

Pitfalls of Using Multinomial Regression Analysis to Identify Class-Structure-Relevant Variables in Biomedical Data Sets: Why a Mixture of Experts (MOE) Approach Is Better



Abstract
:1. Introduction
Introductory Example Case
2. Materials and Methods
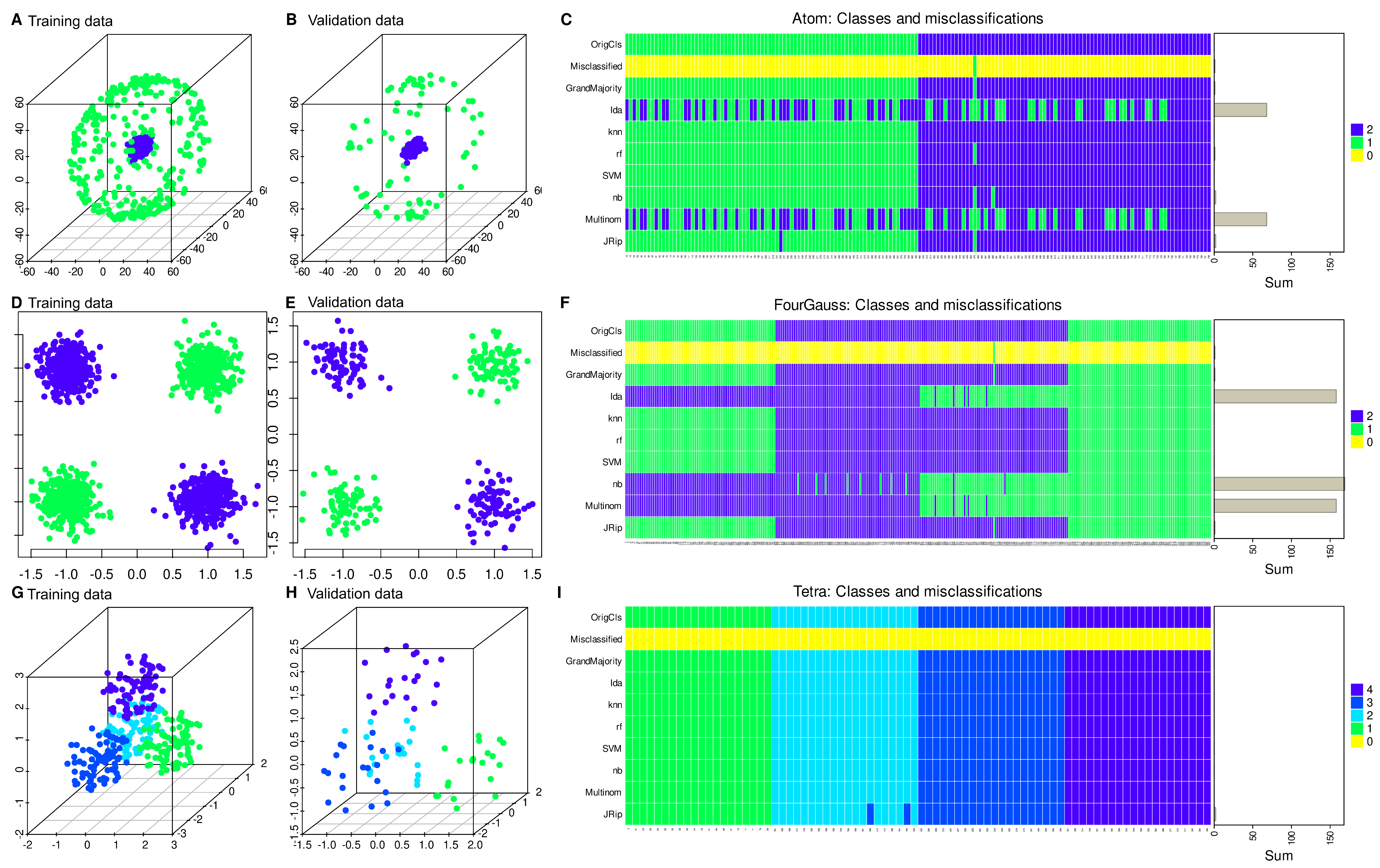
2.1. Sample Data Sets
2.2. Experimentation
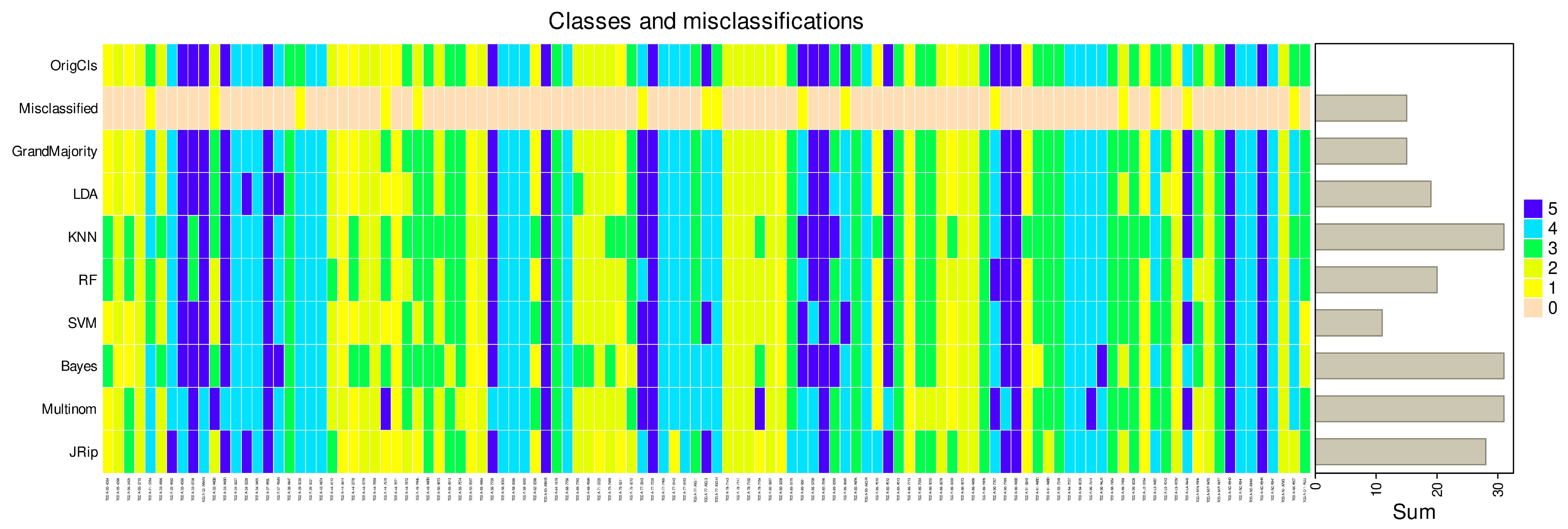
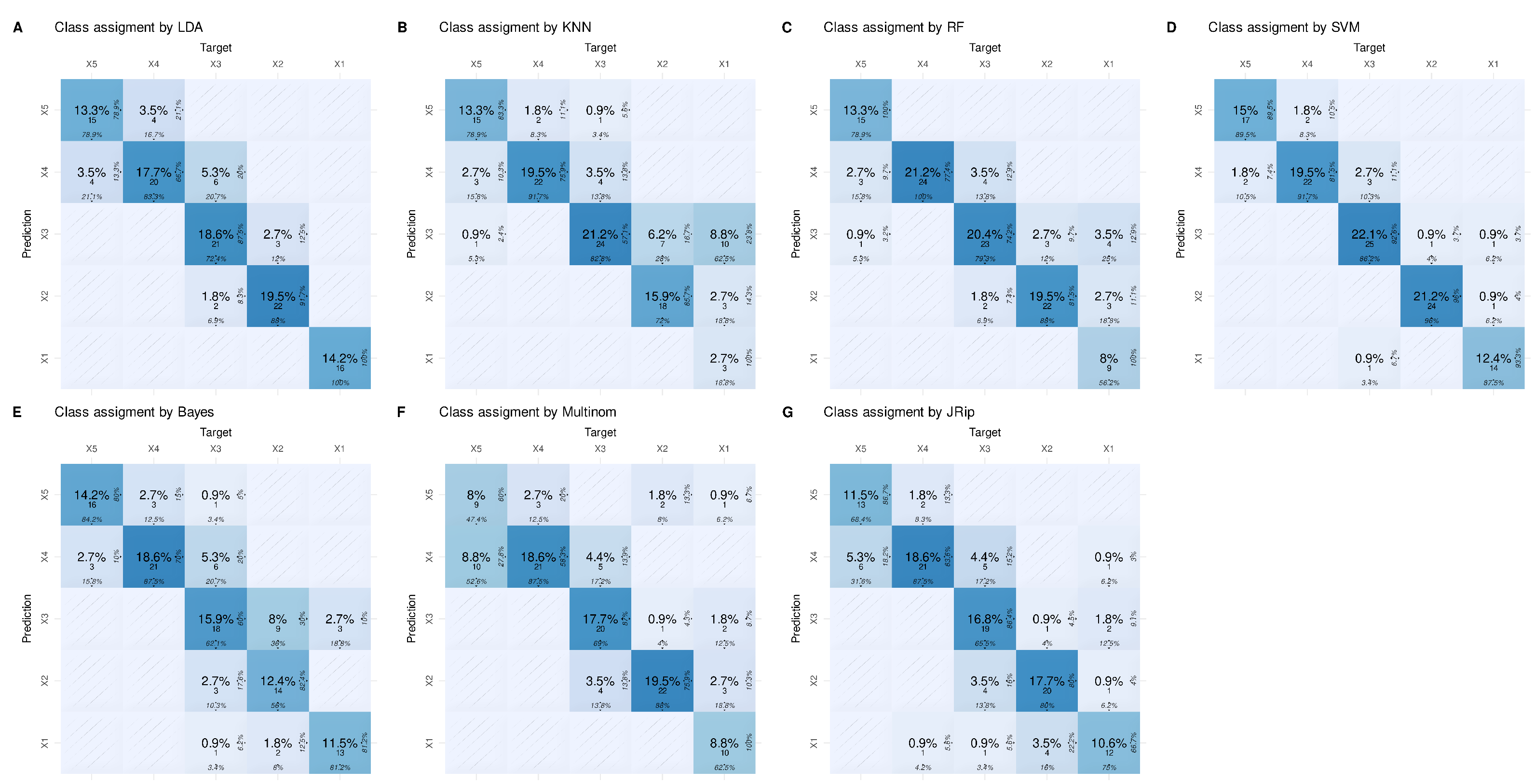
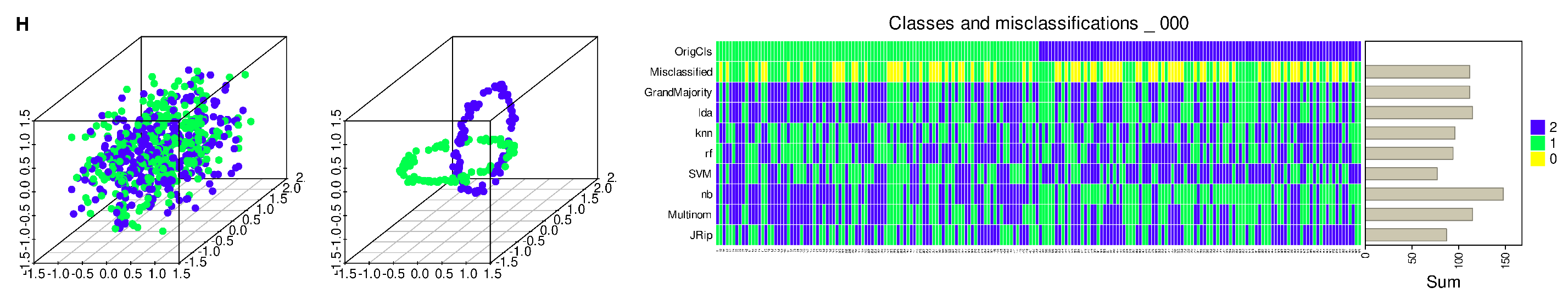
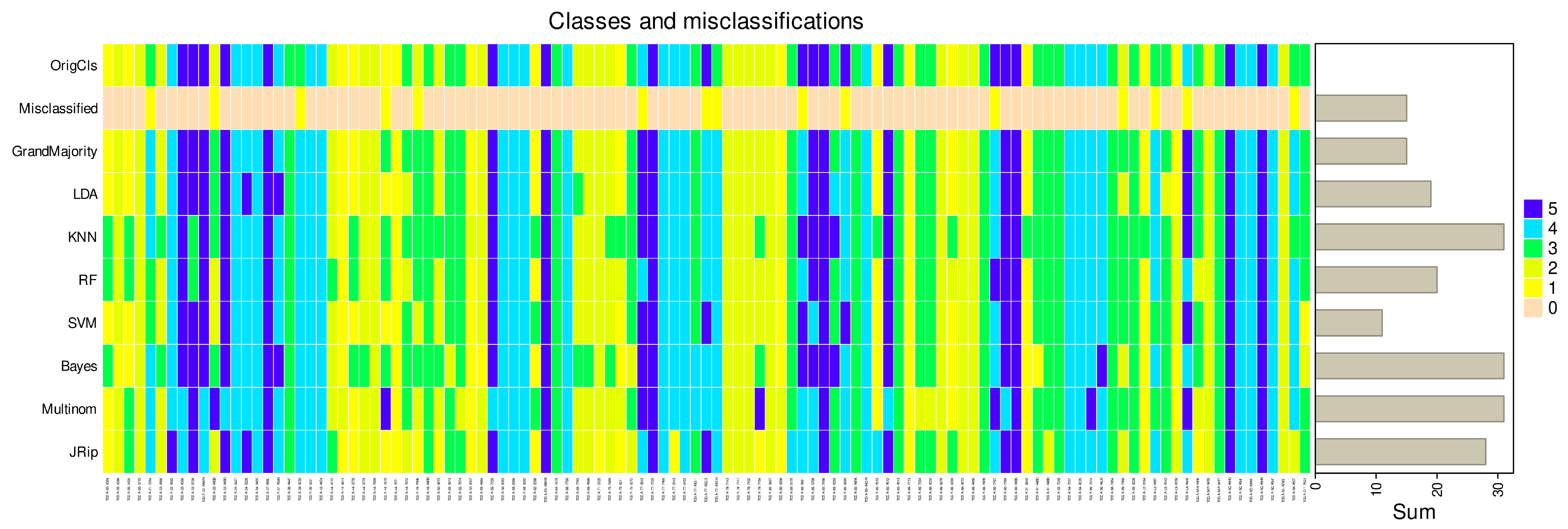
3. Results
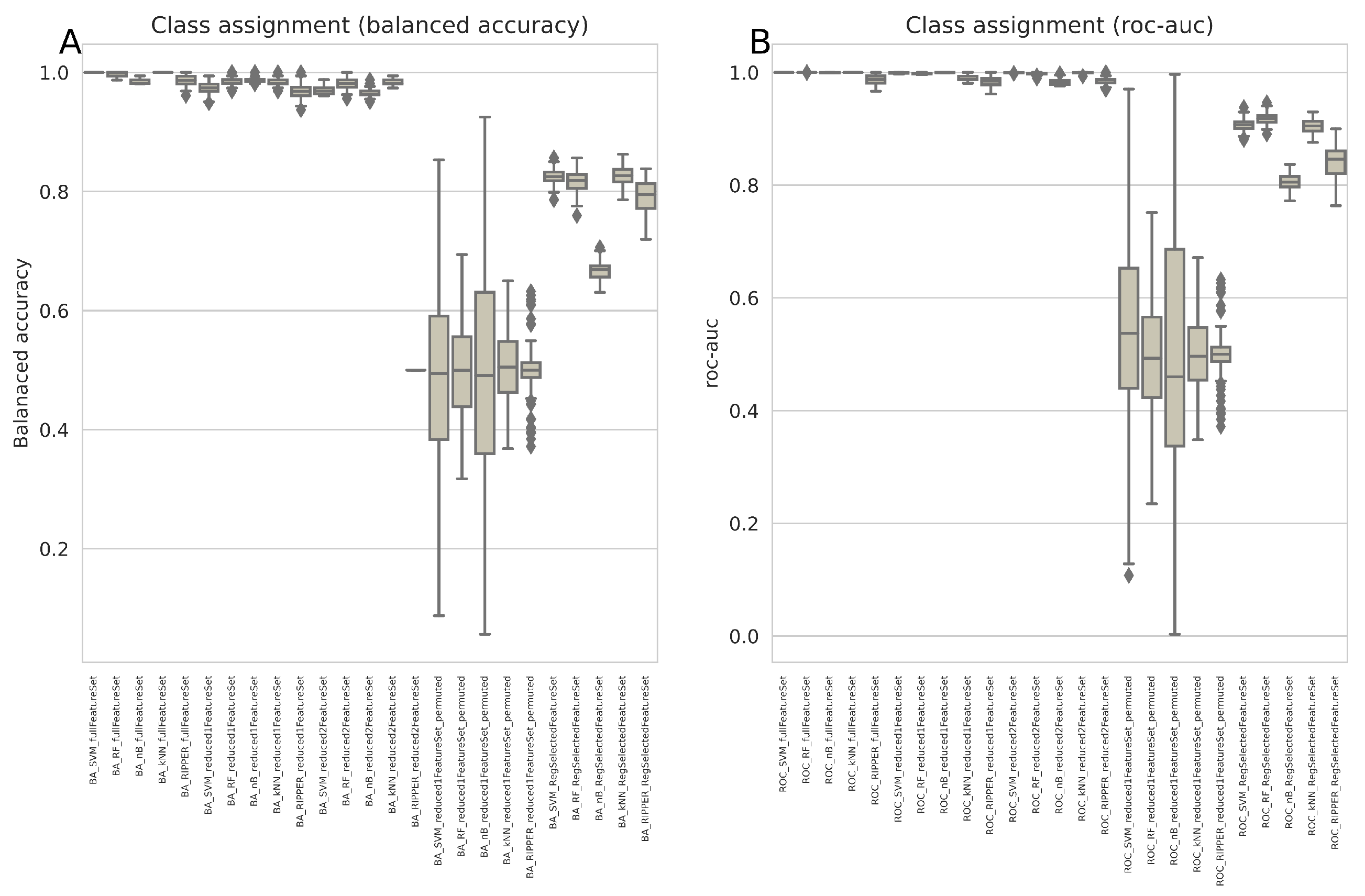
3.1. Regression Occasionally Generalizes Poorly Compared to Alternative Methods
3.2. Regression Inadequately Captures the Structural Characteristics of Certain Data Sets
3.3. Variables Chosen by the Most Successful Algorithms Are More Generalizable
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lo, A.; Chernoff, H.; Zheng, T.; Lo, S.H. Why significant variables are not automatically good predictors. Proc. Natl. Acad. Sci. USA 2015, 112, 13892–13897. [Google Scholar] [CrossRef] [PubMed]
- Ultsch, A.; Lötsch, J. The Fundamental Clustering and Projection Suite (FCPS): A Dataset Collection to Test the Performance of Clustering and Data Projection Algorithms. Data 2020, 5, 13. [Google Scholar] [CrossRef]
- Pearson, K. LIII. On lines and planes of closest fit to systems of points in space. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1901, 2, 559–572. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Thrun, M.; Stier, Q. Fundamental clustering algorithms suite. SoftwareX 2021, 13, 100642. [Google Scholar]
- Minsky, M.; Papert, S. Perceptrons: An Introduction to Computational Geometry; MIT Press: Cambridge, MA, USA, 1969. [Google Scholar]
- Khadirnaikar, S.; Shukla, S.; Prasanna, S.R.M. Machine learning based combination of multi-omics data for subgroup identification in non-small cell lung cancer. Sci. Rep. 2023, 13, 4636. [Google Scholar] [CrossRef]
- Ihaka, R.; Gentleman, R. R: A Language for Data Analysis and Graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar] [CrossRef]
- Van Rossum, G.; Drake, F.L., Jr. Python Tutorial; Centrum voor Wiskunde en Informatica Amsterdam: Amsterdam, The Netherlands, 1995; Volume 620. [Google Scholar]
- Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Ligges, U.; Mächler, M. Scatterplot3d–An R Package for Visualizing Multivariate Data. J. Stat. Softw. 2003, 8, 1–20. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Olsen, L.R.; Zachariae, H.B. cvms: Cross-Validation for Model Selection. 2023. Available online: https://cran.r-project.org/package=cvms (accessed on 14 August 2023).
- Venables, W.N.; Ripley, B.D. Modern Applied Statistics with S, 4th ed.; Springer: New York, NY, USA, 2002; ISBN 0-387-95457-0. [Google Scholar]
- Waskom, M.L. Seaborn: Statistical data visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- Fisher, R.A. The use of multiple measurements in taxonomic problems. Ann. Eugen. 1936, 7, 179–188. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-Vector Networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Cover, T.; Hart, P. Nearest neighbor pattern classification. IEEE Trans. Inf. Theor. 1967, 13, 21–27. [Google Scholar] [CrossRef]
- Bayes, M.; Price, M. An Essay towards Solving a Problem in the Doctrine of Chances. By the Late Rev. Mr. Bayes, F. R. S. Communicated by Mr. Price, in a Letter to John Canton, A. M. F. R. S. Philos. Trans. 1763, 53, 370–418. [Google Scholar] [CrossRef]
- Cohen, W.W. Fast Effective Rule Induction. In Machine Learning Proceedings 1995, Proceedings of the Twelfth International Conference on Machine Learning, Tahoe City, California, 9–12 July 1995; Prieditis, A., Russell, S., Eds.; Morgan Kaufmann: San Francisco, CA, USA, 1995; pp. 115–123. [Google Scholar] [CrossRef]
- Brodersen, K.H.; Ong, C.S.; Stephan, K.E.; Buhmann, J.M. The Balanced Accuracy and Its Posterior Distribution. In Proceedings of the 2010 20th International Conference on Pattern Recognition (ICPR), Istanbul, Turkey, 23–26 August 2010; pp. 3121–3124. [Google Scholar] [CrossRef]
- Peterson, W.; Birdsall, T.; Fox, W. The theory of signal detectability. Trans. Ire Prof. Group Inf. Theory 1954, 4, 171–212. [Google Scholar] [CrossRef]
- Ultsch, A.; Lötsch, J. Computed ABC Analysis for Rational Selection of Most Informative Variables in Multivariate Data. PLoS ONE 2015, 10, e0129767. [Google Scholar] [CrossRef]
- Juran, J.M. The non-Pareto principle; Mea culpa. Qual. Prog. 1975, 8, 8–9. [Google Scholar]
- Guyon, I. An introduction to variable and feature selection. J. Mach. Learn. Res. 2003, 3, 1157–1182. [Google Scholar]
- Hosmer, D.; Lemeshow, S.; Sturdivant, R. Applied Logistic Regression; Wiley Series in Probability and Statistics; Wiley: Hoboken, NJ, USA, 2013. [Google Scholar]
- Fahrmeir, L.; Kneib, T.; Lang, S.; Marx, B. Regression: Models, Methods and Applications; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Rosenblatt, F. The perceptron: A probabilistic model for information storage and organization in the brain. Psychol. Rev. 1958, 65, 386–408. [Google Scholar] [CrossRef]
- Elizondo, D. The linear separability problem: Some testing methods. IEEE Trans. Neural Netw. 2006, 17, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Verikas, A.; Bacauskiene, M. Feature selection with neural networks. Pattern Recognit. Lett. 2002, 23, 1323–1335. [Google Scholar] [CrossRef]
- Lötsch, J.; Mayer, B. A Biomedical Case Study Showing That Tuning Random Forests Can Fundamentally Change the Interpretation of Supervised Data Structure Exploration Aimed at Knowledge Discovery. BioMedInformatics 2022, 2, 544–552. [Google Scholar] [CrossRef]
- Hu, Y.H.; Palreddy, S.; Tompkins, W.J. A patient-adaptable ECG beat classifier using a mixture of experts approach. IEEE Trans. Biomed. Eng. 1997, 44, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, M.; Vittrant, B.; Martin-Magniette, M.L.; Scott Boyer, M.P.; Perin, O.; Bergeron, A.; Fradet, Y.; Droit, A. Large-Scale Automatic Feature Selection for Biomarker Discovery in High-Dimensional OMICs Data. Front. Genet. 2019, 10, 452. [Google Scholar] [CrossRef]
- Miettinen, T.; Nieminen, A.I.; Mäntyselkä, P.; Kalso, E.; Lötsch, J. Machine Learning and Pathway Analysis-Based Discovery of Metabolomic Markers Relating to Chronic Pain Phenotypes. Int. J. Mol. Sci. 2022, 23, 5085. [Google Scholar] [CrossRef]
- Kringel, D.; Kaunisto, M.A.; Kalso, E.; Lötsch, J. Machine-learned analysis of global and glial/opioid intersection-related DNA methylation in patients with persistent pain after breast cancer surgery. Clin. Epigenetics 2019, 11, 167. [Google Scholar] [CrossRef]
- Lötsch, J.; Schiffmann, S.; Schmitz, K.; Brunkhorst, R.; Lerch, F.; Ferreiros, N.; Wicker, S.; Tegeder, I.; Geisslinger, G.; Ultsch, A. Machine-learning based lipid mediator serum concentration patterns allow identification of multiple sclerosis patients with high accuracy. Sci. Rep. 2018, 8, 14884. [Google Scholar] [CrossRef]
- Statnikov, A.; Henaff, M.; Narendra, V.; Konganti, K.; Li, Z.; Yang, L.; Pei, Z.; Blaser, M.J.; Aliferis, C.F.; Alekseyenko, A.V. A comprehensive evaluation of multicategory classification methods for microbiomic data. Microbiome 2013, 1, 11. [Google Scholar] [CrossRef]
- Li, K.; Wang, F.; Yang, L.; Liu, R. Deep feature screening: Feature selection for ultra high-dimensional data via deep neural networks. Neurocomputing 2023, 538, 126186. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Regression | ||||
|---|---|---|---|---|---|
| Estimate | Std. Error | Z-Value | Pr(>|z|) | Signif. | |
| (Intercept) | 0.01541 | 0.08657 | 0.178 | 0.859 | |
| X | 0.03846 | 0.0827 | 0.465 | 0.642 | |
| Y | 1.56726 | 0.11461 | 13.674 | <2 × 10−16 | *** |
| Z | −0.06873 | 0.0824 | −0.834 | 0.404 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lötsch, J.; Ultsch, A. Pitfalls of Using Multinomial Regression Analysis to Identify Class-Structure-Relevant Variables in Biomedical Data Sets: Why a Mixture of Experts (MOE) Approach Is Better. BioMedInformatics 2023, 3, 869-884. https://doi.org/10.3390/biomedinformatics3040054
Lötsch J, Ultsch A. Pitfalls of Using Multinomial Regression Analysis to Identify Class-Structure-Relevant Variables in Biomedical Data Sets: Why a Mixture of Experts (MOE) Approach Is Better. BioMedInformatics. 2023; 3(4):869-884. https://doi.org/10.3390/biomedinformatics3040054
Chicago/Turabian StyleLötsch, Jörn, and Alfred Ultsch. 2023. "Pitfalls of Using Multinomial Regression Analysis to Identify Class-Structure-Relevant Variables in Biomedical Data Sets: Why a Mixture of Experts (MOE) Approach Is Better" BioMedInformatics 3, no. 4: 869-884. https://doi.org/10.3390/biomedinformatics3040054
APA StyleLötsch, J., & Ultsch, A. (2023). Pitfalls of Using Multinomial Regression Analysis to Identify Class-Structure-Relevant Variables in Biomedical Data Sets: Why a Mixture of Experts (MOE) Approach Is Better. BioMedInformatics, 3(4), 869-884. https://doi.org/10.3390/biomedinformatics3040054