
- Upon workflow completion, the execution front end presents the user with the URL(s) to retrieve the output from the workflow, with the options to also retrieve the results from the intermediate processing steps.
3.2. Comparison to Similar Tools
3.2.1. Criteria
- Learning Curve: Considering users with no prior knowledge of programming, how much IT knowledge would they need to acquire in order to use the tool proficiently to design and execute their own workflow? A Low learning curve indicates a system that is very intuitive to the user without specialized IT knowledge, e.g., just being able to use a browser and a mouse; a Medium learning curve indicates that the system is relatively intuitive to use but the user will have to learn some IT or programming concepts to use the tool effectively; a High learning curve denotes that the user will essentially have to learn to write code in order to use the tool.
- DSL Type: Does the user model the workflows within the system via a graphical DSL or a textual DSL? This is synonymous with whether the system reduces the amount of code, which needs to be written (low-code) or eliminates coding altogether (no-code).
- Control/Data Flow: This criterion addresses the level of expressiveness of the PL-DSL for modelling within the system. Can the user model both the control flow and the data flow of the workflow, or just the data flow?
- Semantic Typing: This criterion concerns the data modelling facilities offered by the tool: is the data model composed of solely primitive and generic data types, or are domain-specific concepts captured in the data model? The rationale behind this is that a semantically typed modelling language presents concepts directly familiar to the user (i.e., lowers the learning curve) and secondly, it facilitates semantic data-flow analysis for compatibility checks of types that would be syntactically indistinguishable.
- Design Validation: This criterion captures the level of checks that the system applies to the user-defined workflows before submitting them to be executed. Extensive refers to both syntactic and static semantic checks, limited refers to simple checks to ensure that there are no cycles in the workflow, etc.
- Interactive Services: This criterion pertains to how well (if at all) a system caters for services with user interaction at run-time.
- Deployment: This criterion covers how and where the system can be deployed. For example, is it a desktop application or a web application that can be deployed on the cloud? The deployment options have knock-on effects for the system scalability, support of interaction and collaboration and ease of set-up.
3.2.2. Comparison
- Galaxy: https://usegalaxy.eu/ [36] (accessed on 1 August 2024) is a browser-based workbench for scientific computing that supports the execution of biomedical analysis workflows through a graphical LCNC manner. Galaxy has a mature ecosystem that supports an extensive number of use cases. It has a unified front end with high flexibility over the execution back end. However, the Galaxy modelling language is data-flow only; as AI use increases in this context, modelling control flow will be essential when deciding whether AI can be used to automate decisions (i.e., which direction a workflow should take) or for use cases similar to the one we described in Section 2.1. Galaxy provides only minimal support for interactive tools, and, as described in their own documentation, integrating new interactive tools into Galaxy is a non-trivial task. Finally, Galaxy uses generic typing (json, list, TIFF, etc.), so it does not capture data concepts familiar to a user’s domain, and it permits invalid workflows to execute. For example, a list of images and a list of text are treated the same in both cases: the type is a list, design time. Galaxy does not prevent a user from inputting a list of textual data into a service that expects a list of images. There is accordingly a significant burden of knowledge and checks on the users, who need training and carefulness at every step.
- KNIME: https://hub.knime.com/ [37] (accessed on 1 August 2024) is a desktop application for the execution of data analytics workflows. It supports users to model workflows graphically with both control and data flow, where the control flow is modelled using control nodes for if-else branching, etc. Third-party services, which support Representational State Transfer (REST) communication, can be integrated into applications by users. The tools make use of a generic data model and do not natively capture any concepts specific to the biomedical domain. Owing to the fact that it is a desktop application it lacks scalability. It has an extension to work with Apache Spark; however, that would require the user to deploy a spark cluster separately before linking it to KNIME. The KNIME Server version of the tool has a free community version, but the vast majority of the functionality is reserved for the paid version and, therefore, is not discussed here.
- QuPath: https://qupath.github.io/ (accessed on 1 August 2024) is an open-source desktop application designed for digital pathology, specifically for the analysis of TMAs and WSIs. It is primarily an image viewer but offers limited support for data processing and executing workflows. A user can use a variety of built-in tools to process an image, these then show up in a command history widget, and that history can be exported as a script to re-execute it as an automated workflow. Even though many tools support interaction (i.e., manual annotation, etc.) it is not recommended by the developers to use such tools in a workflow with automated tools. Given that QuPath is a desktop application, it has limited scalability. Also, due to it being designed from the ground up, specifically for digital pathology, the system can not be easily repurposed for processing/analysis of other biomedical images. It also does not support the integration of many of the state-of-the-art open-source tools.
- CellProfiler: https://cellprofiler.org/ [38] (accessed on 1 August 2024) is a desktop application for processing Biomedical Images. The interaction is mainly conducted through a Graphical User Interface (GUI). To define a workflow (or pipeline, as it is called in the application), users drag-n-drop modules such as Crop Image, then input parameters for each module using forms displayed in the application. The workflow is data-flow only and limited checks are applied to the workflow before it can be executed. The data model does not include semantic types as it is tailored for general biomedical image processing. It does not support any interactive services as a part of a workflow. Being a desktop application, it is limited in terms of scalability. It also does not support the integration of many of the state-of-the-art open-source tools.
- NextFlow: https://www.nextflow.io/ (accessed on 1 August 2024) [14] is a Groovy-based textual DSL and workflow management system for the creation and execution of data analysis workflows. It is highly expressive and supports a variety of runtime environments for services, like Docker, Shifter [39], Singularity [40], Podman and Conda. Whilst Nextflow eases the deployment of workflows, the technical expertise required to use it is far beyond what could be expected from a typical domain researcher.
- SnakeMake: https://snakemake.github.io/(accessed on 1 August 2024) [15] is a Python-based textual DSL and workflow management system for the creation and execution of data analysis workflows. It is highly expressive and supports multiple runtime environments for service execution (Singularity and Conda). Similar to Nextflow, while SnakeMake eases the deployment of workflows, the technical expertise required to use it is far beyond what could be expected from a typical domain researcher.
- CdB: In comparison, CdB is superior in many comparison dimensions and equivalent in others: it offers a powerful modelling environment that covers both dataflow and control flow with associated syntactic correctness checks and it supports semantic types that are intuitive for the domain experts, with associated semantic compatibility checks. With the purely graphical modelling environment, it has a low learning curve, especially if connected with a cloud-based hosting, which eliminates the need for installation. The graphical models are stored in a JSON format, so they are actually also available in a textual form. The support for interactive services is a core requirement of CdB, so, in fact, we designed the execution environment in such a way as to maximally simplify this capability. Finally, the entire environment is designed for future growth, with ease of integration and extension in mind.
4. Conclusions and Future Work
- Easy access: installation-free, ubiquitous, access via a Web Browser;
- Easy use: domain-specific LCNC modelling support that requires no technical knowledge.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Goltsev, Y.; Samusik, N.; Kennedy-Darling, J.; Bhate, S.; Hale, M.; Vazquez, G.; Black, S.; Nolan, G.P. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell 2018, 174, 968–981. [Google Scholar] [CrossRef]
- Mund, A.; Brunner, A.D.; Mann, M. Unbiased spatial proteomics with single-cell resolution in tissues. Mol. Cell 2022, 82, 2335–2349. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.J. ImageJ for microscopy. Biotechniques 2007, 43, S25–S30. [Google Scholar] [CrossRef] [PubMed]
- Leipzig, J. A review of bioinformatic pipeline frameworks. Briefings Bioinform. 2017, 18, 530–536. [Google Scholar] [CrossRef]
- Mangul, S.; Mosqueiro, T.; Abdill, R.J.; Duong, D.; Mitchell, K.; Sarwal, V.; Hill, B.; Brito, J.; Littman, R.J.; Statz, B.; et al. Challenges and recommendations to improve the installability and archival stability of omics computational tools. PLoS Biol. 2019, 17, e3000333. [Google Scholar] [CrossRef] [PubMed]
- Blankenberg, D.; Kuster, G.V.; Coraor, N.; Ananda, G.; Lazarus, R.; Mangan, M.; Nekrutenko, A.; Taylor, J. Galaxy: A web-based genome analysis tool for experimentalists. Curr. Protoc. Mol. Biol. 2010, 89, 10–19. [Google Scholar] [CrossRef]
- Hunter, A.A.; Macgregor, A.B.; Szabo, T.O.; Wellington, C.A.; Bellgard, M.I. Yabi: An online research environment for grid, high performance and cloud computing. Source Code Biol. Med. 2012, 7, 1. [Google Scholar] [CrossRef]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Thiel, K.; Wiswedel, B. KNIME-the Konstanz information miner: Version 2.0 and beyond. AcM SIGKDD Explor. Newsl. 2009, 11, 26–31. [Google Scholar] [CrossRef]
- Lamprecht, A.L.; Margaria, T.; Steffen, B. Seven Variations of an Alignment Workflow—An Illustration of Agile Process Design and Management in Bio-jETI. In Proceedings of the Bioinformatics Research and Applications, Atlanta, Georgia, 6–9 May 2008; Lecture Notes in Bioinformatics; Springer: Berlin/Heidelberg, Germany, 2008; Volume 4983, pp. 445–456. [Google Scholar] [CrossRef]
- Lamprecht, A.L.; Margaria, T.; Steffen, B.; Sczyrba, A.; Hartmeier, S.; Giegerich, R. GeneFisher-P: Variations of GeneFisher as processes in Bio-jETI. BMC Bioinform. 2008, 9, S13. [Google Scholar] [CrossRef] [PubMed]
- Margaria, T. From computational thinking to constructive design with simple models. In Proceedings, Part I 8, Proceedings of the Leveraging Applications of Formal Methods, Verification and Validation. Modeling: 8th International Symposium, ISoLA 2018, Limassol, Cyprus, 5–9 November 2018; Springer: Berlin/Heidelberg, Germany, 2018; pp. 261–278. [Google Scholar]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Köster, J.; Rahmann, S. Snakemake—A scalable bioinformatics workflow engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef] [PubMed]
- Bourgey, M.; Dali, R.; Eveleigh, R.; Chen, K.C.; Letourneau, L.; Fillon, J.; Michaud, M.; Caron, M.; Sandoval, J.; Lefebvre, F.; et al. GenPipes: An open-source framework for distributed and scalable genomic analyses. Gigascience 2019, 8, giz037. [Google Scholar] [CrossRef] [PubMed]
- Sadedin, S.P.; Pope, B.; Oshlack, A. Bpipe: A tool for running and managing bioinformatics pipelines. Bioinformatics 2012, 28, 1525–1526. [Google Scholar] [CrossRef] [PubMed]
- Novella, J.A.; Emami Khoonsari, P.; Herman, S.; Whitenack, D.; Capuccini, M.; Burman, J.; Kultima, K.; Spjuth, O. Container-based bioinformatics with Pachyderm. Bioinformatics 2019, 35, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Lampa, S.; Dahlö, M.; Alvarsson, J.; Spjuth, O. SciPipe: A workflow library for agile development of complex and dynamic bioinformatics pipelines. GigaScience 2019, 8, giz044. [Google Scholar] [CrossRef] [PubMed]
- Lampa, S.; Alvarsson, J.; Spjuth, O. Towards agile large-scale predictive modelling in drug discovery with flow-based programming design principles. J. Cheminform. 2016, 8, 67. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.P. Flow-based programming. In Proceedings of the Proceedings 1st International Workshop on Software Engineering for Parallel and Distributed Systems, Hsinchu, Taiwan, 19–21 December 1994; pp. 25–29. [Google Scholar]
- Rentsch, T. Object oriented programming. ACM Sigplan Not. 1982, 17, 51–57. [Google Scholar] [CrossRef]
- Naur, P.; Randell, B. (Eds.) Software Engineering: Report of a Conference Sponsored by the NATO Science Committee, Garmisch, Germany, 7–11 October 1968; Scientific Affairs Division, NATO: Brussels, Belgium, 1969. [Google Scholar]
- Dorai, C.; Venkatesh, S. Bridging the semantic gap with computational media aesthetics. IEEE MultiMedia 2003, 10, 15–17. [Google Scholar] [CrossRef]
- Hein, A.M. Identification and Bridging of Semantic Gaps in the Context of Multi-Domain Engineering. Proc. Forum Philos. Eng. Technol. 2010. Available online: https://mediatum.ub.tum.de/1233138 (accessed on 1 August 2024).
- Steffen, B.; Gossen, F.; Naujokat, S.; Margaria, T. Language-Driven Engineering: From General-Purpose to Purpose-Specific Languages. In Computing and Software Science: State of the Art and Perspectives; Steffen, B., Woeginger, G., Eds.; LNCS; Springer: Berlin/Heidelberg, Germany, 2019; Volume 10000. [Google Scholar] [CrossRef]
- Zweihoff, P.; Tegeler, T.; Schürmann, J.; Bainczyk, A.; Steffen, B. Aligned, Purpose-Driven Cooperation: The Future Way of System Development. In Proceedings of the Leveraging Applications of Formal Methods, Verification and Validation; Margaria, T., Steffen, B., Eds.; Springer: Cham, Switzerland, 2021; pp. 426–449. [Google Scholar] [CrossRef]
- Mussbacher, G.; Amyot, D.; Breu, R.; Bruel, J.M.; Cheng, B.H.C.; Collet, P.; Combemale, B.; France, R.B.; Heldal, R.; Hill, J.; et al. The Relevance of Model-Driven Engineering Thirty Years from Now. In Proceedings of the 17th International Conference on Model Driven Engineering Languages and Systems (MODELS’14); number 8767 in LNCS. Springer International Publishing: Cham, Switzerland, 2014; pp. 183–200. [Google Scholar] [CrossRef]
- Mellor, S.J.; Balcer, M.J. Executable UML: A Foundation for Model-Driven Architecture; Addison-Wesley Professional: Boston, MA, USA, 2002. [Google Scholar]
- Steffen, B.; Margaria, T.; Nagel, R.; Jörges, S.; Kubczak, C. Model-Driven Development with the jABC. In Hardware and Software, Verification and Testing; Lecture Notes in Computer, Science; Bin, E., Ziv, A., Ur, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; Volume 4383, pp. 92–108. [Google Scholar] [CrossRef]
- Neubauer, J.; Frohme, M.; Steffen, B.; Margaria, T. Prototype-driven development of web applications with DyWA. In Proceedings, Part I 6, Proceedings of the Leveraging Applications of Formal Methods, Verification and Validation. Technologies for Mastering Change: 6th International Symposium, ISoLA 2014, Imperial, Corfu, Greece, 8–11 October 2014; Springer: Berlin/Heidelberg, Germany, 2014; pp. 56–72. [Google Scholar]
- Boßelmann, S.; Frohme, M.; Kopetzki, D.; Lybecait, M.; Naujokat, S.; Neubauer, J.; Wirkner, D.; Zweihoff, P.; Steffen, B. DIME: A programming-less modeling environment for web applications. In Proceedings, Part II 7, Proceedings of the Leveraging Applications of Formal Methods, Verification and Validation: Discussion, Dissemination, Applications: 7th International Symposium, ISoLA 2016, Imperial, Corfu, Greece, 10–14 October 2016; Springer: Berlin/Heidelberg, Germany, 2016; pp. 809–832. [Google Scholar]
- Naujokat, S.; Lybecait, M.; Kopetzki, D.; Steffen, B. CINCO: A simplicity-driven approach to full generation of domain-specific graphical modeling tools. Int. J. Softw. Tools Technol. Transf. 2018, 20, 327–354. [Google Scholar] [CrossRef]
- Bainczyk, A.; Busch, D.; Krumrey, M.; Mitwalli, D.S.; Schürmann, J.; Tagoukeng Dongmo, J.; Steffen, B. CINCO cloud: A holistic approach for web-based language-driven engineering. In Proceedings of the International Symposium on Leveraging Applications of Formal Methods; Springer: Cham, Switzerland, 2022; pp. 407–425. [Google Scholar]
- Luksa, M. Kubernetes in Action; Simon and Schuster: New York, NY, USA, 2017. [Google Scholar]
- The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. 2022, 50, W345–W351. [CrossRef] [PubMed]
- Fillbrunn, A.; Dietz, C.; Pfeuffer, J.; Rahn, R.; Landrum, G.A.; Berthold, M.R. KNIME for reproducible cross-domain analysis of life science data. J. Biotechnol. 2017, 261, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Stirling, D.R.; Swain-Bowden, M.J.; Lucas, A.M.; Carpenter, A.E.; Cimini, B.A.; Goodman, A. CellProfiler 4: Improvements in speed, utility and usability. BMC Bioinform. 2021, 22, 433. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, L.; Bhimji, W.; Canon, S.; Fasel, M.; Jacobsen, D.; Mustafa, M.; Porter, J.; Tsulaia, V. Shifter: Containers for hpc. J. Phys. 2017, 898, 082021. [Google Scholar] [CrossRef]
- Kurtzer, G.M.; Sochat, V.; Bauer, M.W. Singularity: Scientific containers for mobility of compute. PLoS ONE 2017, 12, e0177459. [Google Scholar] [CrossRef]
- Naujokat, S.; Lamprecht, A.L.; Steffen, B. Loose Programming with PROPHETS. In Proceedings of the 15th International Conference on Fundamental Approaches to Software Engineering (FASE 2012), Tallinn, Estonia, 24 March 2012–1 April 2012; de Lara, J., Zisman, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 7212, pp. 94–98. [Google Scholar] [CrossRef]
- Lamprecht, A.L.; Margaria, T.; Steffen, B. Bio-jETI: A framework for semantics-based service composition. BMC Bioinform. 2009, 10 (Suppl. S10). [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Platform | Learning Curve | DSL Type | Expressiveness | Semantic Typing | Design Validation | Interactive Services | Deployment |
|---|---|---|---|---|---|---|---|
| Cinco de Bio | Low | Graphical | Control and Data flow | Yes | Extensive | Extensive | Local/Cloud |
| Galaxy | Medium | Graphical | Data Flow | No | Limited | Limited | Local/Cloud |
| Knime | Medium | Graphical | Control and Data flow | No | Limited | Limited | Desktop |
| Qupath | Medium | Graphical and Textual | Data Flow | Yes | Limited | Limited | Desktop |
| CellProfiler | Medium | Graphical | Data Flow | No | Limited | No | Desktop |
| Nextflow | High | Textual | Data and Control Flow | No | No | No | Local/Cloud |
| Snakemake | High | Textual | Data and Control Flow | No | No | No | Local/Cloud |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brandon, C.; Boßelmann, S.; Singh, A.; Ryan, S.; Schieweck, A.; Fennell, E.; Steffen, B.; Margaria, T. Cinco de Bio: A Low-Code Platform for Domain-Specific Workflows for Biomedical Imaging Research. BioMedInformatics 2024, 4, 1865-1883. https://doi.org/10.3390/biomedinformatics4030102
Brandon C, Boßelmann S, Singh A, Ryan S, Schieweck A, Fennell E, Steffen B, Margaria T. Cinco de Bio: A Low-Code Platform for Domain-Specific Workflows for Biomedical Imaging Research. BioMedInformatics. 2024; 4(3):1865-1883. https://doi.org/10.3390/biomedinformatics4030102
Chicago/Turabian StyleBrandon, Colm, Steve Boßelmann, Amandeep Singh, Stephen Ryan, Alexander Schieweck, Eanna Fennell, Bernhard Steffen, and Tiziana Margaria. 2024. "Cinco de Bio: A Low-Code Platform for Domain-Specific Workflows for Biomedical Imaging Research" BioMedInformatics 4, no. 3: 1865-1883. https://doi.org/10.3390/biomedinformatics4030102
APA StyleBrandon, C., Boßelmann, S., Singh, A., Ryan, S., Schieweck, A., Fennell, E., Steffen, B., & Margaria, T. (2024). Cinco de Bio: A Low-Code Platform for Domain-Specific Workflows for Biomedical Imaging Research. BioMedInformatics, 4(3), 1865-1883. https://doi.org/10.3390/biomedinformatics4030102