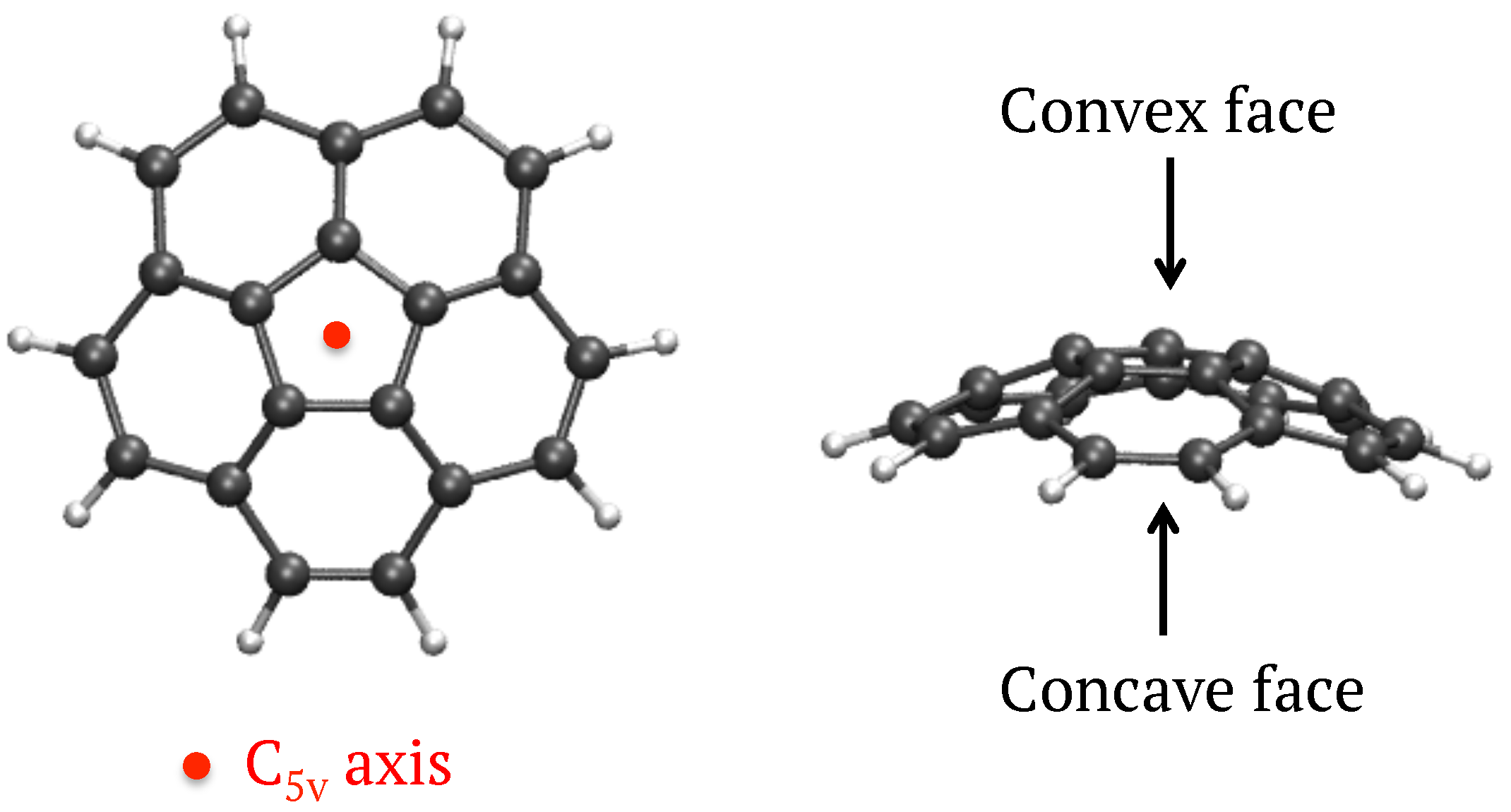
3.1.1. Structures and Energetics
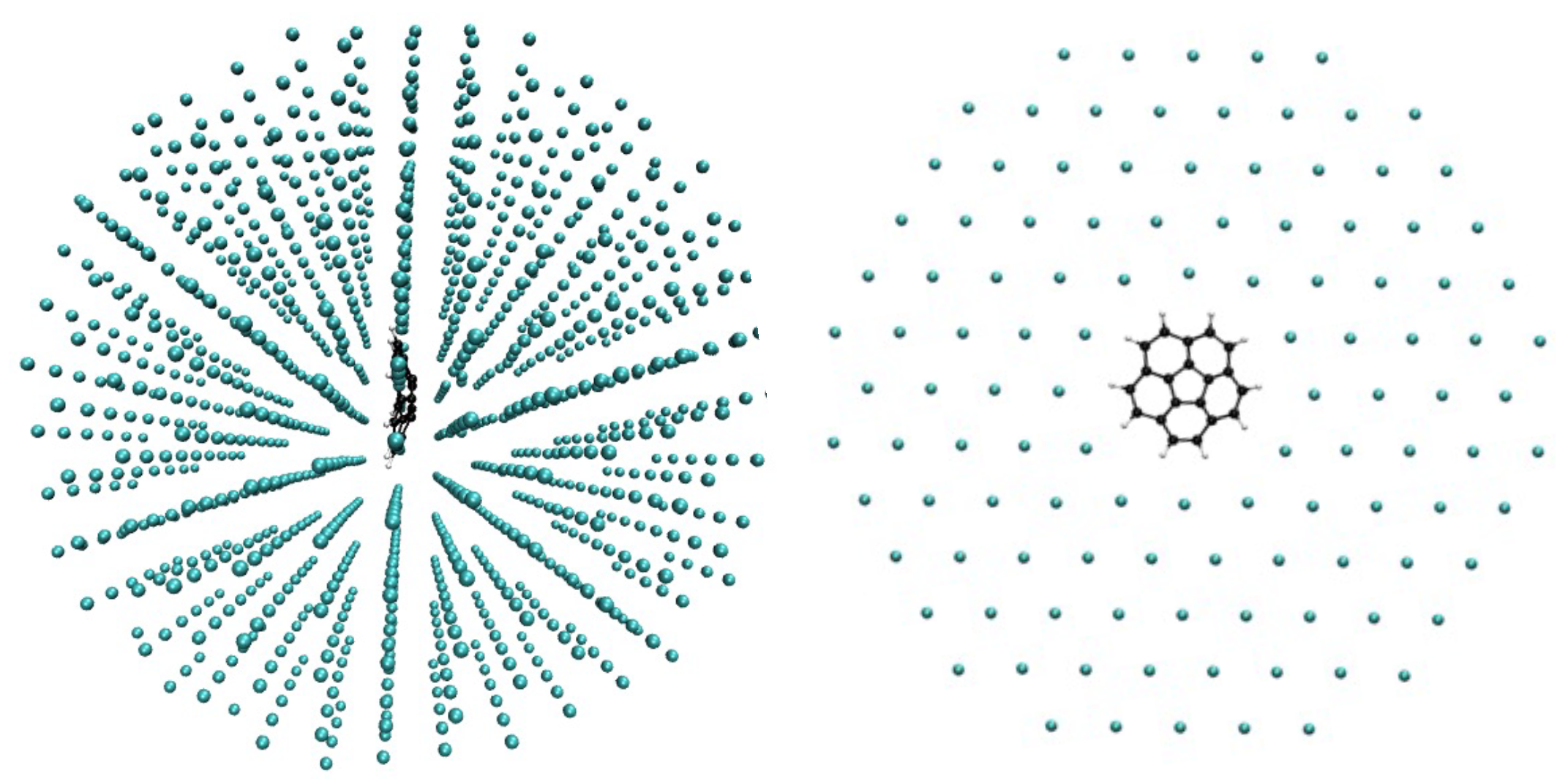
Corannulene was inserted in different symmetry planes of the Ar matrix represented by a finite size fcc cluster initially of 1139 Ar atoms. After removing the minimum number of Ar atoms we found that the most stable situation corresponded to an insertion of the corannulene molecule in the [111] plane with a vacancy of 7 Ar atoms (see
Figure 2,
E = −2470 cm
, also see
Appendix A,
Table A1 for the results of the other symmetry planes). We obtained the same result as for coronene [
18] and other PAHs such as naphthalene [
57]. The substitution energy is smaller than that computed for coronene using the same model (
E = −4810 cm
[
18]) which can be understood as the non planearity of corannulene leads to more perturbation of the Ar layers close to corannulene.
In
Table 1 are reported the substitution energies obtained using Equation (
1) for the most stable isomers of (C
20H
10)(H
2O)
:Ar (
n = 1–3) of different types i.e., depending whether the water molecules interact with the concave or convex faces of corannulene or with its H atoms. The geometries of the most stable isomers are reported in
Figure 3,
Figure 4 and
Figure 5 for
n = 1, 2 and 3 respectively.
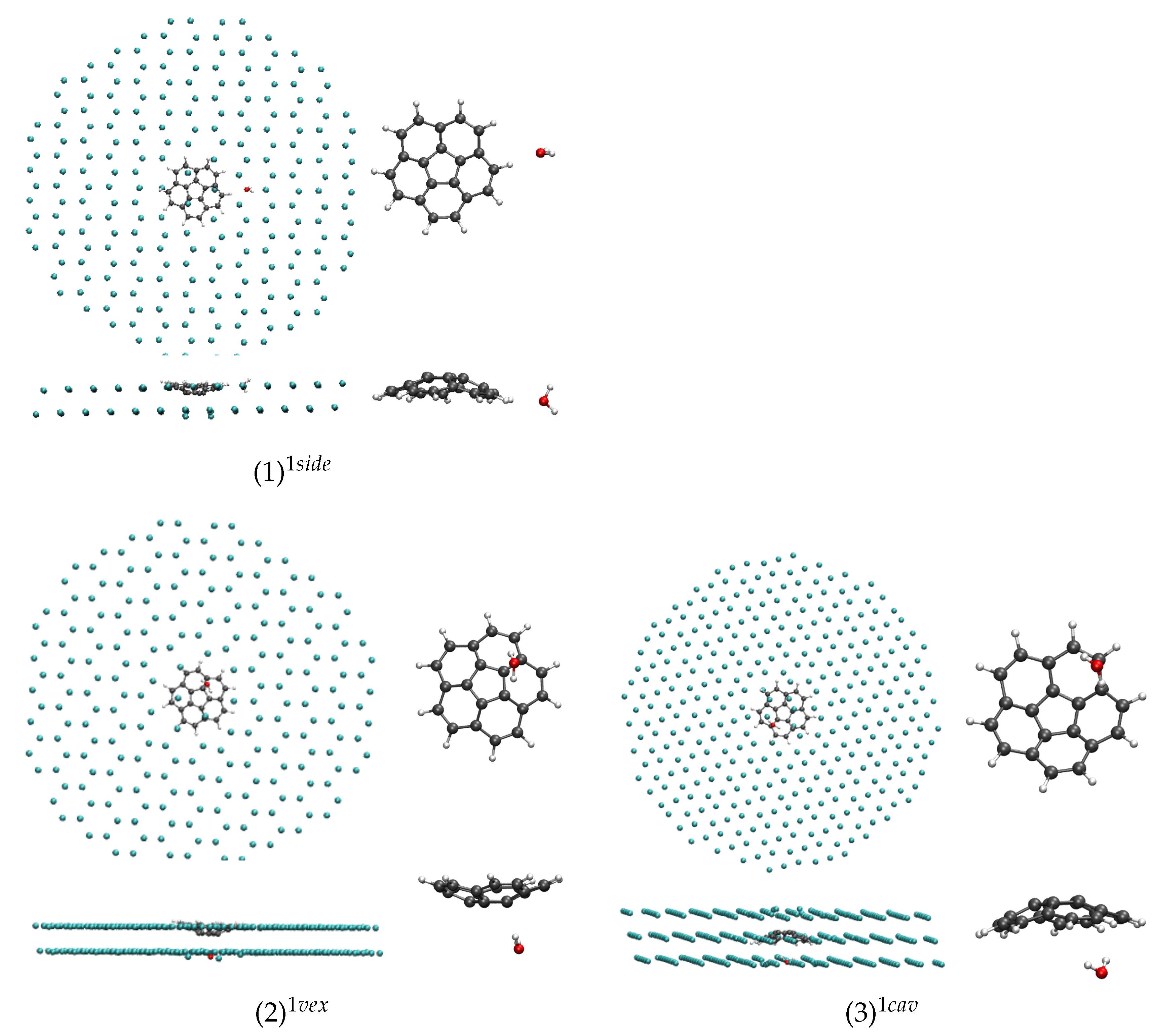
In the case of
n = 1, the three conformers (1)
, (2)
and (3)
lie in an energy range of 150 cm
(1.8 kJ mol
), the most stable one corresponding to the water molecule interacting with an H atom of corannulene in the [111] symmetry plane ((1)
). Interestingly, the conformer in which the water molecule interacts with the concave face, known to be the most stable isomer in the gas phase [
43], is the highest energy one ((3)
), although the energy difference is small. These subtle differences from the gas phase originate from the constraints of the fcc lattice. For instance, in the (3)
isomer, the water molecule substitutes an Ar atom whose position is not aligned with the C
axis of corannulene, and this prevents the water molecule from having the most stabilizing interaction with the corannulene as in the gas phase. When relaxing the geometry of this isomer after removing the matrix environment, the water molecule migrates towards the center of the “cup” and becomes the lowest energy isomer (see last column of
Table 1).
The perturbation of the [111] Ar layers adjacent to that containing corannulene due to the presence of the impurity can be visualized on the side views of the systems in
Figure 3,
Figure 4 and
Figure 5, a few Ar atoms being moved out of their original plane. Now comparing with the results obtained for coronene (C
H
, planar PAH) inside an Ar matrix using the same computational approach, the isomer similar to the (1)
isomer of the present work, designated as
isomer in Simon et al. [
18], was found more stable than the
isomer by ∼800 cm
in the case of coronene. This means that the stabilization of the “side” (or “
”) isomer is enhanced in the case of a planar PAH with respect to the case of corannulene where it appears in competition with the two “
” isomers (2)
and (3)
.
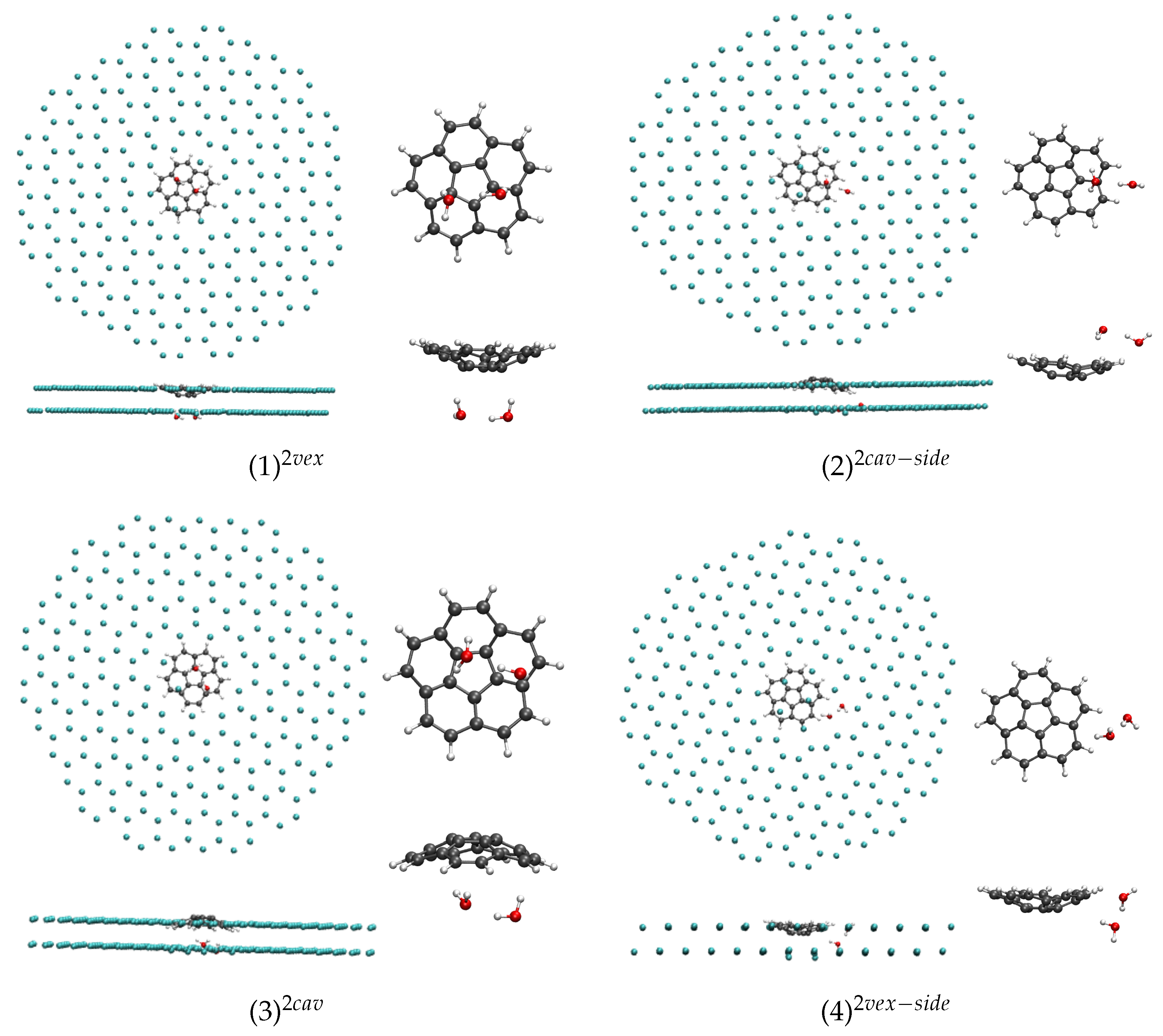
In the case of
n = 2, the most stable isomer corresponds to the water dimer interacting through two H atoms with the convex face of corannulene ((1)
, see
Figure 4), the two H atoms pointing towards two central C atoms of corannulene. The corresponding isomer in which the water dimer interacts with the concave face of corannulene ((3)
) is found 210 cm
(or 2.5 kJ·mol
) higher in energy. The isomer in which the water dimer interacts with both the concave (resp. convex) face and side H of corannulene is found 190 cm
or 2.3 kJ·mol
(resp. 240 cm
or 2.9 kJ·mol
) higher in energy. As in the case of
n = 1, when the geometries of (C
20H
10)(H
2O)
are further relaxed after removal of the Ar matrix, the water dimer tends to move towards the center of the cup when it interacts with the concave face of corannulene and it leads to the stabilisation of such conformers (see last column of
Table 1).
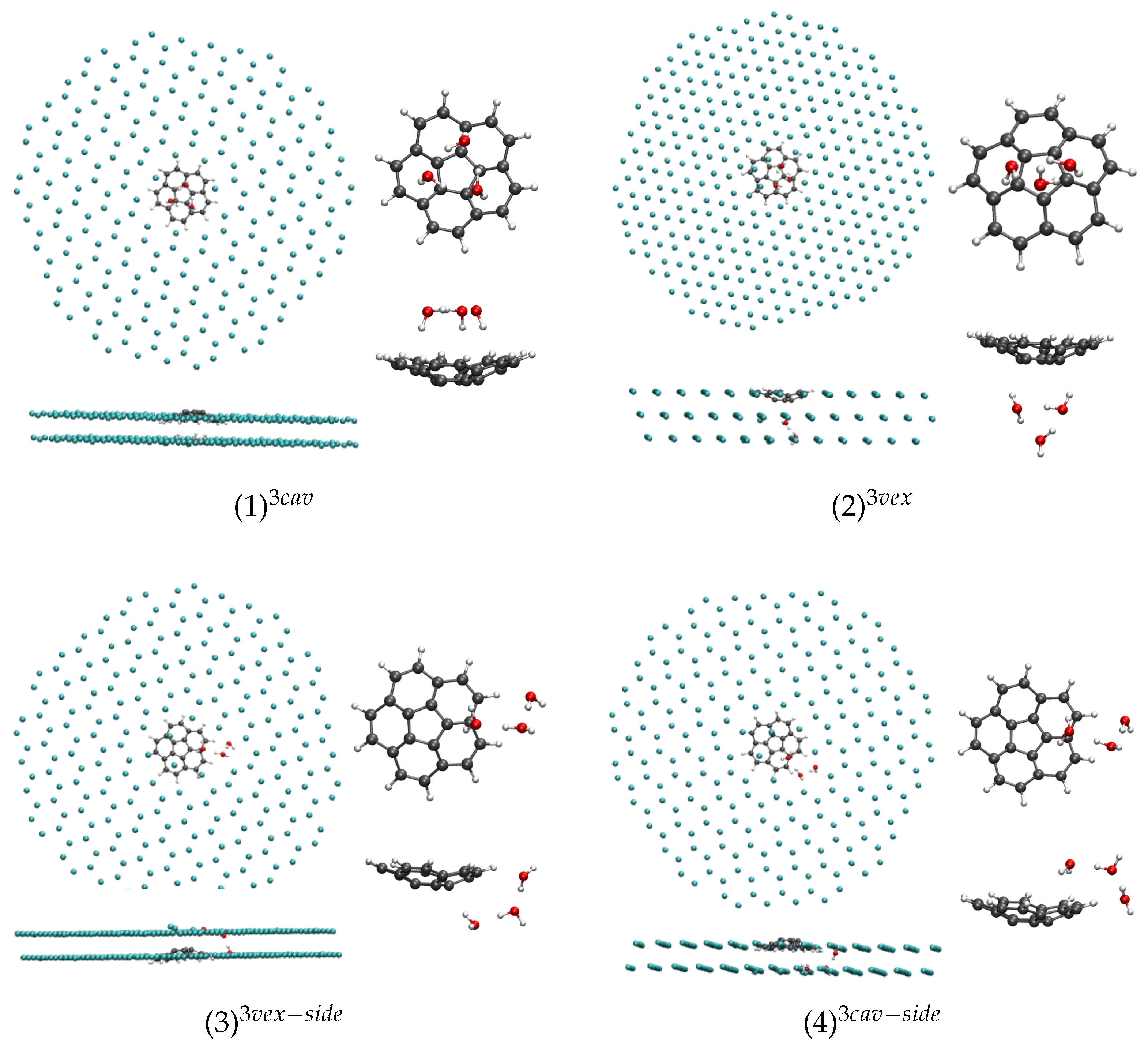
In the case of
n = 3, the isomer in which the water cyclic trimer, located in the [111] plane adjacent to that of corannulene, with its center approximately on the C
axis of corannulene, interacts with the concave face of corannulene through three H atoms ((1)
, see
Figure 5), is shown to be particularly stable with our model. Contrary to the smaller clusters (
n = 1, 2), it is also the case after relaxation of the cluster after removing the Ar matrix as this conformer remains the most stable one in the gas phase. Next isomer, (2)
, in which the cyclic trimer interacts with the convex face of corannulene through two H atoms, is found 340 cm
higher in energy. The next two isomers in which the water trimer adopts a “linear form” interacting with both one face (convex or concave) and an H atom of corannulene, namely the (3)
and (4)
are found respectively 590 and 780 cm
(7.1 and 9.3 kJ·mol
) higher in energy than the stable (1)
isomer.
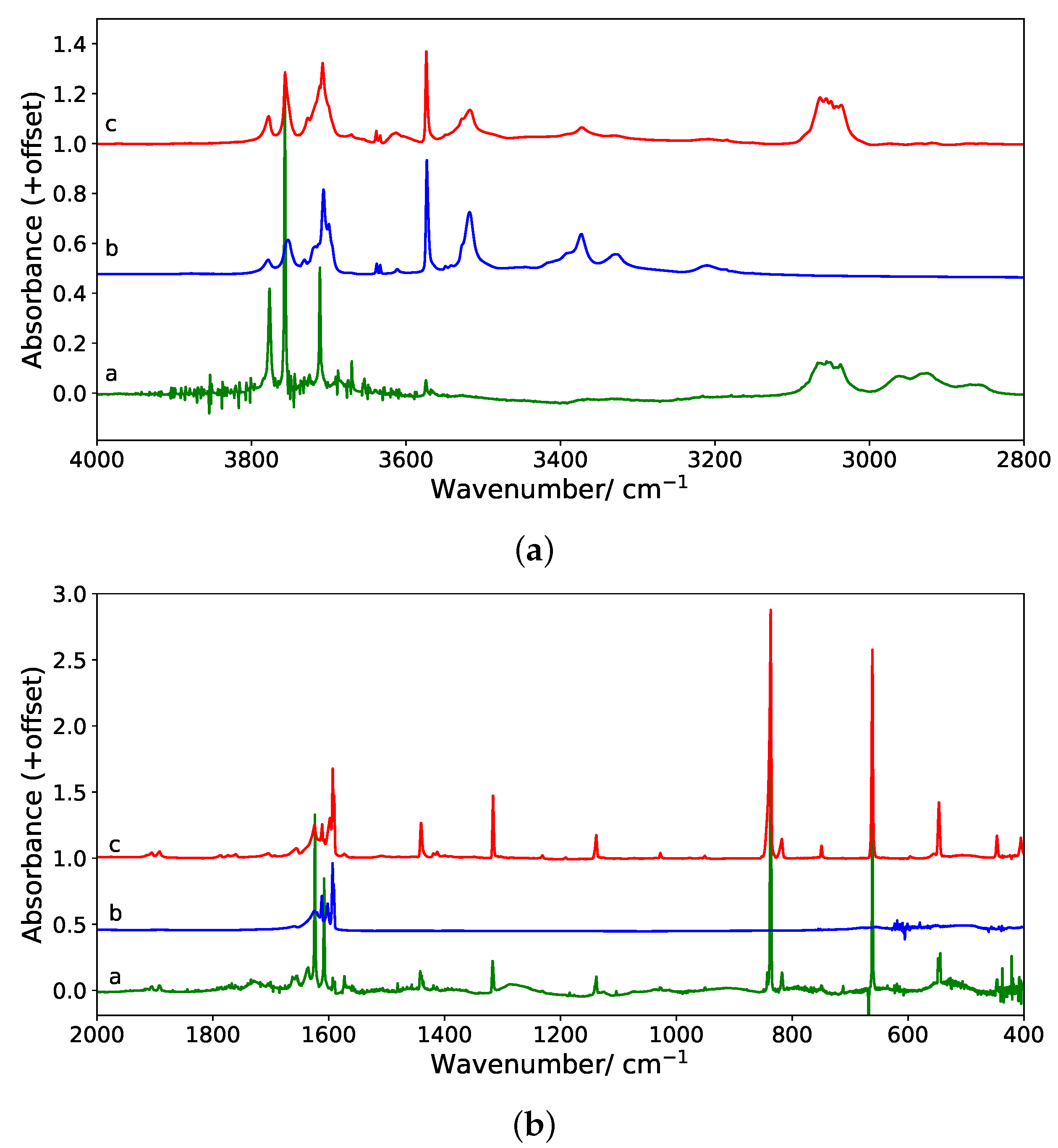
3.1.2. IR Spectra
This section reports the computed IR spectra of all the (C
20H
10)(H
2O)
n:Ar isomers for
n = 1–3 and the comparison with those of C
20H
10 and of the corresponding (H
2O)
n clusters determined at the same level of theory. These simulations aim at getting insights into the influence of the coordination site (concave, convex or side) and of the geometry of the water cluster (linear or cycle)—in the case of the trimer—on the IR spectrum of the water molecule/cluster and corannulene upon coordination. The values of the positions and the shifts of the bands of interest upon coordination are reported in
Table A2,
Table A3 and
Table A4 for the harmonic spectra (
n = 1 to 3 respectively), whereas
Table 2,
Table 3 and
Table 4 report the data for finite-temperature (10 K) spectra. For the latter, the maxima were determined using the full width at half maximum (FWHM). Due to the fluctuations of the simulated IR cross sections and the significant broadening and merge of bands in some cases, we can estimate a human error of ±5 cm
on the reported wavenumbers. The 10 K dynamic spectra of all the isomers reported in
Figure 3,
Figure 4 and
Figure 5 are displayed in
Figure 6,
Figure 7 and
Figure 8.
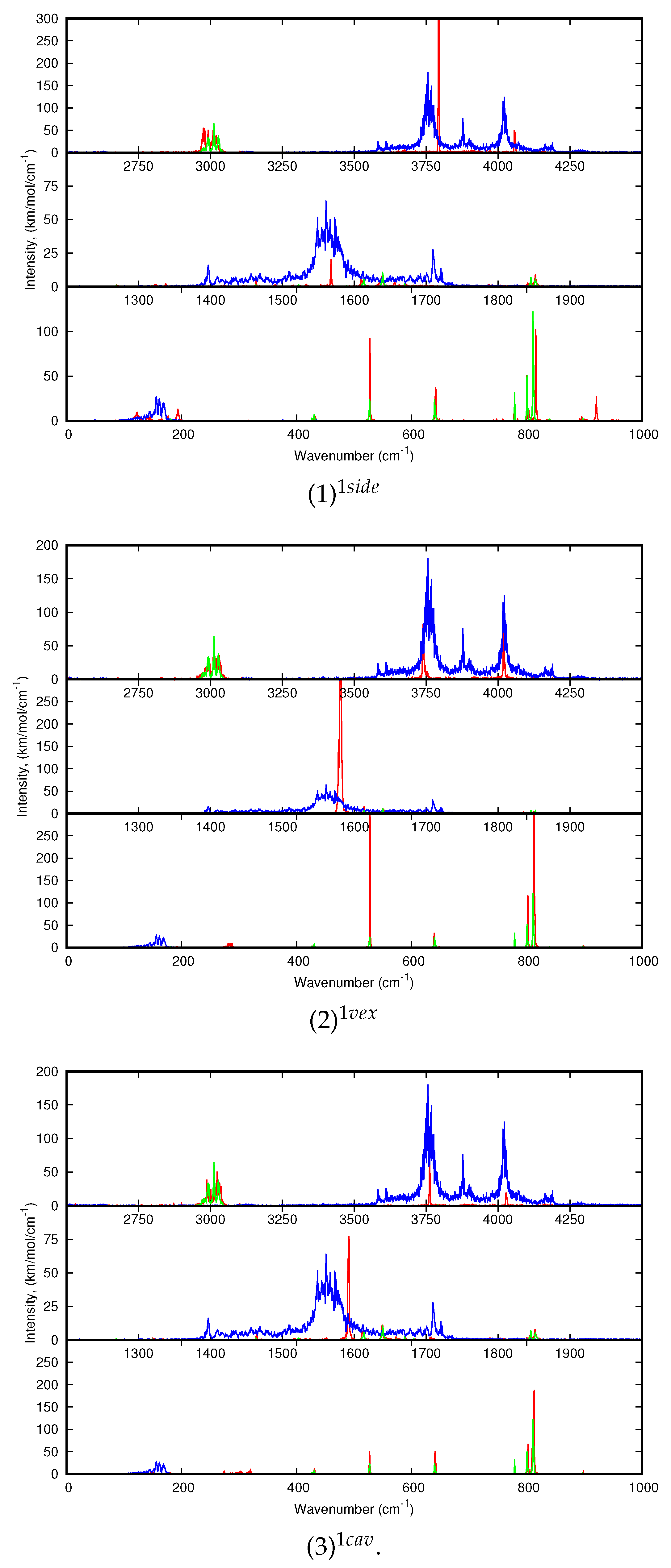
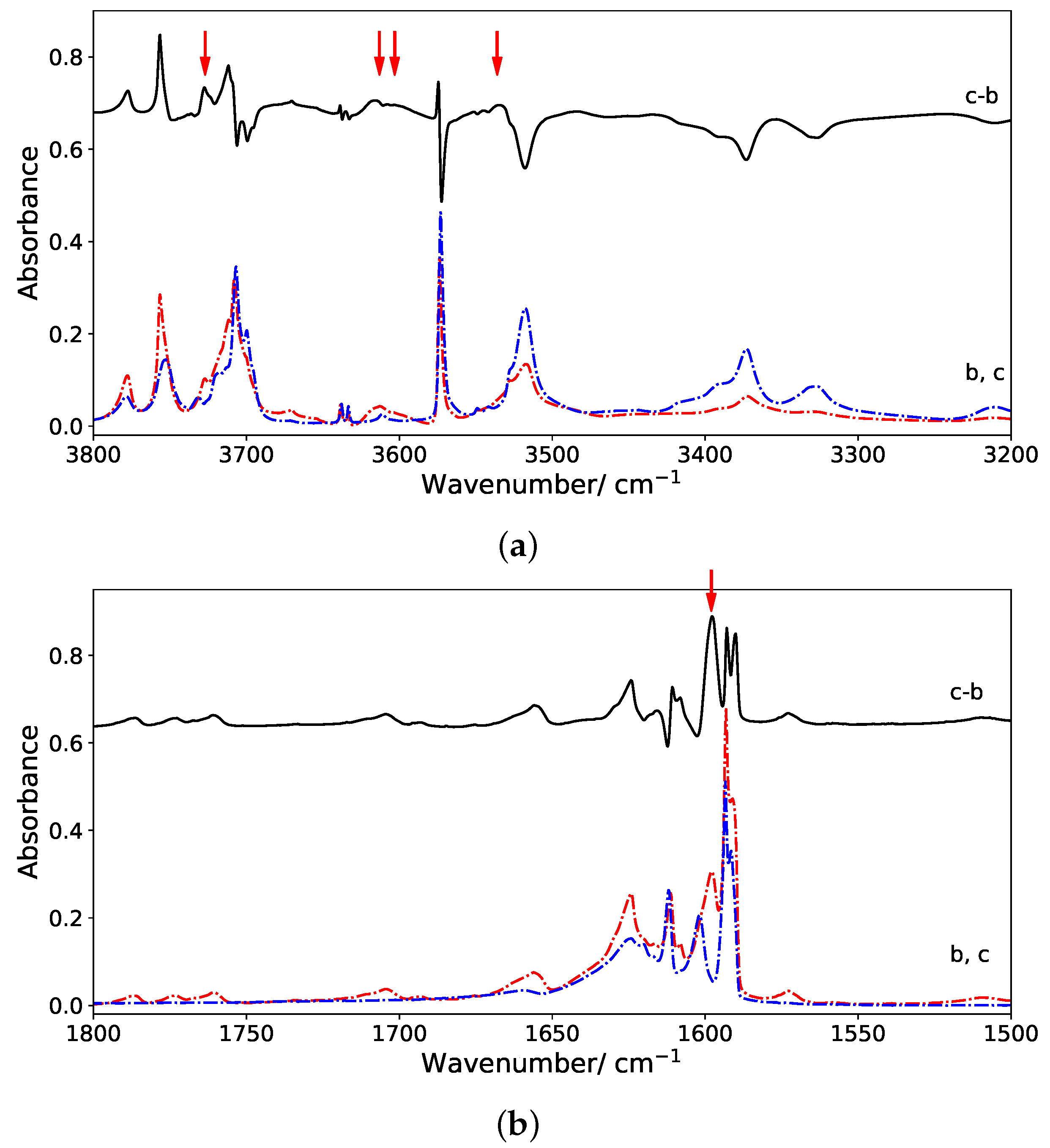
First of all, we found that, for all clusters, the bands of corannulene were hardly affected upon coordination, and it is in line with the experimental results. This is clearly the case of the
(C-H stretch) bands, and for the
(CH) +
(CC) bands (combination of in plane C-H bending and C-C stretches) that have very weak intensities in the computed spectra. Regarding the
(C-H out-of-plane) most modes, their energy can be slightly blue-shifted for the “side” isomers all the more as the cluster size increases. The most intense
band is shifted by +5 cm
for (1)
, +9 cm
for (2)
, +8 and +14 cm
for (3)
and (4)
respectively using the harmonic data. This small blue shift was also observed for the
isomer the coronene-water complex [
18]. The computed dynamic shifts were found similar to the harmonic ones. Therefore a side isomer may be distinguished from a convex or concave isomer through the position of the
band as in the case of coronene [
18]. It should be noticed that these shift values are small with respect to the accuracy of the computed absolute positions. However, the description of intermolecular interactions has been carefully benchmarked against wavefunction results, insuring their accuracy [
48].
In the following, we focus on the influence of the interaction with corannulene on the IR spectra of (H2O)n:Ar (n = 1–3).
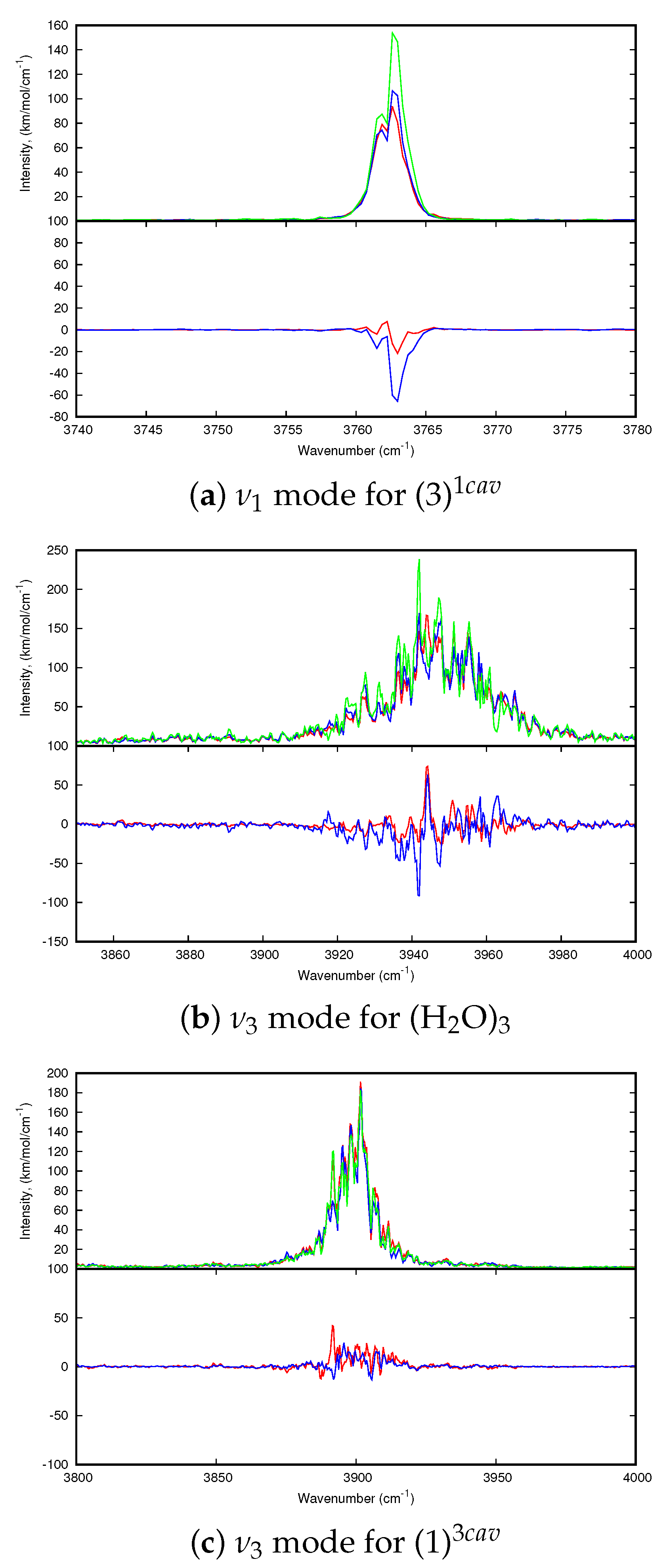
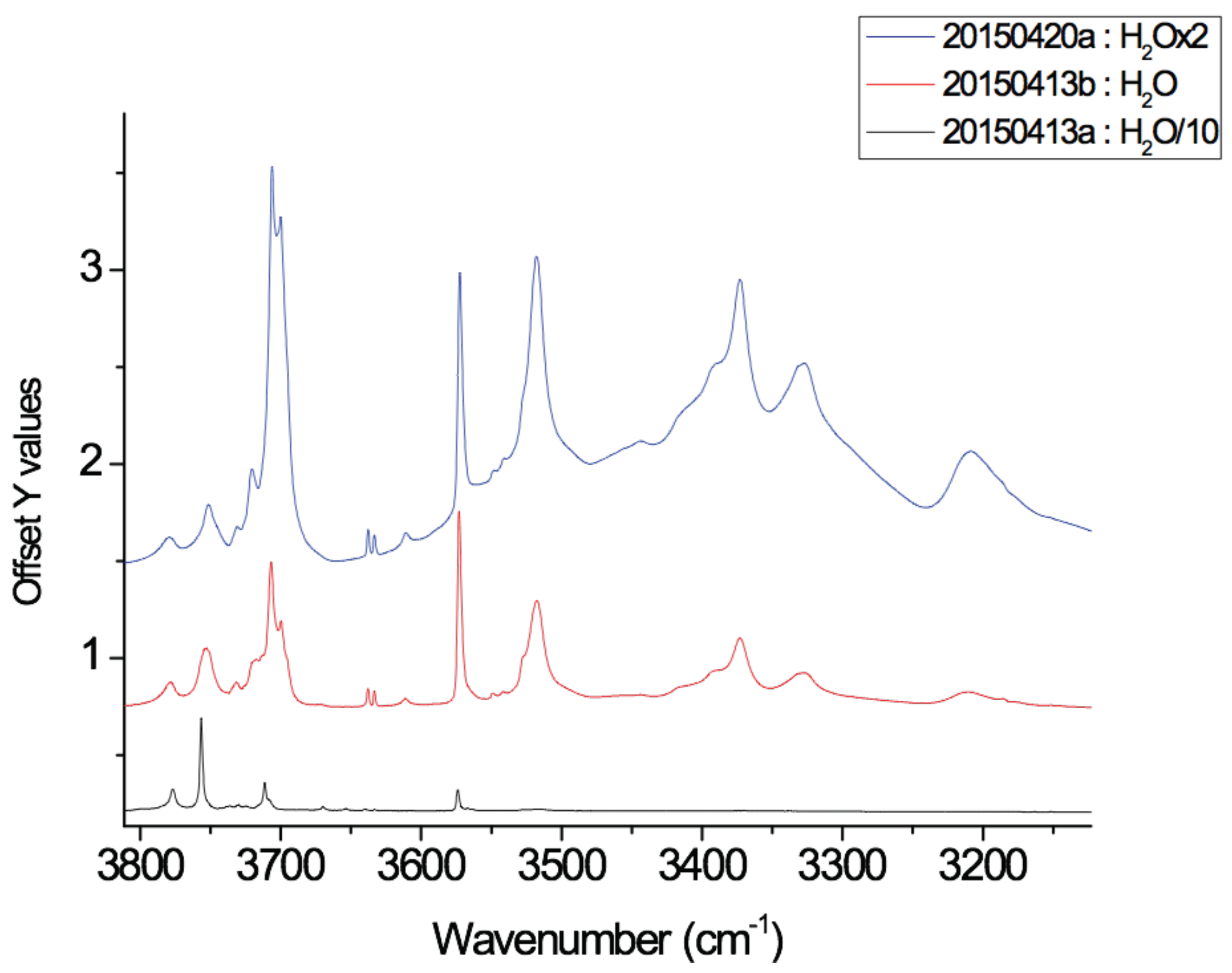
Regarding the dynamic spectra of the isomers with one water molecule (
Figure 6), the first striking feature is the narrowing of the water bands upon coordination. Indeed, in our simulations of H
2O:Ar at 10 K, the water molecule vibrates and rotates in the matrix, leading to the broadening of the resonant symmetric and assymmetric stretching modes (
and
) as well as the bending mode (
). Besides, the stretching modes are significantly redshifted with respect to the harmonic ones, as we reported in our previous work focusing on the IR spectra of the water molecules and clusters inside a rare gas matrix [
56]. As can be seen in
Table A2 and
Table 2, the shifts are quite variable depending on the isomer. For the (1)
isomer, the
and
modes are blueshifted upon coordination, the blue shift being more pronounced in the anharmonic spectrum (+38 and +35 vs. +4 and +2 cm
for the anharmonic and harmonic
and
modes respectively). On the opposite, in the case of the (2)
isomer, the
and
bands are redshifted, the shifts being less pronounced in the dynamic spectra (−36 and −43 cm
vs. −17 and −1 cm
). The case of (3)
is intermediate as the
and
modes are redshifted at the harmonic level and become slightly blueshifted in the 10 K dynamic spectra (
Figure 6). Regarding the
mode, it it blueshifted (except in the (1)
harmonic spectrum) increasing from the side to convex and concave isomers. However, the significant narrowing of the water bands upon coordination may lead to the impossibility to detect the (C
20H
10)(H
2O):Ar clusters in the matrix as they can be hindered by the bands of the water molecule.
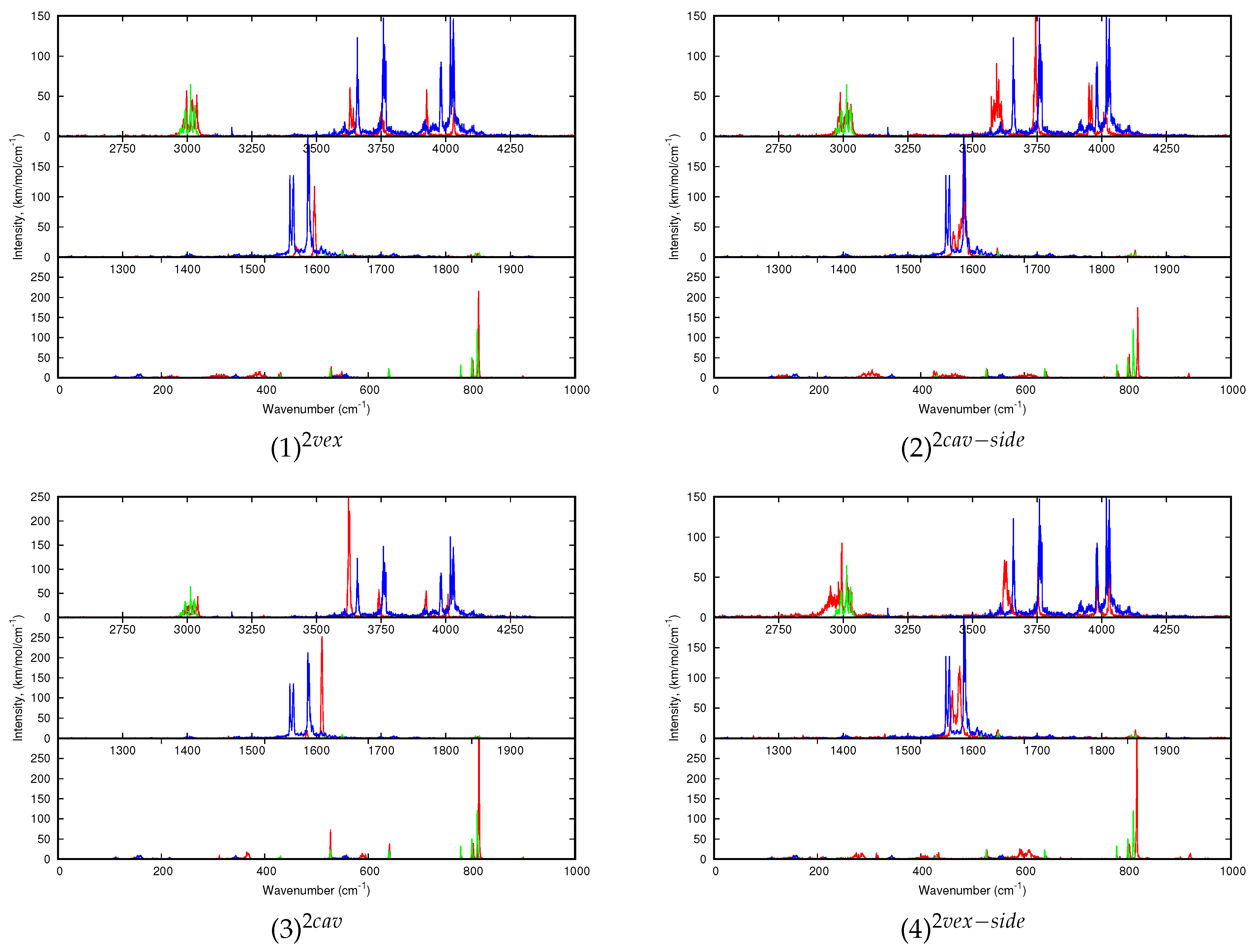
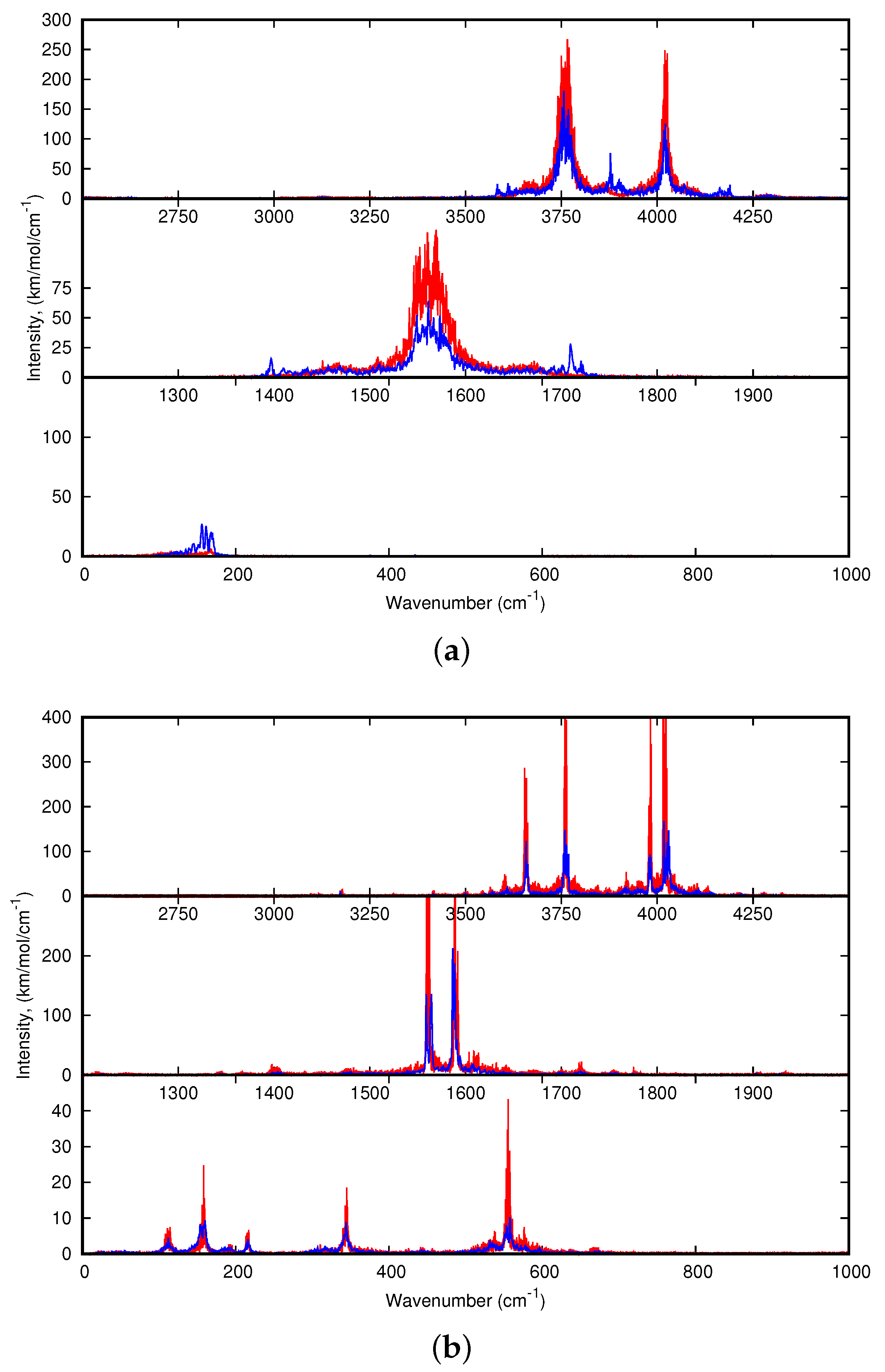
For larger clusters, only the dynamic spectra will be discussed. Regarding (C
20H
10)
0,1 (H
2O)
2:Ar, let us just specify that, regarding the 10 K spectrum of the water dimer, the
and
modes are redshifted with respect to the harmonic spectra. The shifts obtained for the O-H modes for the H atoms interacting with the Ar atoms of the matrix (the
and
most energetic modes, usually designated as
and
, “a” standing for “acceptor” ) are the most shifted (both by 48 cm
) whereas the lowest energetic ones (
and
, “d” standing for “donor”) are only shifted by 7 and 9 cm
respectively. It can be understood as the former interact with the Ar atoms of the matrix and therefore are more sensitive to the loose interaction with Ar expected to lead to anharmonic effects at low temperature. Extremely weak intensity bands also accompany the intense ones, to a lesser extent than for the water monomer because the motion is more hindered by the matrix environment. The water dimer vibrates around its equilibrium position with the possible occurence of the rotation of the H-acceptor water monomer around the OO axis. Interestingly, the water dimer motion in our simulations appears similar to the experimental evidence of the “acceptor switching and rotation of the water dimer around its O-O axis” by Ceponkus et al. [
58].
Regarding the spectra of all C
20H
10)(H
2O)
2:Ar isomers reported
Figure 7, the differences in the shifts of the bands with respect to those of the bare water dimer might help identifying the type of isomer (cav, vex, cav-side or vex-side) formed in the experiments. Upon interaction with corannulene, the (H
2O)
2 modes are globally redshifted and the shifts values are smaller in the 10 K spectra than in the harmonic ones. The shifts values are dependent on the isomer being smaller for the (1)
and (4)
isomers (see
Table 3). The
bands are redshifted for (2)
and (3)
, but is it not the case for the two other isomers for which the higher energy
band is blueshifted. Finally, a difference between the shifts of the
bands between the different isomers is noticeable. For instance, those for (1)
and (3)
are both blueshifted, the blue shift being larger for the latter.
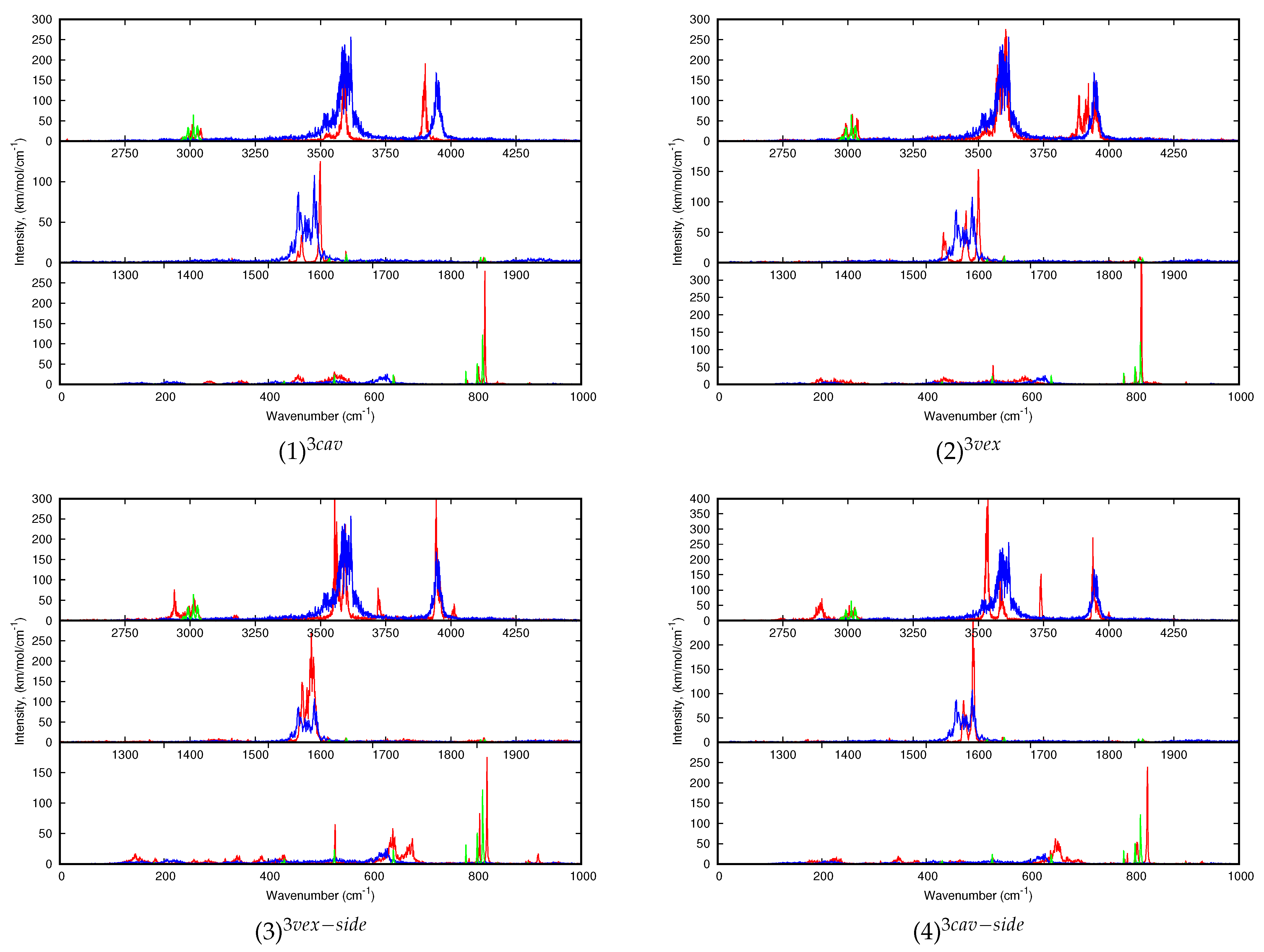
Finally, the dynamic spectra of (C
20H
10)
0,1(H
2O)
0,3:Ar are reported in
Figure 8. Regarding the water modes of (H
2O)
3, each type of mode leads to three broad bands more or less structured centered at 3596, 1569 and 3948 cm
for
to
. During the simulation, the water trimer vibrates around its equilibrium position with occasional flipping of the O-H bonds- not involved in intermolecular O–H bonds- from one side of the plane of the OOO cycle to the other side. As in the case of one and two water molecules, the water IR bands become narrower due to a hindrance of their motion upon interaction with corannulene, although it is not as relevant in the case of (2)
where the water trimer globally maintains its structure. It is all the more verified for the “cav” isomers (1)
and (4)
, reflecting stronger corannulene-water interactions than for the “vex” isomers. A significant difference is noticeable between the “side” isomers and the others. In the former, a band can be found at ∼3720 and 3740 cm
, that was assigned to the O-H stretching mode for the bond interacting with the PAH [
51]. In contrast with the other stoichiometries, the stretching modes do not appear systematically reshifted. The
mode is hardly redshifted in the case of the most stable (1)
isomers whereas is it slightly blueshifted in the (2)
spectrum. The
mode is redshifted for (1)
but leads to mostly blueshifted bands for the three other isomers. The
modes are also globally blueshifted for all isomers, the amplitude of the shifts being dependent on the isomer. As in the case of the other stoichiometries, the differences in the spectra may allow us to distinguish specific isomers formed in the experiment.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










