
A Comprehensive Evaluation of a Chalcone Derivative: Structural, Spectroscopic, Computational, Electrochemical, and Pharmacological Perspectives

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Experimental Methods
2.2.2. Computational Studies
Theoretical Study
Structure Preprocessing, Validation, and Molecular Docking
Molecular Dynamic (MD) Simulation and Trajectory Analysis
Free Energy and Decomposition
2.3. Cytotoxicity Studies and Apoptosis Using Flow Cytometer
3. Results and Discussions
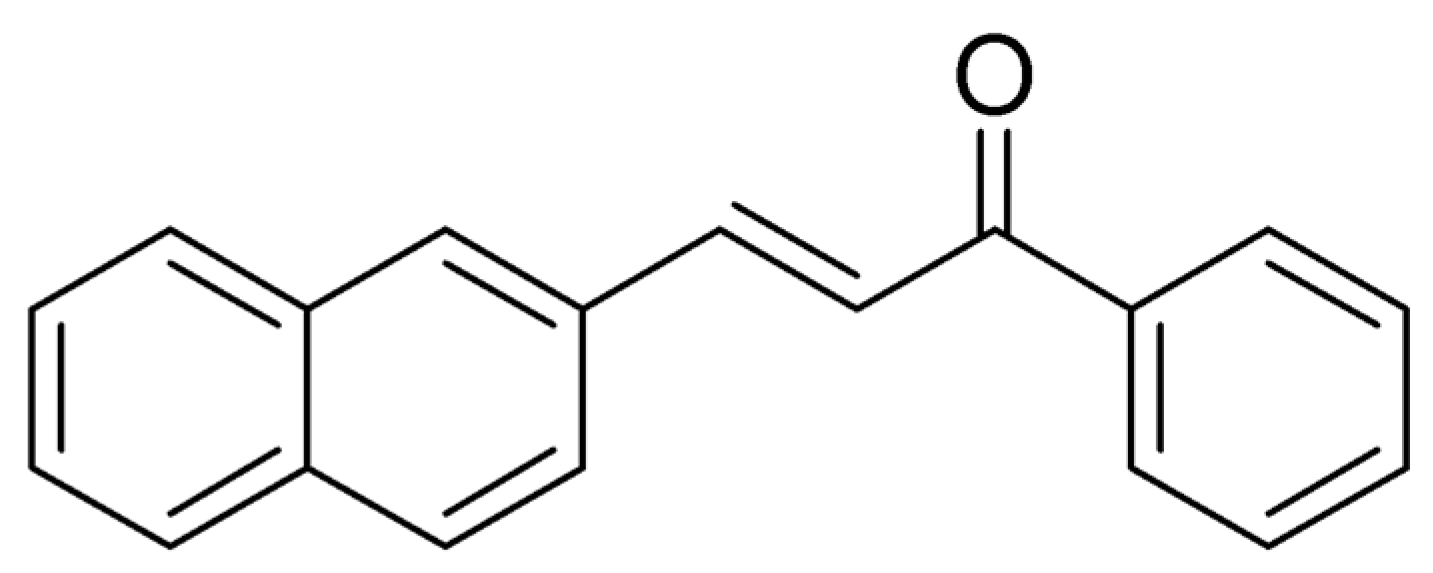
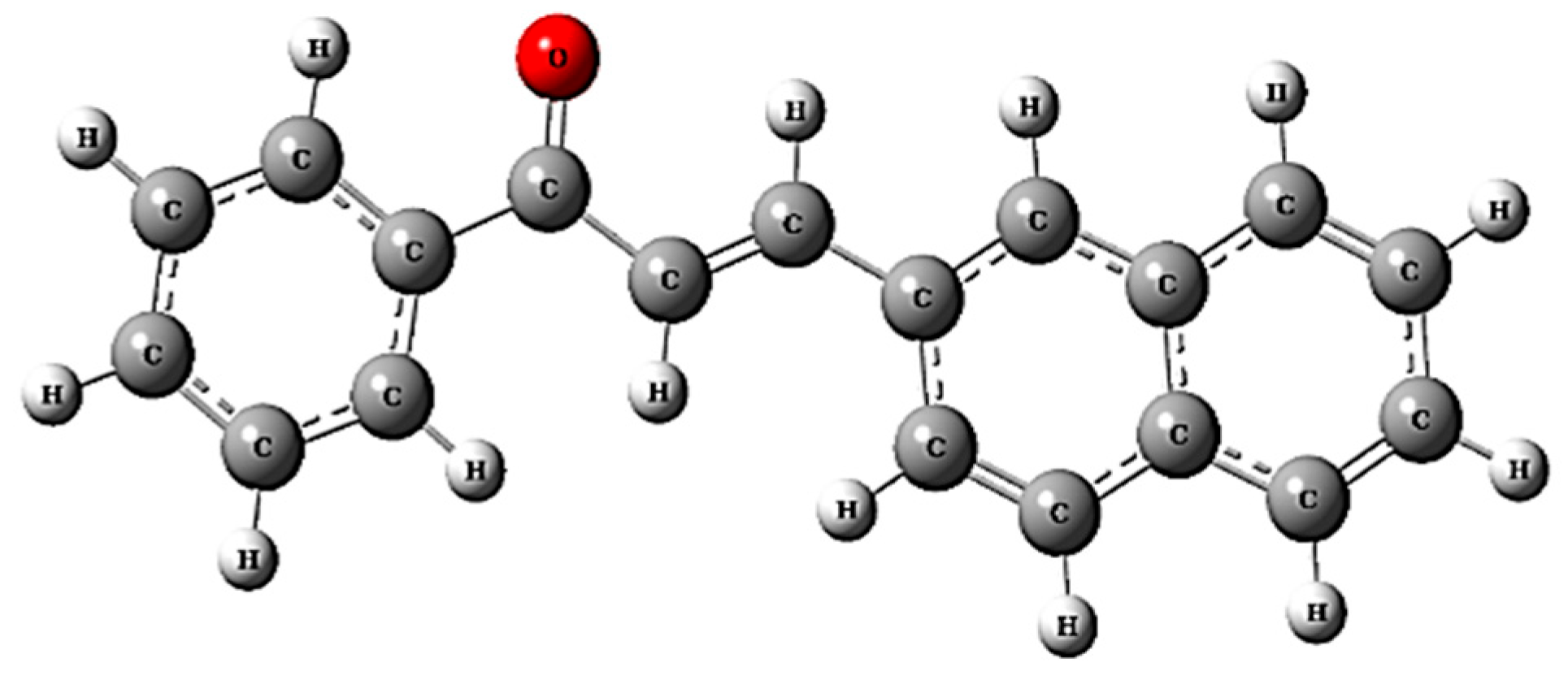
3.1. Structural Analysis
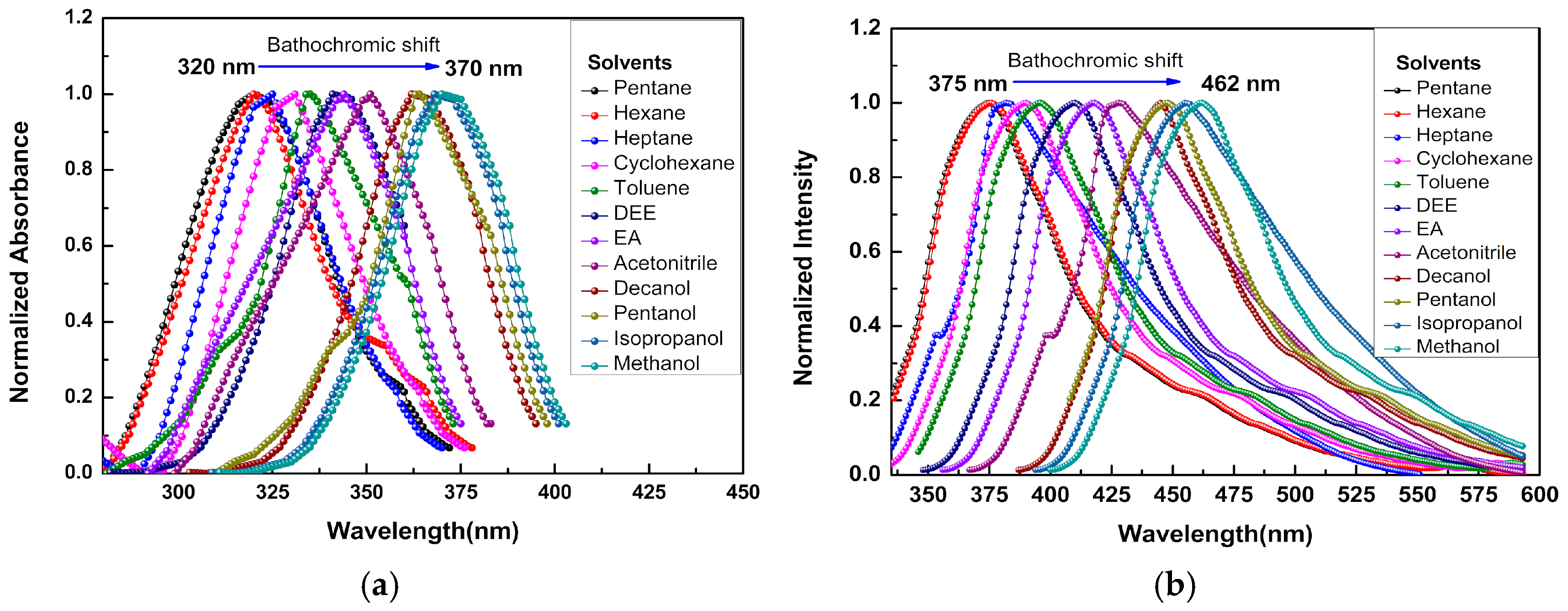
3.2. Solvent Effect on Photophysical Properties of 3NPEO
3.3. Fluorescence Quantum Yield (Photonic Efficiency)
3.4. Nonlinear Optical Studies
3.4.1. Third Order Nonlinear Optical Properties
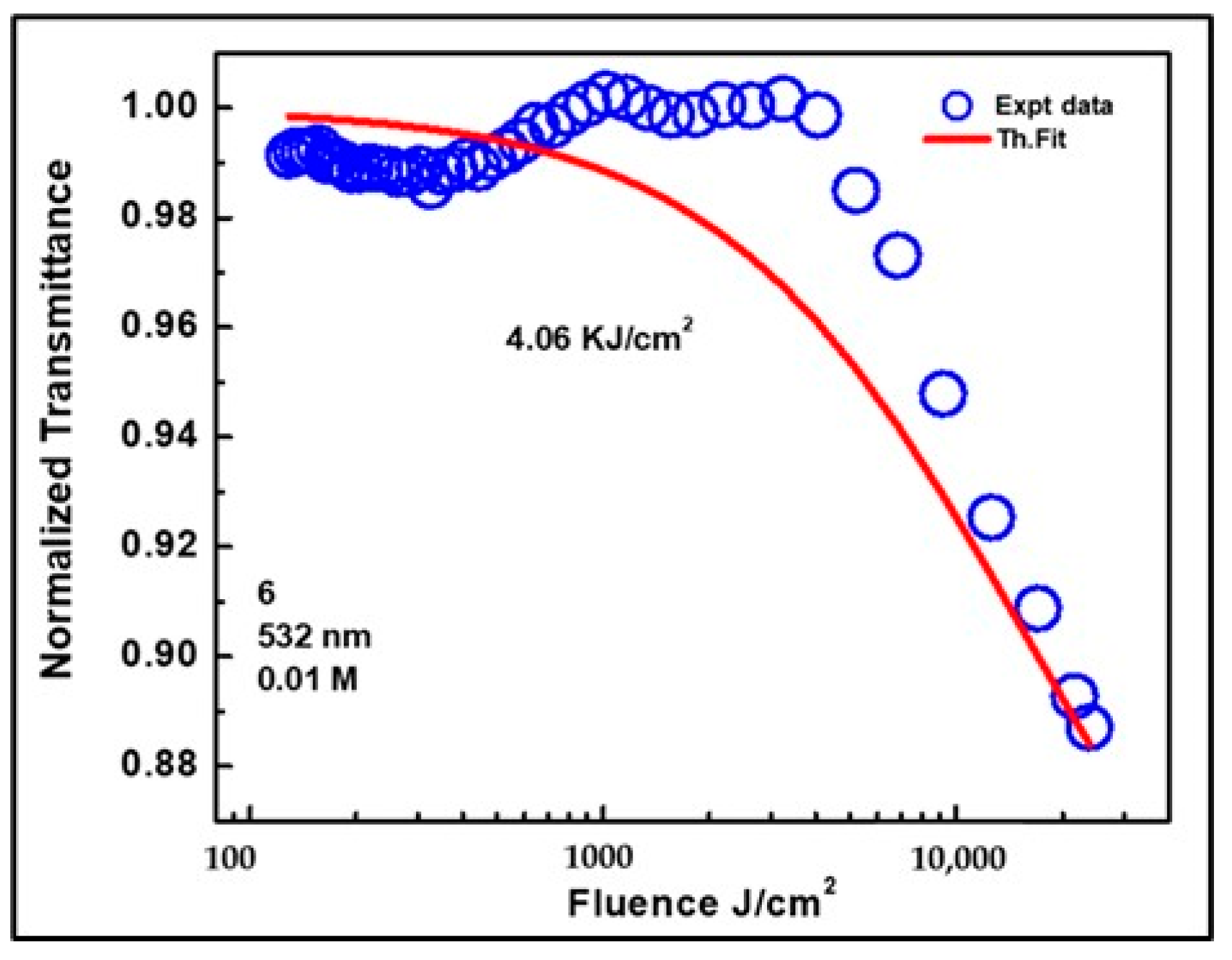
3.4.2. Optical Limiting
3.5. Electrochemical Property
3.6. Computational Analysis
3.6.1. Theoretical Study
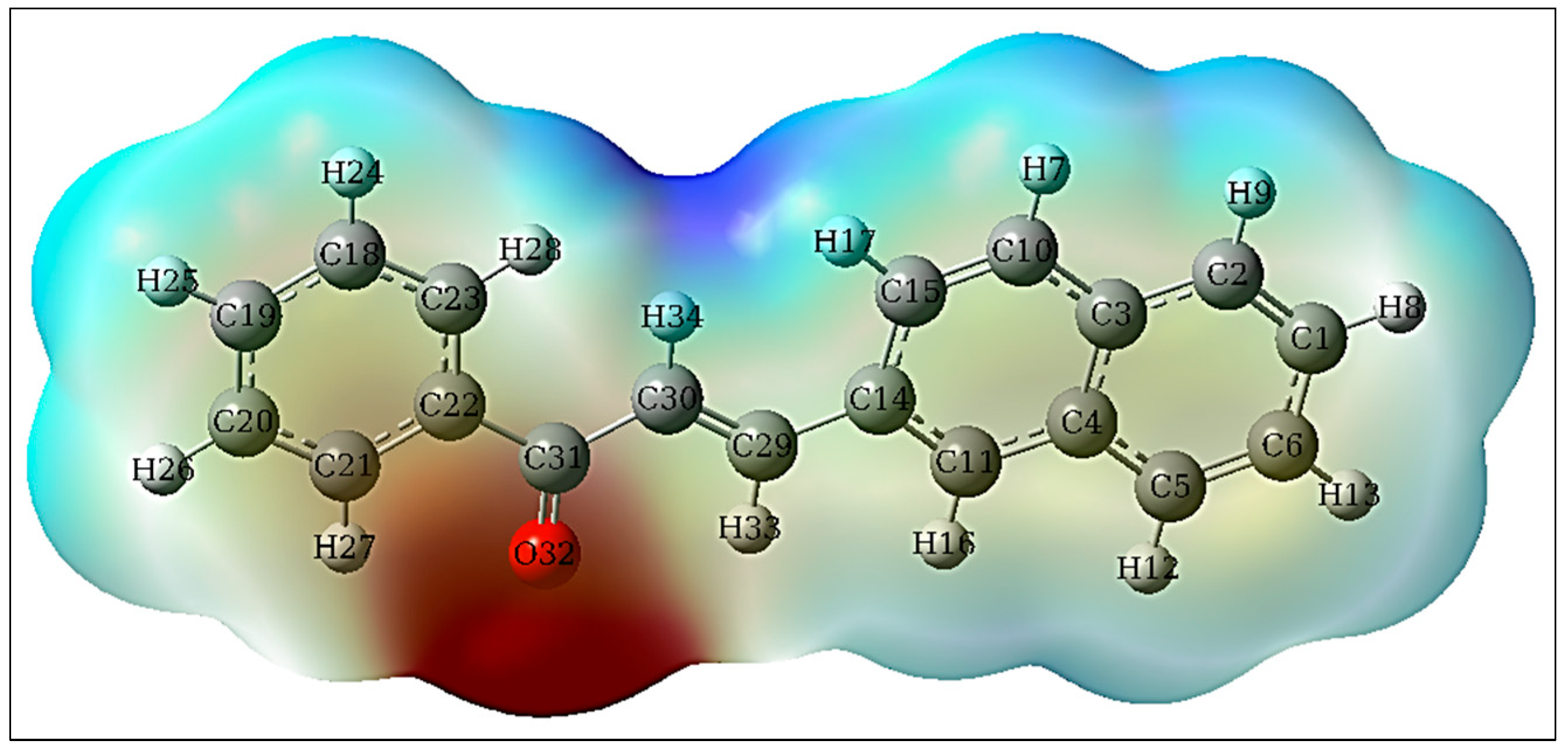
3.6.2. HOMO-LUMO, GCRD and MEP
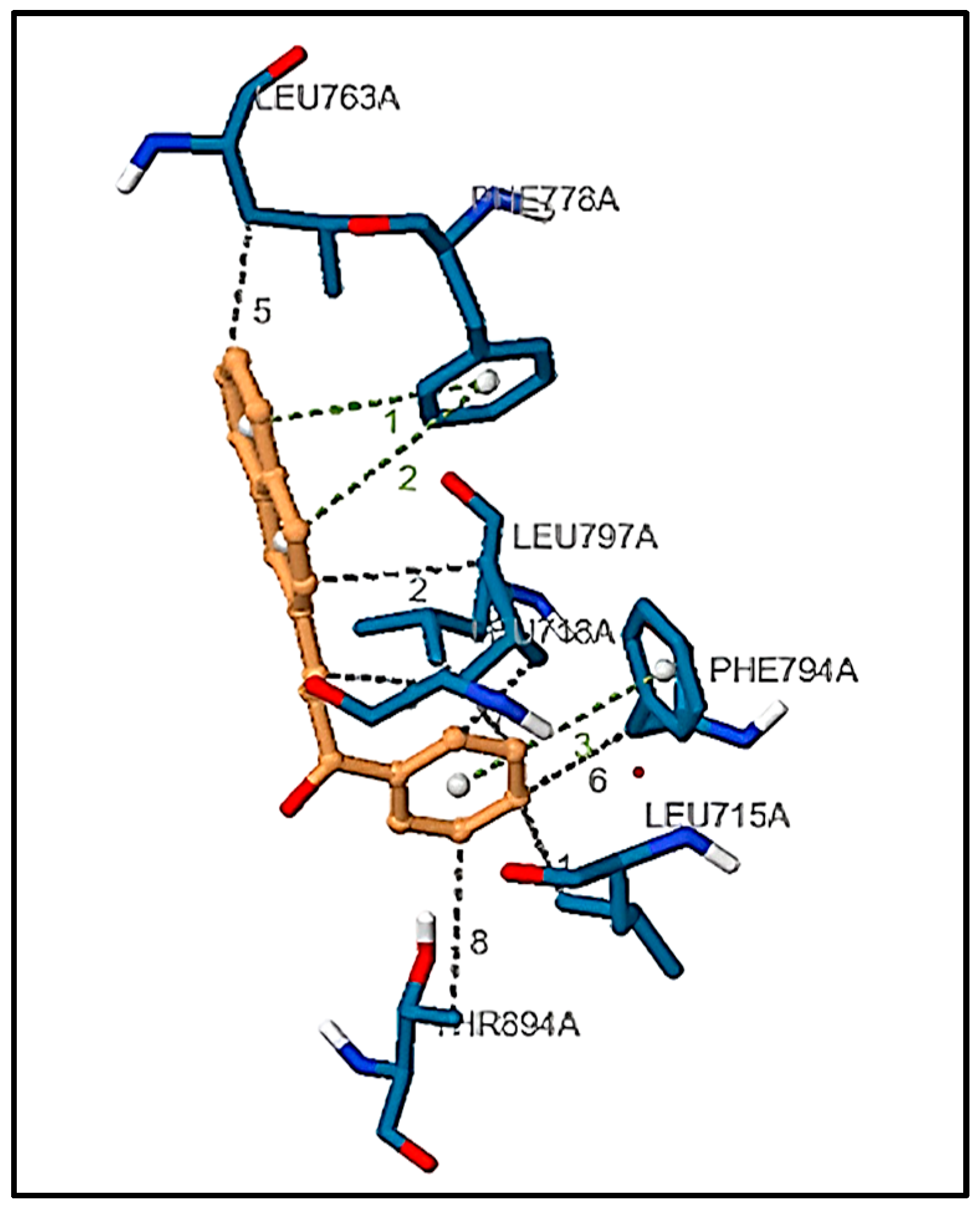
3.6.3. Structure Preprocessing, Validation, and Molecular Docking
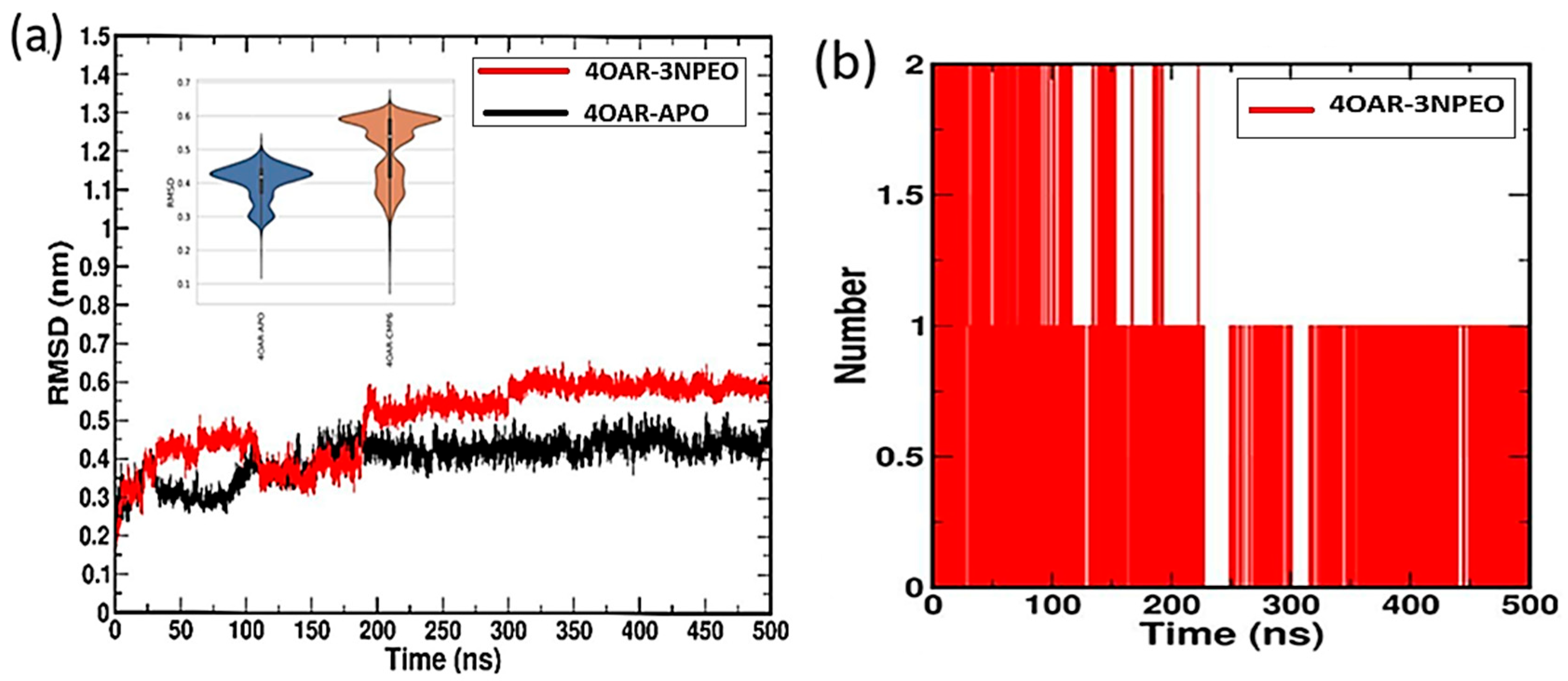
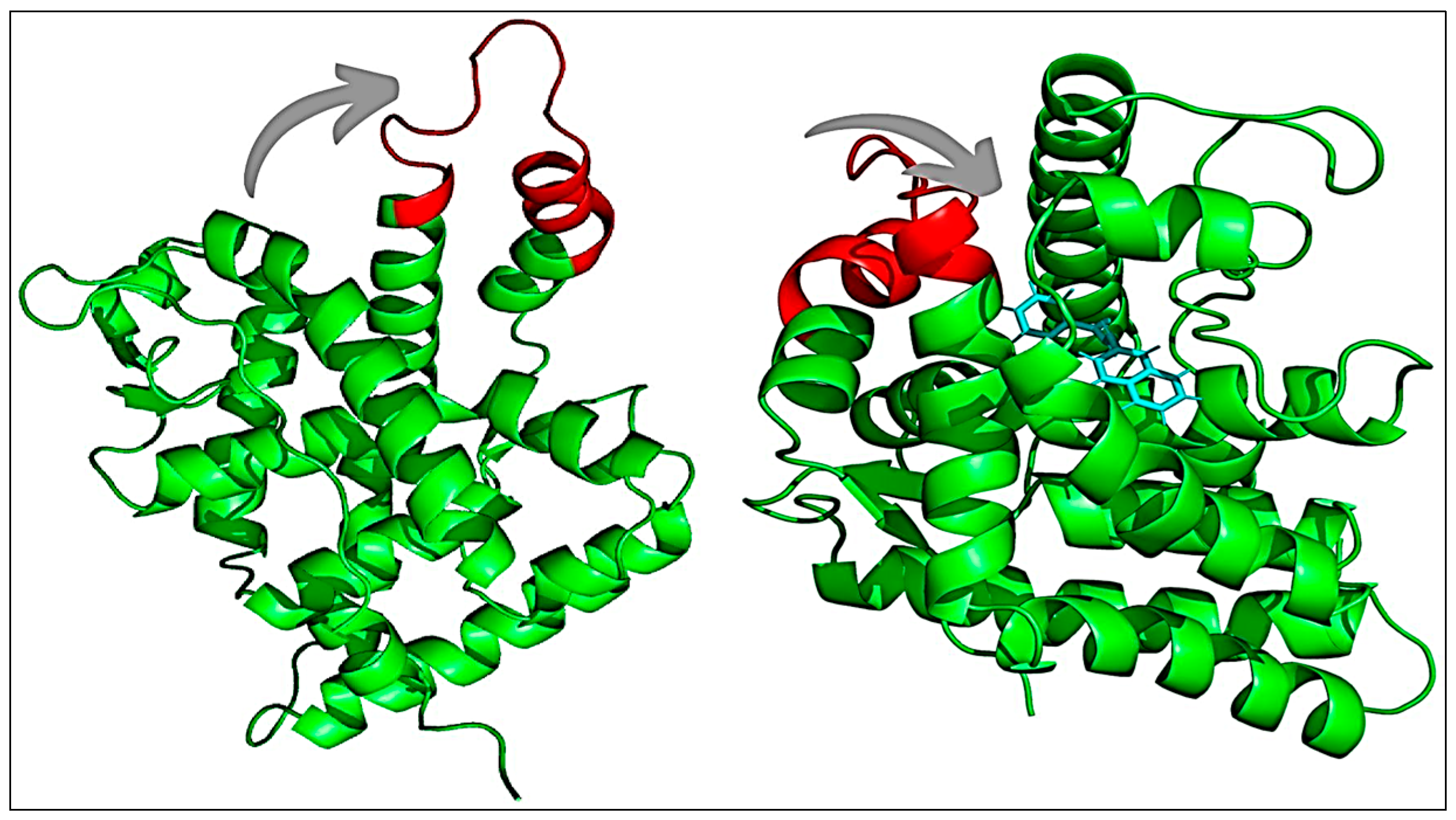
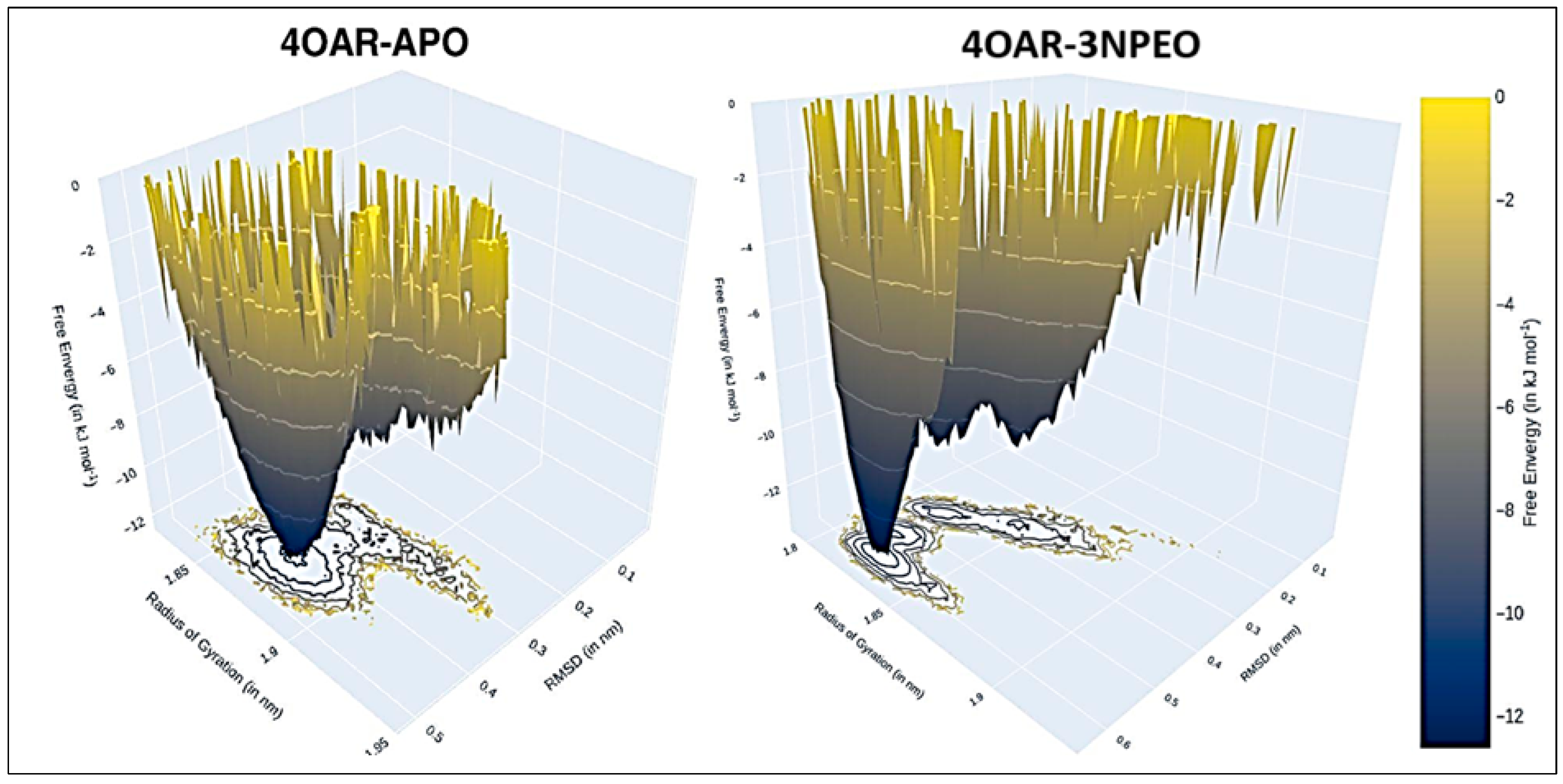
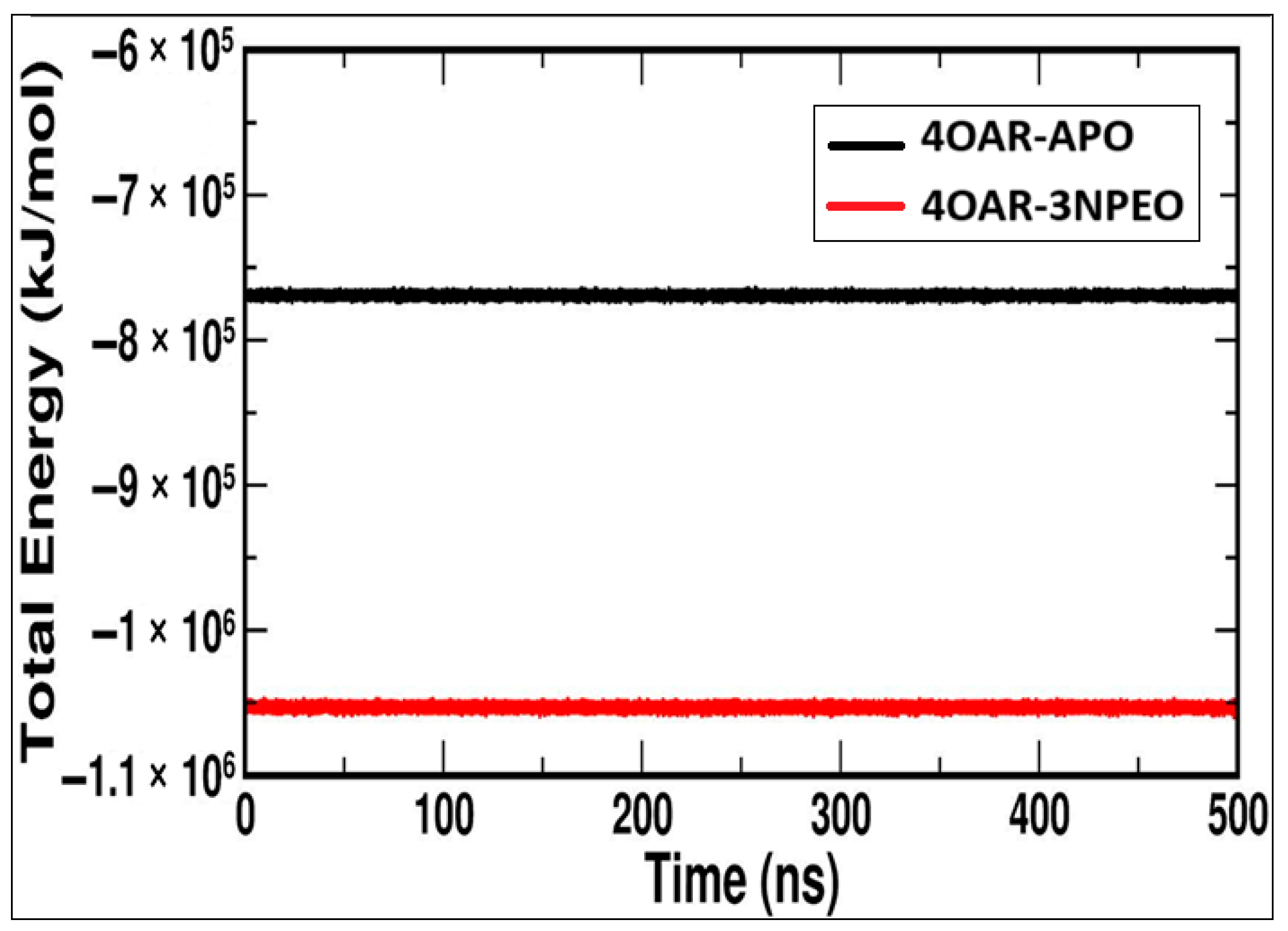
3.6.4. Simulation Trajectory Analysis
3.6.5. Conformational Minima, Binding Energy, and Total System Energy
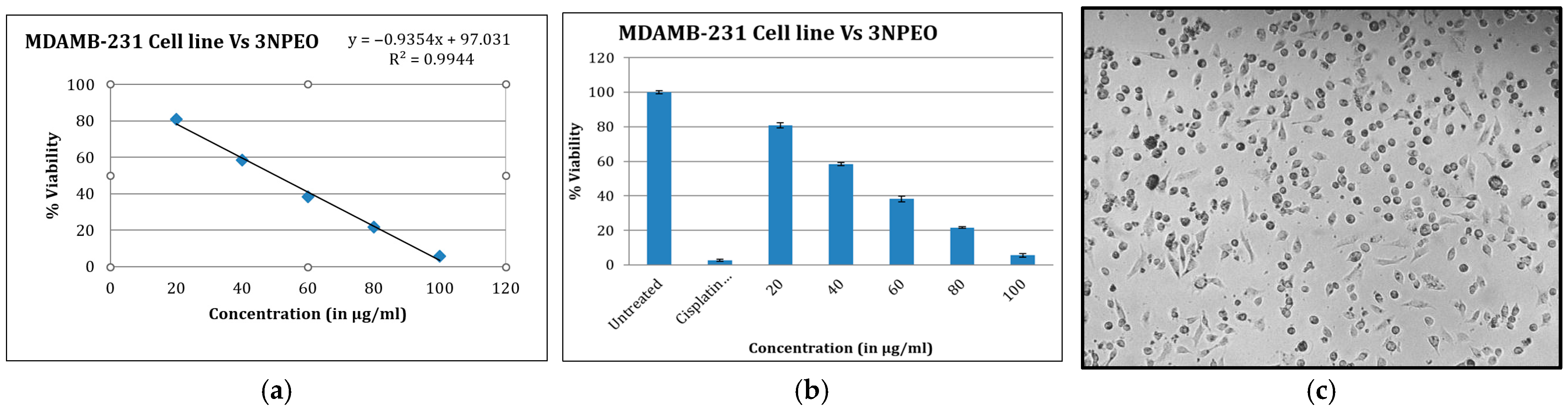
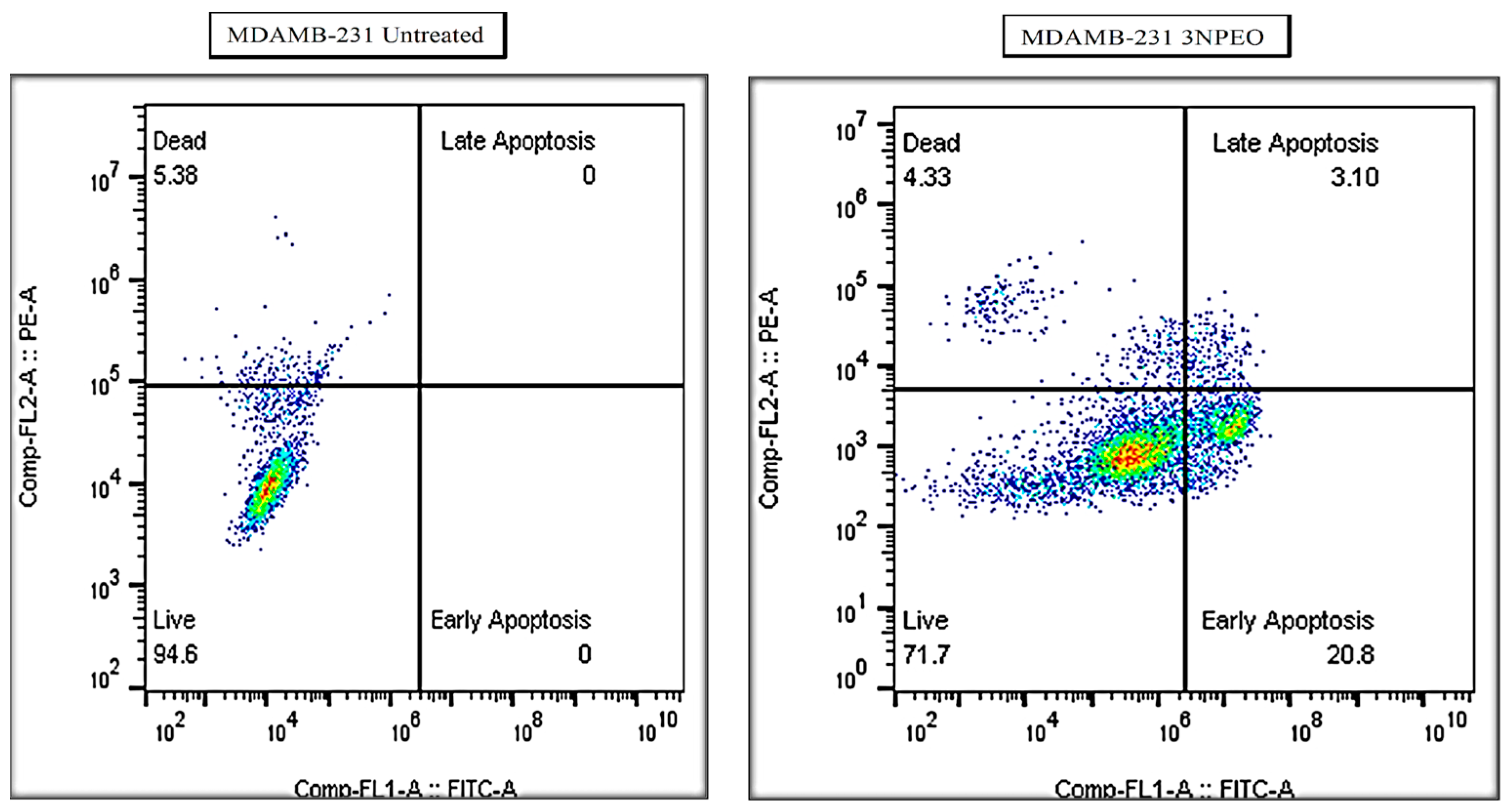
3.7. Cytotoxicity Studies and Detection of Early and Late Apoptosis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dhar, D.N. The Chemistry of Chalcones and Related Compounds; John Wiley & Sons: Hoboken, NJ, USA, 1981. [Google Scholar]
- da Costa, R.G.M.; de Queiroz Garcia, R.; Fiuza, R.M.D.R.; Maqueira, L.; Pazini, A.; De Boni, L.; Limberger, J. Synthesis, photophysical properties and aggregation-induced enhanced emission of bischalcone-benzothiadiazole and chalcone-benzothiadiazole hybrids. J. Lumin. 2021, 239, 118367. [Google Scholar] [CrossRef]
- Irfan, R.; Mousavi, S.; Alazmi, M.; Saleem, R.S.Z. A comprehensive review of amino chalcones. Molecules 2020, 25, 5381. [Google Scholar] [CrossRef]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A privileged structure in medicinal chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef]
- More, V.S.; Panchgalle, S.P.; Gayake, R.A.; Jagrut, V.B.; Kodape, M.M.; Birhade, D.R.; Lokhande, M.N.N. Synthesis, Molecular Docking, and Biological Evaluation of Some New Naphthalene-Chalcone Derivatives as Potential Anticancer Agent on MCF-7 Cell Line by MTT Assay. Asian J. Green Chem. 2024, 8, 234–246. [Google Scholar]
- Lee, M.S.; Koh, D.; Kim, G.S.; Lee, S.E.; Noh, H.J.; Kim, S.Y.; Lee, Y.H.; Lim, Y.; Shin, S.Y. 2-Hydroxy-3,4-naphthochalcone (2H-NC) inhibits TNFα-induced tumor invasion through the downregulation of NF-κB-mediated MMP-9 gene expression. Bioorg. Med. Chem. Lett. 2015, 25, 128–132. [Google Scholar] [CrossRef]
- Ali, M.K.M.; Elzupir, A.O.; Ibrahem, M.A.; Suliman, I.I.; Modwi, A.; Idriss, H.; Ibnaouf, K.H. Characterization of optical and morphological properties of chalcone thin films for optoelectronics applications. Optik 2017, 145, 529–533. [Google Scholar] [CrossRef]
- Sharma, V.S.; Sharma, A.S.; Agarwal, N.K.; Shah, P.A.; Shrivastav, P.S. Self-assembled blue-light emitting materials for their liquid crystalline and OLED applications: From a simple molecular design to supramolecular materials. Mol. Syst. Des. Eng. 2020, 5, 1691–1705. [Google Scholar] [CrossRef]
- Fayed, T.A. A novel chalcone-analogue as an optical sensor based on ground and excited states intramolecular charge transfer: A combined experimental and theoretical study. Chem. Phys. 2006, 324, 631–638. [Google Scholar] [CrossRef]
- Custodio, J.M.F.; Gotardo, F.; Vaz, W.F.; D’Oliveira, G.D.C.; de Almeida, L.R.; Fonseca, R.D.; Cocca, L.H.Z.; Perez, C.N.; Oliver, A.G.; de Boni, L.; et al. Benzenesulfonyl incorporated chalcones: Synthesis, structural and optical properties. J. Mol. Struct. 2020, 1208, 127845. [Google Scholar] [CrossRef]
- Anandkumar, D.; Ganesan, S.; Rajakumar, P.; Maruthamuthu, P. Synthesis, photophysical and electrochemical properties and DSSC applications of triphenylamine chalcone dendrimers via click chemistry. New J. Chem. 2017, 41, 11238–11249. [Google Scholar] [CrossRef]
- Constantinescu, T.; Mihis, A.G. Two Important Anticancer Mechanisms of Natural and Synthetic Chalcones. Int. J. Mol. Sci. 2022, 23, 11595. [Google Scholar] [CrossRef]
- Garg, S.; Raghav, N. Spectrophotometric Analysis of Bovine Serum Albumin in Presence of Some 1-(Naphthalen-3-Yl)- 3-Phenylprop-2-En-1-Ones. Int. J. Chem. Sci. 2013, 11, 1137–1145. [Google Scholar]
- Shamsunder Misra, Chalcones XIX: Potenial Germicides Derived from 2-Acetonaphthones. J. Indian Chem. Soc. 1975, 52, 1095–1096. [CrossRef]
- Deshpande, A.M.; Argade, N.P.; Natu, A.A.; Eckman, J. Synthesis and screening of a combinatorial library of naphthalene substituted chalcones: Inhibitors of leukotriene B41. Bioorganic Med. Chem. 1999, 7, 61237–61240. [Google Scholar] [CrossRef]
- Robinson, T.P.; Hubbard, R.B., 4th; Ehlers , T.J.; Arbiser , J.L.; Goldsmith , D.J.; Bowen , J.P. Synthesis and biological evaluation of aromatic enones related to curcumin. Bioorganic Med. Chem. 2005, 13, 4007–4013. [Google Scholar] [CrossRef]
- Lee, J.M.; Lee, M.S.; Koh, D.; Lee, Y.H.; Lim, Y.; Shin, S.Y. A new synthetic 2′-hydroxy-2,4,6-trimethoxy-5′,6′ naphthochalcone induces G2/M cell cycle arrest and apoptosis by disrupting the microtubular network of human colon cancer cells. Cancer Lett. 2014, 354, 348–354. [Google Scholar] [CrossRef]
- Schmitt, F.; Draut, H.; Biersack, B.; Schobert, R. Halogenated naphthochalcones and structurally related naphthopyrazolines with antitumor activity. Bioorganic Med. Chem. Lett. 2016, 26, 5168–5171. [Google Scholar] [CrossRef]
- Anizaim, A.H.; Zainuri, D.A.; Zaini, M.F.; Razak, I.A.; Bakhtiar, H.; Arshad, S. Comparative Analyses of New Donor-π-Acceptor Ferrocenyl-Chalcones Containing Fluoro and Methoxy-Fluoro Acceptor Units as Synthesized Dyes for Organic Solar Cell Material. PLoS ONE 2020, 15, e0241113. [Google Scholar] [CrossRef]
- Lan, H.; Guo, T.; Dan, F.; Li, Y.; Tang, Q. Ratiometric Fluorescence Chemodosimeter for Hydrazine in Aqueous Solution and Gas Phase Based on Quinoline-Malononitrile. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2022, 271, 120892. [Google Scholar] [CrossRef]
- Kagatikar, S.; Sunil, D.; Kekuda, D.; Satyanarayana, M.N.; Kulkarni, S.D.; Sudhakar, Y.N.; Vatti, A.K.; Sadhanala, A. Pyrene-Based Chalcones as Functional Materials for Organic Electronics Application. Mater. Chem. Phys. 2023, 293, 126839. [Google Scholar] [CrossRef]
- Maltais, R.; Roy, J.; Poirier, D. Turning a Quinoline-Based Steroidal Anticancer Agent into Fluorescent Dye for Its Tracking by Cell Imaging. ACS. Med. Chem. Lett. 2021, 12, 822–826. [Google Scholar] [CrossRef]
- Praveen Kumar, C.H.; Katagi, M.S.; Nandeshwarappa, B.P. Novel Synthesis of Quinoline Chalcone Derivatives—Design, Synthesis, Characterization and Antimicrobial Activity. Chem. Data Collect. 2022, 42, 100955. [Google Scholar] [CrossRef]
- Sharma, S.; Singh, S. Synthetic Routes to Quinoline-Based Derivatives Having Potential Anti-Bacterial AndAnti-Fungal Properties. Curr. Org. Chem. 2022, 26, 1453–1469. [Google Scholar] [CrossRef]
- Kaddah, M.M.; Morsy, A.R.I.; Fahmi, A.A.; Kamel, M.M.; Elsafty, M.M.; Rizk, S.A.; Ramadan, S.K. Synthesis and Biological Activity on IBD Virus of Diverse Heterocyclic Systems Derived from 2-Cyano-N-((2-Oxo-1,2-Dihydroquinolin-3-Yl)Methylene)-Acetohydrazide. Synth. Commun. 2021, 51, 3366–3378. [Google Scholar] [CrossRef]
- Radini, I.A.M.; Elsheikh3, T.M.Y.; El-Telbani, E.M.; Khidre, R.E. New Potential Antimalarial Agents: Design, Synthesis and Biological Evaluation of Some Novel Quinoline Derivatives as Antimalarial Agents. Molecules 2016, 21, 909. [Google Scholar] [CrossRef]
- Valencia, J.; Rubio, V.; Puerto, G.; Vasquez, L.; Bernal, A.; Mora, J.R.; Cuesta, S.A.; Paz, J.L.; Insuasty, B.; Abonia, R.; et al. QSAR Studies, Molecular Docking, Molecular Dynamics, Synthesis, and Biological Evaluation of Novel Quinolinone-Based Thiosemicarbazones against Mycobacterium Tuberculosis. Antibiotics 2023, 12, 61. [Google Scholar] [CrossRef]
- Bacci, A.; Corsi, F.; Runfola, M.; Sestito, S.; Piano, I.; Manera, C.; Saccomanni, G.; Gargini, C.; Rapposelli, S. Design, Synthesis, and In Vitro Evaluation of Novel 8-Amino-Quinoline Combined with Natural Antioxidant Acids. Pharmaceuticals 2022, 15, 688. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.F.A.; Abuo-Rahma, G.E.A. Molecular Targets and Anticancer Activity of Quinoline–Chalcone Hybrids: Literature Review. RSC Adv. 2020, 10, 31139–31155. [Google Scholar] [CrossRef]
- Kivshar, Y.S. Nonlinear optics: The next decade. Opt. Exp. 2008, 16, 22126. [Google Scholar] [CrossRef] [PubMed]
- Custodio, J.M.F.; D’Oliveira, G.D.C.; Gotardo, F.; Cocca, L.H.Z.; Boni, L.D.; Perez, C.N.; Maia, L.J.Q.; Valverde, C.; Osório, F.A.P.; Napolitano, H.B. Chalcone as Potential Nonlinear Optical Material: A Combined Theoretical, Structural, and Spectroscopic Study. J. Phys. Chem. C 2019, 123, 5931–5941. [Google Scholar] [CrossRef]
- Prasad, P.N.; Williams, D.J. Introduction to Nonlinear Optical Effects in Organic Molecules and Polymers; Wiley: New York, NY, USA, 1991. [Google Scholar]
- Sutherland, R.L. Hand book of Nonlinear Optics, 2nd ed.; Marcel Dekker Inc.: New York, NY, USA, 2003. [Google Scholar]
- Baig, H.; Iqbal, A.; Rasool, A.; Hussain, S.Z.; Iqbal, J.; Alazmi, M.; Alshammari, N.; Alazmi, A.; AlGhadhban, A.; Abdel Sulieman, M.E.; et al. Synthesis and Photophysical, Electrochemical, and DFT Studies of Piperidyl and Pyrrolidinyl Chalcones. ACS Omega 2023, 8, 28499–28510. [Google Scholar] [CrossRef]
- Insuasty, D.; Mutis, M.; Trilleras, J.; Illicachi, L.A.; Rodríguez, J.D.; Ramos-Hernández, A.; San-Juan-Vergara, H.G.; Cadena-Cruz, C.; Mora, J.R.; Paz, J.L.; et al. Synthesis, Photophysical Properties, Theoretical Studies, and Living Cancer Cell Imaging Applications of New 7-(Diethylamino)quinolone Chalcones. ACS Omega 2024, 9, 18786–18800. [Google Scholar] [CrossRef]
- Gomes, M.N.; Muratov, E.N.; Pereira, M.; Peixoto, J.C.; Rosseto, L.P.; Cravo, P.V.L.; Andrade, B.J. Neves, Chalcone Derivatives: Promising Starting Points for Drug Design. Molecules 2017, 22, 1210. [Google Scholar] [CrossRef] [PubMed]
- Chiaradia, L.D.; Mascarello, A.; Purificação, M.; Vernal, J.; Sechini Cordeiro, M.N.; Zenteno, M.E.; Villarino, A.; Nunes, R.J.; Yunes, R.A.; Terenzi, H. Synthetic chalcones as efficient inhibitors of Mycobacterium tuberculosis protein tyrosine phosphatase PtpA. Bioorganic Med. Chem. Lett. 2008, 23, 6227–6230. [Google Scholar] [CrossRef]
- Damazio, R.G.; Zanatta, A.P.; Cazarolli, L.H.; Chiaradia, L.D.; Mascarello, A.; Nunes, R.J.; Yunes, R.A.; Barreto Silva, F.R.M. Antihyperglycemic activity of naphthylchalcones. Eur. J. Med. Chem. 2010, 4, 1332–1337. [Google Scholar] [CrossRef]
- Kusanur, R.A.; Kulkarni, M.V. A Novel Route for the Synthesis of Recemic 4-(Coumaryl) Alanines and Their Antimicrobial Activity. Asian J. Chem. 2014, 26, 1077. [Google Scholar] [CrossRef]
- Kusanur, R.; Ghate, M.; Kulkarni, M. Synthesis and biological activities of some substituted 4-{4-(1, 5-diphenyl-1H-pyrazol-3-yl) phenoxymethyl} coumarins. Indian J. Heterocycl. Chem. 2004, 13, 201–204. [Google Scholar]
- Patagar, D.; Uttarkar, A.; Patra, S.M.; Patil, J.H.; Kusanur, R.; Niranjan, V.; Ashok Kumar, H.G. Spiro benzodiazepine substituted fluorocoumarins as potent anti-anxiety agents. Russ. J. Bioorganic Chem. 2021, 47, 390–398. [Google Scholar] [CrossRef]
- Sheik-Bahae, M.; Said, A.A.; Wei, T.H.; Hagan, D.J.; Van Stryland, E.W. Sensitive measurement of optical nonlinearities using a single beam. IEEE J. Quantum Electron. 1990, 26, 760–769. [Google Scholar] [CrossRef]
- Sheik Bahae, M.; Hutchings, D.C.; Hagan, D.J.; Van Stryland, E.W. Dispersion of bound electronic nonlinear refraction in solids. IEEE J. Quantum Electron. 1991, 27, 1296–1309. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016.
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView 6.0.16, Semichem Inc.: Shawnee Mission, KS, USA, 2016.
- Zarezade, V.; Abolghasemi, M.; Rahim, F.; Veisi, A.; Behbahani, M. In silico assessment of new progesterone receptor inhibitors using molecular dynamics: A new insight into breast cancer treatment. J. Mol. Model. 2018, 24, 337. [Google Scholar] [CrossRef]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.; Pieper, U.; Sali, A. Comparative Protein Structure Modeling Using Modeller. Curr. Protoc. Bioinform. 2006, 54, 5–6. [Google Scholar] [CrossRef]
- Ahmad, M.; Bauer, W.; Katz, S. Ulcerative colitis, hyperamylasemia, and asymptomatic pancreatic calcifications: Making the case for pancreatitis as an extra luminal manifestation. Am. J. Gastroenterol. 1997, 92, 2307–2309. [Google Scholar] [PubMed]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef] [PubMed]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Tuble, S.C.; Anwar, J.; Gale, J.D. An Approach to Developing a Force Field for Molecular Simulation of Martensitic Phase Transitions between Phases with Subtle Differences in Energy and Structure. J. Am. Chem. Soc. 2004, 126, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Gummagol, N.B.; Yaraguppi, D.A.; Patil, S.B.; Patil, P.S.; Patil, N.R.; Ayachit, N.H. Exploring the anticancer potential of novel chalcone derivatives: Synthesis, characterization, computational analysis, and biological evaluation against breast cancer. J. Mol. Struct. 2025, 1320, 139586. [Google Scholar] [CrossRef]
- Al-Rubaye, H.I.; Abbas, Z.M.; Jinzeel, N.A.; Askar, F.W. Synthesis and Preliminary Biological study of New Chalcone Derivatives Containing Isoxazoline Moiety. J. Kufa Chem. Sci. 2024, 3, 304–319. [Google Scholar] [CrossRef]
- El-Naggar, M.; Rashdan, H.R.M.; Abdelmonsef, A.H. Cyclization of Chalcone Derivatives:Design, Synthesis, InSilico Docking Study, and Biological Evaluation of New Quinazolin-2,4 diones Incorporating Five-,Six-,and Seven-Membered Ring Moieties as Potent Antibacterial Inhibitors. ACS Omega 2023, 8, 27216–27230. [Google Scholar] [CrossRef]
- Siddique, I.M. Exploring Functional Groups and Molecular Structures: A Comprehensive Analysis using FTIR Spectroscopy. Chem. Res. J. 2024, 9, 70–76. [Google Scholar] [CrossRef]
- Muddapur, G.V.; Patil, N.R.; Patil, S.S.; Melavanki, R.M.; Kusanur, R.A. Estimation of Ground and Excited State Dipole Moments of aryl Boronic acid Derivative by Solvatochromic Shift Method. J. Fluoresc. 2014, 24, 1651–165949. [Google Scholar] [CrossRef]
- Patil, S.S.; Muddapur, G.V.; Patil, N.R.; Melavanki, R.M.; Kusanur, R.A. Fluorescence characteristics of aryl boronic acid derivative (PBA). Pectrochimica Acta Part A Mol. Biomol. Spectrosc. 2015, 138, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Muddapur, G.V.; Melavanki, R.M.; Patil, P.G.; Nagaraja, D.; Patil, N.R. Photophysical properties of 3MPBA: Evaluation and co-relation between solvatochromism and quantum yield in different solvents. J. Mol. Liq. 2016, 224, 201–210. [Google Scholar] [CrossRef]
- Muddapur, G.V.; Melavanki, R.M.; Sharma, K.; Srinivasa, H.T.; Thipperudrappa, J. Fluorescence properties of aromatic asymmetric di-ketone compound in polar and non-polar solvents. J. Phys. Conf. Ser. 2020, 1473, 012044. [Google Scholar] [CrossRef]
- Melavanki, R.M.; Muddapur, G.V.; Srinivasa, H.T.; Honnanagoudar, S.S.; Patil, N.R. Solvation, rotational dynamics, photophysical properties study of aromatic asymmetric di-ketones: An experimental and theoretical approach. J. Mol. Liq. 2021, 337, 116456. [Google Scholar] [CrossRef]
- Wangngae, S.; Chansaenpak, K.; Nootem, J.; Ngivprom, U.; Aryamueang, S.; Lai, R.; Kamkaew, A. Photophysical Study and Biological Applications of Synthetic Chalcone-Based Fluorescent Dyes. Molecules 2021, 26, 2979. [Google Scholar] [CrossRef]
- Baig, H.; Irfan, R.; Rasool, A.; Hussain, S.Z.; Siddique, S.A.; Iqbal, J.; Alazmi, M.; Alshammari, N.; Alazmi, A.; AlGhadhban, A.; et al. Synthesis, photophysical, voltammetric, and DFT studies of 4-aminochalones. J. Photochem. Photobiol. A Chem. 2023, 442, 114790. [Google Scholar] [CrossRef]
- Sachdeva, T.; Milton, M.D. AIEE active novel red-emitting D-π-A phenothiazine chalcones displaying large Stokes shift, solvatochromism and “turn-on” reversible mechano fluorochromism. Dye. Pigment. 2020, 181, 108539. [Google Scholar] [CrossRef]
- Wang, S.; Li, B.; Zhang, F. Molecular Fluorophores for Deep-Tissue Bioimaging. ACS Cent. Sci. 2020, 6, 1302–1316. [Google Scholar] [CrossRef]
- Bilot, L.; Kawski, A. Der Einfluß des Lösungsmittels auf die Elektronenspektren lumineszierender Moleküle. Z. Für Naturforschung A 1963, 18, 10–15. [Google Scholar] [CrossRef]
- Lippert, E. Spektroskopische Bestimmung des Dipolmomentes aromatischer Verbindungen im ersten angeregten Singulettzustand. Z. Elektrochem. Berichte Bunsenges. Phys. Chem. 1955, 10, 541. [Google Scholar] [CrossRef]
- Bakshiev, N.G. Universal Intermolecular Interactions and Their Effect on the Position of the Electronic Spectra of Molecules in Two-Component Solutions. VII. Theory (General Case of an Isotropic Solution). Opt. Spektrosk. 1964, 16, 821. [Google Scholar]
- Chamma, A.; Viallet, P. Determination du moment dipolaire d’une molecule dans un etat excite singulet. CR Acad. Sci. Paris. Ser. C 1970, 270, 1901–1904. [Google Scholar]
- Reichardt, C. Solvatochromic dyes as solvent polarity indicators. Chem. Rev. 1994, 94, 2319–2358. [Google Scholar] [CrossRef]
- Patil, N.R.; Melavanki, R.M.; Kapatkar, S.B.; Ayachit, N.H.; Saravanan, J. Solvent Effect on Absorption and Fluorescence Spectra of Three Biologically Active Carboxamides (C1, C2 and C3). Estimation of Ground and Excited State Dipole Moment from Solvatochromic Method Using Solvent Polarity Parameters. J. Fluoresce. 2011, 21, 1213–1222. [Google Scholar] [CrossRef]
- Melavanki, R.M.; Patil, N.R.; Kapatkar, S.B.; Ayachit, N.H.; Umapathy, S.; Thipperudrappa, J.; Nataraju, A.R. Solvent effect on the spectroscopic properties of 6MAMC and 7MAMC. J. Mol. Liq. 2011, 158, 105–110. [Google Scholar] [CrossRef]
- Mallik, T.; Banerjee, D. Solvation and Solvatochromism: An Overview. Indian J. Chem. 2022, 61, 472–481. [Google Scholar] [CrossRef]
- Bhavya, P.; Melavanki, R.M.; Manjunatha, M.N.; Koppal, V.V.; Patil, N.R.; Muttannavar, V.T. Solvent Effects on the Photophysical Properties of Coumarin Dye. AIP Conf. Proc. 2018, 1953, 080022. [Google Scholar] [CrossRef]
- Gaur, A.; Gaur, P.; Sharma, D.; Sharma, D.K.; Singh, N.; Malik, B.P. Study of transmittance dependence closed-aperture Z-scan curves in the materials with nonlinear refraction and strong absorption. Optik 2012, 123, 1583–1587. [Google Scholar] [CrossRef]
- Yan, X.Q.; Liu, Z.B.; Zhang, X.L.; Zhou, W.Y.; Tian, J.G. Polarization dependence of Z-scan measurement: Theory and experiment. Opt Express 2009, 17, 6397–6406. [Google Scholar] [CrossRef]
- Amore, F.D.; Osmond, J.; Destri, S.; Pasini, M.; Rossi, V.; Porzio, W. Effects of backbone modification on the linear and third order nonlinear optical properties in fluorene-based copolymers. Synth. Met. 2005, 149, 123–127. [Google Scholar]
- Koppal, V.V.; Muddapur, G.V.; Patil, N.R.; Melavanki, R.M. Spectroscopic studies of biologically active coumarin laser dye: Evaluation of dipole moments by solvatochromic shift method. AIP Conf. Proc. 2016, 1728, 020411. [Google Scholar] [CrossRef]
- Vasu, K.K.; Ingawale, H.D.; Sagar, S.R.; Sharma, J.A.; Pandya, D.H.; Agarwal, M. 2-((1H-1,2,3-triazol-1-yl) methyl)-3-phenylquinazolin-4(3H)-ones: Design, Synthesis and Evaluation as Anti-cancer Agents. Curr. Bioact. Compd. 2018, 14, 254–263. [Google Scholar] [CrossRef]
- Kiran, A.J.; Udayakumar, D.; Chandrashekaran, K.; Adhikari, A.V.; Shashikala, H.D. Z-Scan and degenerate four wave mixing studies on newly synthesized copolymers containing alternating substituted thiophene and 1,3,4-oxadiazole units. J. Phys. B 2006, 39, 3747–3756. [Google Scholar] [CrossRef]
- Ekbote, A.; Patil, P.S.; Maidur, S.R.; Chia, T.S.; Quah, C.K. Structural, third-order optical nonlinearities and figures of merit (E)-1-(3-substituted phenyl)-3-(4-fluorophenyl) prop2-en-1-one under CW regime: New Chalcone derivatives for optical limiting applications. Dye. Pigment. 2017, 139, 720–729. [Google Scholar] [CrossRef]
- Klus, S.; Nüske, F.; Peitz, S. Koopman analysis of quantum systems. J. Phys. A Math. Theor. 2022, 55, 314002. [Google Scholar] [CrossRef]
- Murray, J.S.; Sen, K. Molecular Electrostatic Potentials: Concepts and Applications. In Theoretical and Computational Chemistry; Elsevier Science: Amsterdam, The Netherlands, 1996; Volume 3, pp. 1–665. [Google Scholar]
- Scrocco, E.; Tomasi, J. Electronic Molecular structure, Reactivity and Intermolecular Forces: An Heuristic interpretation by means of Electronic Molecular Potentials. In Advances in Quantum Chemistry; Lowdin, P., Ed.; Academic Press: New York, NY, USA, 1978; Volume 2, p. 115. [Google Scholar]
- Nanjundaswamy, S.; Hema, M.K.; Karthik, C.S.; Rajabathar, J.R.; Arokiyaraj, S.; Lokanath, N.K.; Mallu, P. Synthesis, crystal structure, in-silico ADMET, molecular docking and dynamics simulation studies of thiophene-chalcone analogues. J. Mol. Struct. 2022, 1247, 131365. [Google Scholar] [CrossRef]
- Thillainayagam, M.; Malathi, K.; Ramaiah, S. In-Silico molecular docking and simulation studies on novel chalcone and flavone hybrid derivatives with 1, 2, 3-triazole linkage as vital inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J. Biomol. Struct. Dyn. 2018, 36, 3993–4009. [Google Scholar] [CrossRef]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [PubMed]
- Thillainayagam, M.; Malathi, K.; Anbarasu, A.; Singh, H.; Bahadur, R.; Ramaiah, S. Insights on inhibition of Plasmodium falciparum plasmepsin I by novel epoxyazadiradione derivatives—Molecular docking and comparative molecular field analysis. J. Biomol. Struct. Dyn. 2019, 37, 3168–3182. [Google Scholar] [CrossRef]
- Abdullahi, S.H.; Uzairu, A.; Shallangwa, G.A.; Uba, S.; Umar, A.B. In-silico activity prediction, structure-based drug design, molecular docking and pharmacokinetic studies of selected quinazoline derivatives for their antiproliferative activity against triple negative breast cancer (MDA-MB231) cell line. Bull. Natl. Res. Cent. 2022, 46, 2. [Google Scholar] [CrossRef]
- Darzynkiewicz, Z.; Bedner, E.; Smolewski, P. Flow cytometry in analysis of cell cycle and apoptosis. Semin. Hematol. 2001, 38, 179–193. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bands | Assignments | Wavenumber (cm−1) | |
|---|---|---|---|
| Experimental | Calculated | ||
| C-H | stretching | 3045, 3302 | 3196 |
| C=O | stretching | 1655 | 1676 |
| C=C | stretching | 1580 | 1572 |
| C-C | stretching | 1521 | 1420 |
| H–C-H | asymmetric bending | 1344 | 1340 |
| H–C-H | symmetric bending | 1302 | 1252 |
| C–C-H | in-p-bend | 1218 | 1220 |
| C–C-H | o-o-p bend | 1082, 1028, 1001, 964, 851 | 1052, 892, 820, 724 |
| Sl. No. | Solvents | λa (nm) | λf (nm) | (cm−1) | (cm−1) | (cm−1) | (cm−1) | (cm−1) |
|---|---|---|---|---|---|---|---|---|
| 1 | Pentane | 320.00 | 375.00 | 31,250.00 | 26,666.67 | 4583.33 | 57,916.67 | 28,958.33 |
| 2 | Hexane | 320.00 | 376.60 | 31,250.00 | 26,553.37 | 4696.63 | 57,803.37 | 28,901.69 |
| 3 | Heptane | 325.00 | 382.00 | 30,769.23 | 26,178.01 | 4591.22 | 56,947.24 | 28,473.62 |
| 4 | Cyclohexane | 331.90 | 390.00 | 30,129.56 | 25,641.03 | 4488.53 | 55,770.58 | 27,885.29 |
| 5 | 1,4-Dioxane | 332.00 | 392.00 | 30,120.48 | 25,510.20 | 4610.28 | 55,630.69 | 27,815.34 |
| 6 | Toluene | 335.20 | 396.00 | 29,832.94 | 25,252.53 | 4580.41 | 55,085.46 | 27,542.73 |
| 7 | TCE | 338.00 | 402.00 | 29,585.80 | 24,875.62 | 4710.18 | 54,461.42 | 27,230.71 |
| 8 | DEE | 341.70 | 410.00 | 29,265.44 | 24,390.24 | 4875.19 | 53,655.68 | 26,827.84 |
| 9 | DCE | 343.00 | 415.00 | 29,154.52 | 24,096.39 | 5058.13 | 53,250.90 | 26,625.45 |
| 10 | EA | 344.90 | 418.00 | 28,993.91 | 23,923.44 | 5070.47 | 52,917.36 | 26,458.68 |
| 11 | THF | 348.00 | 423.00 | 28,735.63 | 23,640.66 | 5094.97 | 52,376.29 | 26,188.15 |
| 12 | Acetonitrile | 351.30 | 428.00 | 28,465.70 | 23,364.49 | 5101.21 | 51,830.18 | 25,915.09 |
| 13 | DMF | 352.00 | 430.00 | 28,409.09 | 23,255.81 | 5153.28 | 51,664.90 | 25,832.45 |
| 14 | DMSO | 355.00 | 435.00 | 28,169.01 | 22,988.51 | 5180.51 | 51,157.52 | 25,578.76 |
| 15 | Water | 360.00 | 441.00 | 27,777.78 | 22,675.74 | 5102.04 | 50,453.51 | 25,226.76 |
| 16 | Decanol | 362.00 | 445.00 | 27,624.31 | 22,471.91 | 5152.40 | 50,096.22 | 25,048.11 |
| 17 | Octanol | 363.00 | 446.00 | 27,548.21 | 22,421.52 | 5126.68 | 49,969.73 | 24,984.87 |
| 18 | Pentanol | 364.00 | 448.00 | 27,472.53 | 22,321.43 | 5151.10 | 49,793.96 | 24,896.98 |
| 19 | Butanol | 367.00 | 452.00 | 27,247.96 | 22,123.89 | 5124.06 | 49,371.85 | 24,685.93 |
| 20 | Isopropanol | 368.00 | 455.00 | 27,173.91 | 21,978.02 | 5195.89 | 49,151.94 | 24,575.97 |
| 21 | Ethanol | 369.00 | 458.00 | 27,100.27 | 21,834.06 | 5266.21 | 48,934.33 | 24,467.17 |
| 22 | Methanol | 370.00 | 462.00 | 27,027.03 | 21,645.02 | 5382.01 | 48,672.05 | 24,336.02 |
| Radius ‘r’ (Ao) | D | D | D | D | D | D | D | D | D | D | D | D | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3.89 | 3.51 | 5.72 | 10.74 | 12.88 | 1.19 | 2.47 | 14.41 | 12.91 | 18.07 | 13.08 | 2.14 | 1.29 | 1.20 | 00 |
| Sl. No. | Solvents | N | OD | Fint | Φ | τ0 (ns) | Kr 109 (S−1) | Knr 109 (S−1) |
|---|---|---|---|---|---|---|---|---|
| 1 | Hexane | 1.375 | 0.240 | 156,847.12 | 0.578 | 1.320 | 0.438 | 0.320 |
| 2 | Heptane | 1.388 | 0.342 | 46,816.213 | 0.123 | 1.630 | 0.076 | 0.538 |
| 3 | Cyclohexane | 1.426 | 0.118 | 113,862.115 | 0.917 | 1.284 | 0.714 | 0.064 |
| 4 | Pentanol | 1.409 | 0.122 | 23,826.105 | 0.181 | 1.575 | 0.115 | 0.520 |
| 5 | Butanol | 1.399 | 0.122 | 49,067.2595 | 0.368 | 1.458 | 0.252 | 0.433 |
| 6 | Iso-Propanol | 1.378 | 0.119 | 84,261.500 | 0.629 | 1.729 | 0.364 | 0.215 |
| 7 | Ethanol | 1.361 | 0.239 | 109,265.055 | 0.396 | 1.591 | 0.249 | 0.380 |
| 8 | Methanol | 1.328 | 0.118 | 109,265.055 | 0.763 | 2.045 | 0.373 | 0.116 |
| 9 | Acetonitrile | 1.344 | 0.140 | 47,916.138 | 0.289 | 1.840 | 0.157 | 0.386 |
| 10 | DMSO | 1.479 | 0.558 | 304,283.865 | 0.558 | 1.964 | 0.284 | 0.225 |
| Molecule | α0 (cm−1) | β (cm W−1) × 10−5 | n2 (cm2 W−1) × 10−9 | Re χ(3) (e.s.u) × 10−6 | Im χ(3) (e.s.u) × 10−6 | χ(3) (e.s.u) × 10−6 | γh (e.s.u) × 10−26 | OL kJ/cm2 | W × 103 | T |
|---|---|---|---|---|---|---|---|---|---|---|
| 3NPEO | 4.52 | 1.82 | −0.62 | 0.84 | 0.44 | 0.94 | 0.39 | 4.06 | 49.4 | 0.15 |
| Compound | HOMO (eV) | LUMO (eV) | E0−0 | λonset | |||
|---|---|---|---|---|---|---|---|
| Theor. | Expl. | Theor. | Expl. | Theor. | Expl. | Ao | |
| 3NPEO | −6.225 | −4.849 | −2.488 | −1.498 | 3.737 | 3.351 | 370 |
| Energy Term | Energy in kJ/mol |
|---|---|
| van der Waal energy | −166.204 +/− 12.377 |
| Electrostatic energy | −25.227 +/− 7.903 |
| Polar solvation energy | 83.791 +/− 5.629 |
| SASA energy | −18.248 +/− 0.662 |
| Binding energy | −125.888 +/− 11.935 |
| Treatments and Cell Line | Early Apoptosis | Late Apoptosis | Total Apoptosis |
|---|---|---|---|
| 3NPEO-(MDAMB-231) | 20.80 | 3.10 | 23.90 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hebasur, R.K.; Koppal, V.V.; Yaraguppi, D.A.; Gummagol, N.B.; Kusanur, R.; Patil, N.R. A Comprehensive Evaluation of a Chalcone Derivative: Structural, Spectroscopic, Computational, Electrochemical, and Pharmacological Perspectives. Photochem 2025, 5, 20. https://doi.org/10.3390/photochem5030020
Hebasur RK, Koppal VV, Yaraguppi DA, Gummagol NB, Kusanur R, Patil NR. A Comprehensive Evaluation of a Chalcone Derivative: Structural, Spectroscopic, Computational, Electrochemical, and Pharmacological Perspectives. Photochem. 2025; 5(3):20. https://doi.org/10.3390/photochem5030020
Chicago/Turabian StyleHebasur, Rekha K., Varsha V. Koppal, Deepak A. Yaraguppi, Neelamma B. Gummagol, Raviraj Kusanur, and Ninganagouda R. Patil. 2025. "A Comprehensive Evaluation of a Chalcone Derivative: Structural, Spectroscopic, Computational, Electrochemical, and Pharmacological Perspectives" Photochem 5, no. 3: 20. https://doi.org/10.3390/photochem5030020
APA StyleHebasur, R. K., Koppal, V. V., Yaraguppi, D. A., Gummagol, N. B., Kusanur, R., & Patil, N. R. (2025). A Comprehensive Evaluation of a Chalcone Derivative: Structural, Spectroscopic, Computational, Electrochemical, and Pharmacological Perspectives. Photochem, 5(3), 20. https://doi.org/10.3390/photochem5030020