
Deciphering the Coarse-Grained Model of Ionic Liquid by Tunning the Interaction Level and Bead Types of Martini 3 Force Field


Abstract
1. Introduction
2. Methods
2.1. All-Atom (AA) Simulation Details
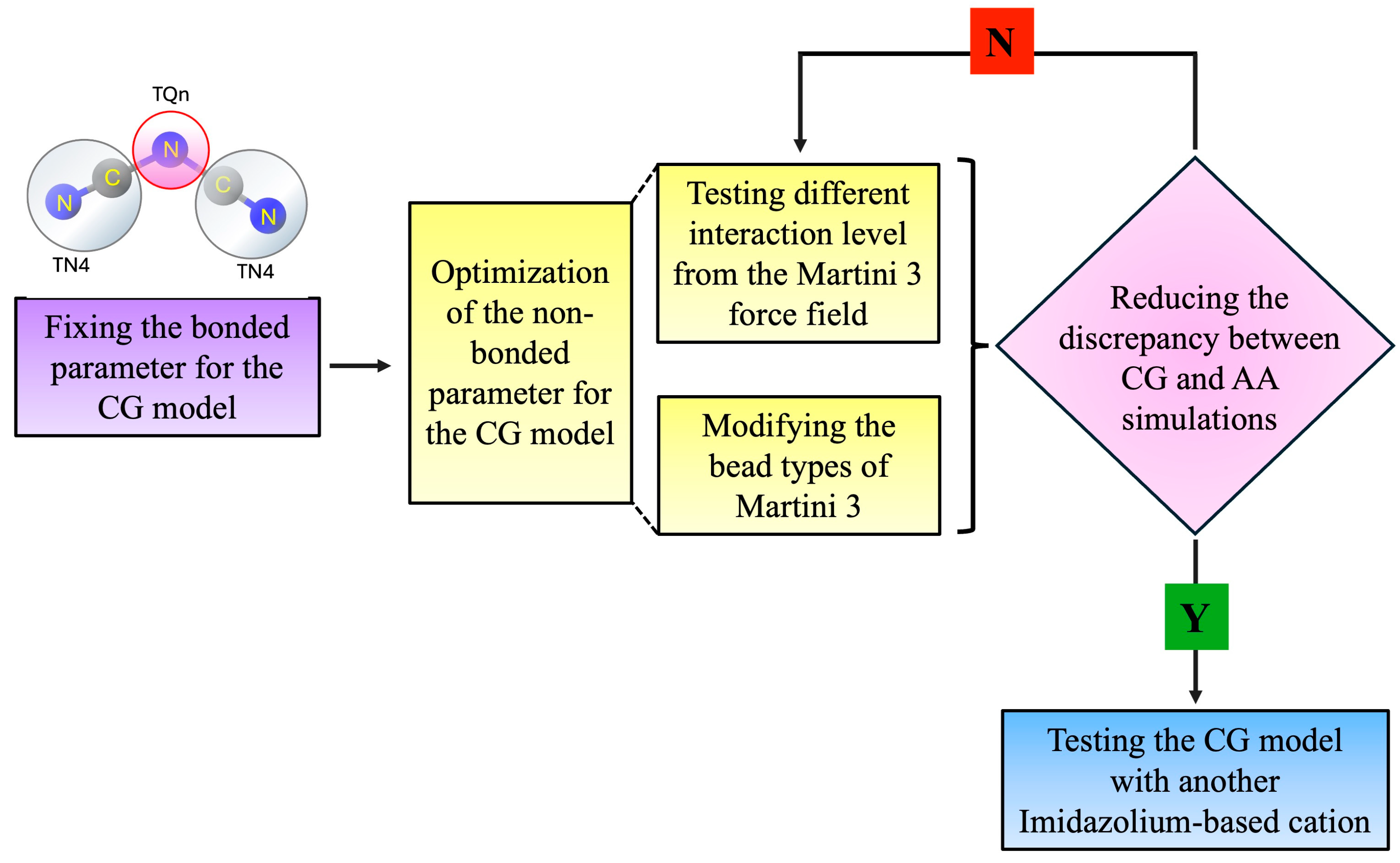
2.2. Coarse-Grained (CG) Model Parameterization
2.2.1. Parameterization of Bonded Interactions
2.2.2. Parameterization of Non-Bonded Interactions
2.3. Coarse-Grained (CG) Simulation Setup
3. Results and Discussion
3.1. Bonded-Interaction Parameterization
3.2. Non-Bonded Parameter for the [DCA]− Anion BEADS
3.2.1. Testing All Types of Charged Beads (TQn) from the MARTINI 3 Force Field
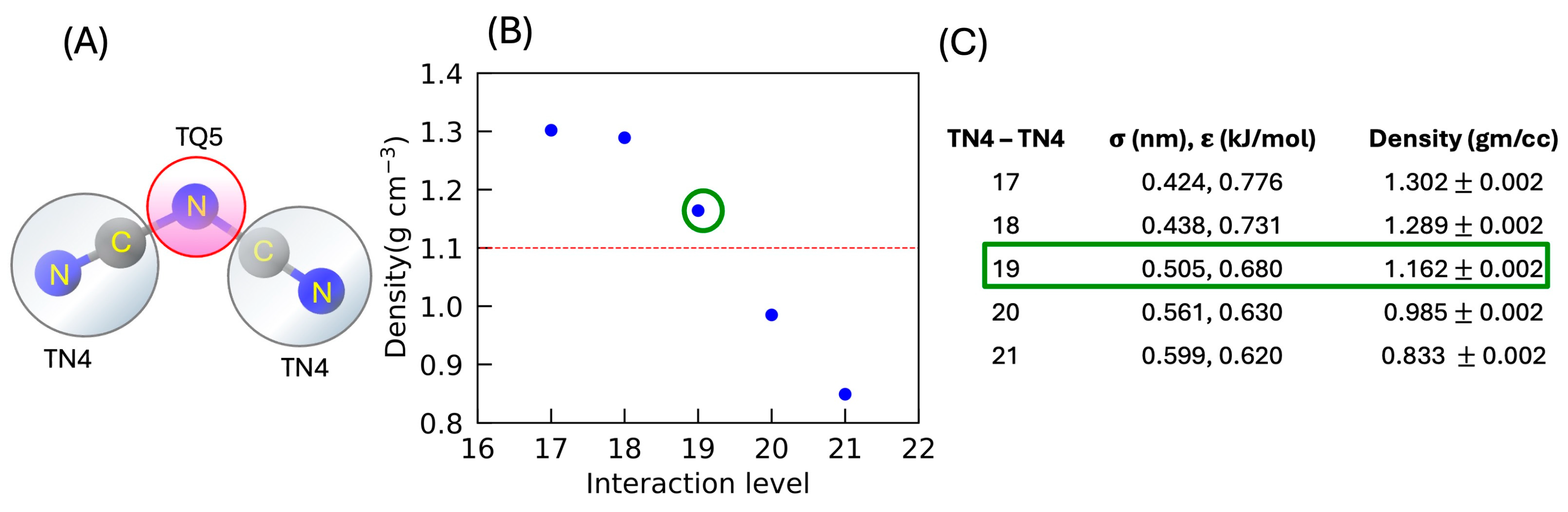
3.2.2. Changing the Interaction Level of the Anion Beads for MARTINI 3 Force Field
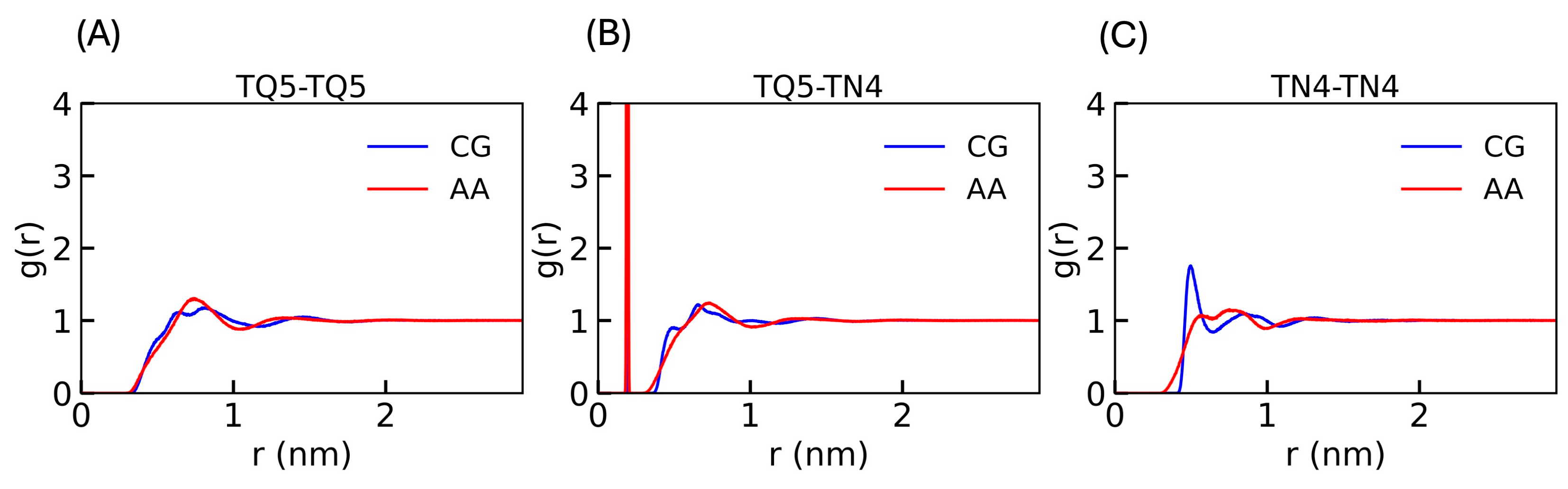
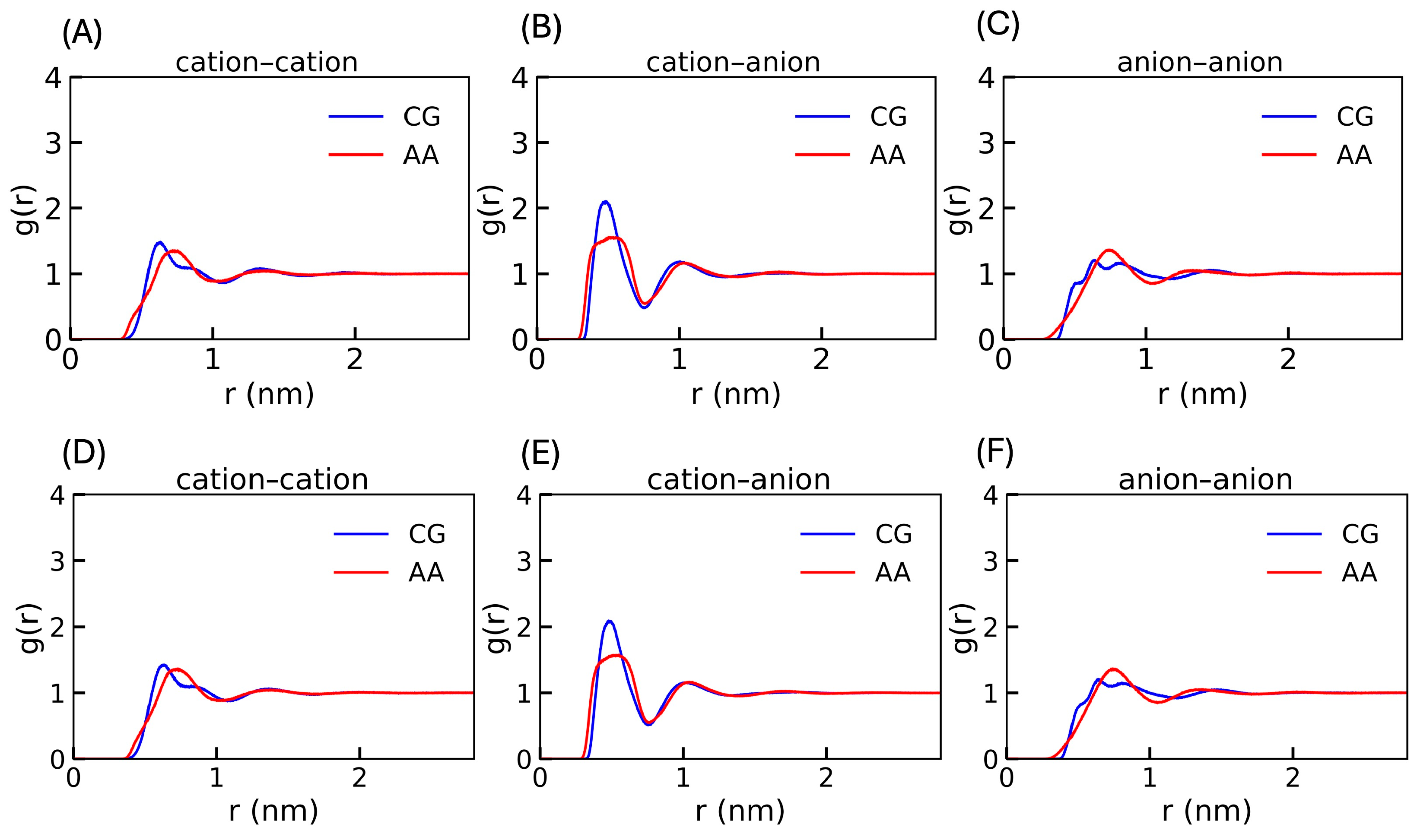
3.3. The Model Validation
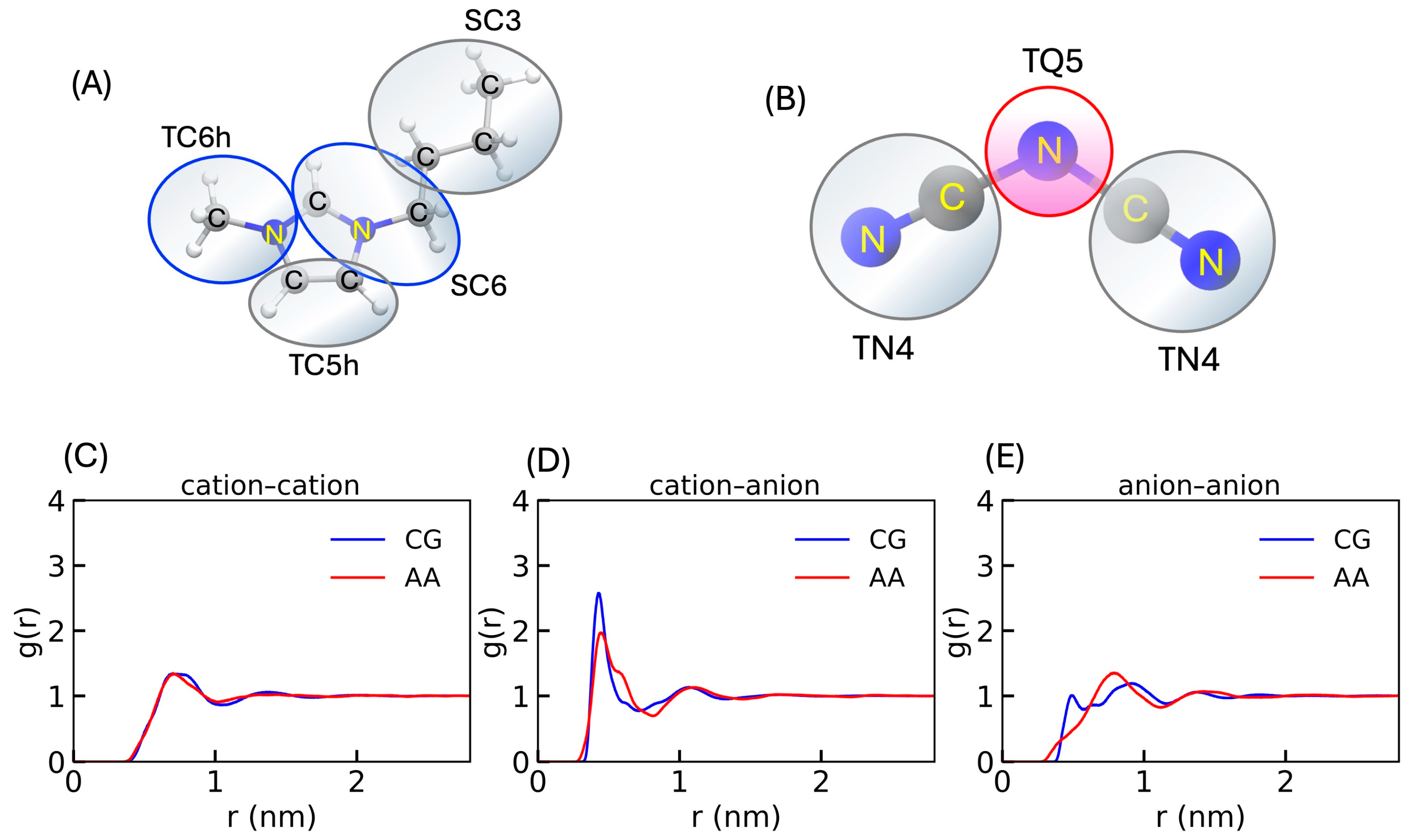
3.4. Validation of the CG Anion Model with Another Cation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chiappe, C.; Pieraccini, D. Ionic Liquids: Solvent Properties and Organic Reactivity. J. Phys. Org. Chem. 2005, 18, 275–297. [Google Scholar] [CrossRef]
- Ueno, K.; Tokuda, H.; Watanabe, M. Ionicity in Ionic Liquids: Correlation with Ionic Structure and Physicochemical Properties. Phys. Chem. Chem. Phys. 2010, 12, 1649. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.-C.; Ho, W.-Y.; Sun, I.-W.; Chou, Y.-K.; Hsieh, H.-H.; Wu, T.-Y. Synthesis and Properties of New Tetrachlorocobaltate (II) and Tetrachloromanganate (II) Anion Salts with Dicationic Counterions. Polyhedron 2011, 30, 497–507. [Google Scholar] [CrossRef]
- Vafaeezadeh, M.; Alinezhad, H. Brønsted Acidic Ionic Liquids: Green Catalysts for Essential Organic Reactions. J. Mol. Liq. 2016, 218, 95–105. [Google Scholar] [CrossRef]
- Fumino, K.; Peppel, T.; Geppert-Rybczyńska, M.; Zaitsau, D.H.; Lehmann, J.K.; Verevkin, S.P.; Köckerling, M.; Ludwig, R. The Influence of Hydrogen Bonding on the Physical Properties of Ionic Liquids. Phys. Chem. Chem. Phys. 2011, 13, 14064. [Google Scholar] [CrossRef]
- Pottkämper, J.; Barthen, P.; Ilmberger, N.; Schwaneberg, U.; Schenk, A.; Schulte, M.; Ignatiev, N.; Streit, W.R. Applying Metagenomics for the Identification of Bacterial Cellulases That Are Stable in Ionic Liquids. Green Chem. 2009, 11, 957–965. [Google Scholar] [CrossRef]
- Pinkert, A.; Marsh, K.N.; Pang, S.; Staiger, M.P. Ionic Liquids and Their Interaction with Cellulose. Chem. Rev. 2009, 109, 6712–6728. [Google Scholar] [CrossRef]
- Köddermann, T.; Paschek, D.; Ludwig, R. Molecular Dynamic Simulations of Ionic Liquids: A Reliable Description of Structure, Thermodynamics and Dynamics. ChemPhysChem 2007, 8, 2464–2470. [Google Scholar] [CrossRef]
- Elaiwi, A.; Hitchcock, P.B.; Seddon, K.R.; Srinivasan, N.; Tan, Y.M.; Welton, T.; Zora, J.A. Hydrogen Bonding in Imidazolium Salts and Its Implications for Ambient-Temperature Halogenoaluminate(III) Ionic Liquids. J. Chem. Soc. Dalton Trans. 1995, 21, 3467–3472. [Google Scholar] [CrossRef]
- Freemantle, M. Designer Solvents. Chem. Eng. News Arch. 1998, 76, 32–37. [Google Scholar] [CrossRef]
- Sun, Z.; Zheng, L.; Zhang, Z.Y.; Cong, Y.; Wang, M.; Wang, X.; Yang, J.; Liu, Z.; Huai, Z. Molecular Modelling of Ionic Liquids: Situations When Charge Scaling Seems Insufficient. Molecules 2023, 28, 800. [Google Scholar] [CrossRef] [PubMed]
- Crespo, E.A.; Schaeffer, N.; Coutinho, J.A.P.; Perez-Sanchez, G. Improved Coarse-Grain Model to Unravel the Phase Behavior of 1-Alkyl-3-Methylimidazolium-Based Ionic Liquids through Molecular Dynamics Simulations. J. Colloid Interface Sci. 2020, 574, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Sánchez, G.; Schaeffer, N.; Lopes, A.M.; Pereira, J.F.B.; Coutinho, J.A.P. Using Coarse-Grained Molecular Dynamics to Understand the Effect of Ionic Liquids on the Aggregation of Pluronic Copolymer Solutions. Phys. Chem. Chem. Phys. 2021, 23, 5824–5833. [Google Scholar] [CrossRef]
- Pérez-Sánchez, G.; Vicente, F.A.; Schaeffer, N.; Cardoso, I.S.; Ventura, S.P.M.; Jorge, M.; Coutinho, J.A.P. Unravelling the Interactions between Surface-Active Ionic Liquids and Triblock Copolymers for the Design of Thermal Responsive Systems. J. Phys. Chem. B 2020, 124, 7046–7058. [Google Scholar] [CrossRef]
- Karimi-Varzaneh, H.A.; Müller-Plathe, F.; Balasubramanian, S.; Carbone, P. Studying Long-Time Dynamics of Imidazolium-Based Ionic Liquids with a Systematically Coarse-Grained Model. Phys. Chem. Chem. Phys. 2010, 12, 4714–4724. [Google Scholar] [CrossRef]
- Bhargava, B.L.; Devane, R.; Klein, M.L.; Balasubramanian, S. Nanoscale Organization in Room Temperature Ionic Liquids: A Coarse Grained Molecular Dynamics Simulation Study. Soft Matter 2007, 3, 1395–1400. [Google Scholar] [CrossRef]
- Kodirov, A.; Abduvokhidov, D.; Mamatkulov, S.; Shahzad, A.; Razzokov, J. The Absorption Mechanisms of CO2, H2S and CH4 Molecules in [EMIM][SCN] and [EMIM][DCA] Ionic Liquids: A Computational Insight. Fluid Phase Equilibria 2024, 581, 114080. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Hockney, R.W.; Goel, S.P.; Eastwood, J.W. Quiet High-Resolution Computer Models of a Plasma. J. Comput. Phys. 1974, 14, 148–158. [Google Scholar] [CrossRef]
- Mayne, C.G.; Saam, J.; Schulten, K.; Tajkhorshid, E.; Gumbart, J.C. Rapid Parameterization of Small Molecules Using the Force Field Toolkit. J. Comput. Chem. 2013, 34, 2757–2770. [Google Scholar] [CrossRef]
- Cadena, C.; Anthony, J.L.; Shah, J.K.; Morrow, T.I.; Brennecke, J.F.; Maginn, E.J. Why Is CO2 so Soluble in Imidazolium-Based Ionic Liquids? J. Am. Chem. Soc. 2004, 126, 5300–5308. [Google Scholar] [CrossRef] [PubMed]
- Cadena, C.; Maginn, E.J. Molecular Simulation Study of Some Thermophysical and Transport Properties of Triazolium-Based Ionic Liquids. J. Phys. Chem. B 2006, 110, 18026–18039. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian, Version 16, Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Canongia Lopes, J.N.; Pádua, A.A.H. Molecular Force Field for Ionic Liquids III: Imidazolium, Pyridinium, and Phosphonium Cations; Chloride, Bromide, and Dicyanamide Anions. J. Phys. Chem. B 2006, 110, 19586–19592. [Google Scholar] [CrossRef] [PubMed]
- Lopes, J.N.C.; Deschamps, J.; Pádua, A.A.H. Modeling Ionic Liquids Using a Systematic All-Atom Force Field. J. Phys. Chem. B 2004, 108, 2038–2047. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A Package for Building Initial Configurations for Molecular Dynamics Simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Nosé, S. A Unified Formulation of the Constant Temperature Molecular Dynamics Methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N Log(N) Method for Ewald Sums in Large Systems. J. Comput. Chem. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Vazquez-Salazar, L.I.; Selle, M.; De Vries, A.H.; Marrink, S.J.; Souza, P.C.T. Martini Coarse-Grained Models of Imidazolium-Based Ionic Liquids: From Nanostructural Organization to Liquid-Liquid Extraction. Green Chem. 2020, 22, 7376–7378. [Google Scholar] [CrossRef]
- Alessandri, R.; Barnoud, J.; Gertsen, A.S.; Patmanidis, I.; de Vries, A.H.; Souza, P.C.T.; Marrink, S.J. Martini 3 Coarse-Grained Force Field: Small Molecules. Adv. Theory Simul. 2022, 5, 2100391. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 1, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 14101. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Tironi, I.G.; Sperb, R.; Smith, P.E.; Van Gunsteren, W.F. A Generalized Reaction Field Method for Molecular Dynamics Simulations. J. Chem. Phys. 1995, 102, 5451–5459. [Google Scholar] [CrossRef]
- Quijada-Maldonado, E.; Van Der Boogaart, S.; Lijbers, J.H.; Meindersma, G.W.; De Haan, A.B. Experimental Densities, Dynamic Viscosities and Surface Tensions of the Ionic Liquids Series 1-Ethyl-3-Methylimidazolium Acetate and Dicyanamide and Their Binary and Ternary Mixtures with Water and Ethanol at T = (298.15 to 343.15 K). J. Chem. Thermodyn. 2012, 51, 51–58. [Google Scholar] [CrossRef]
- Larriba, M.; Navarro, P.; Julián García, J.; Rodríguez, F. Liquid−Liquid Extraction of Toluene from Heptane Using [Emim][DCA], [Bmim][DCA], and [Emim][TCM] Ionic Liquids. Ind. Eng. Chem. Res. 2013, 52, 2341–2720. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Beads Type | Bond (nm) | Kb (kj/mol/nm2) |
|---|---|---|
| TQn-TN4 | 0.1928 | 1,500,000 |
| Beads Type | Angle (°) | Kθ (kj/mol/rad2) |
| TN4-TQn-TN4 | 128.05 | 700 |
| Beads Type (CG) | Density (gm/cc) |
|---|---|
| TQ1 | 1.422 ± 0.003 |
| TQ2 | 1.417 ± 0.003 |
| TQ3 | 1.400 ± 0.003 |
| TQ4 | 1.379 ± 0.003 |
| TQ5 | 1.356 ± 0.004 |
| All-atom (AA) simulations | |
| OPLS-AMBER | 1.104 ± 0.003 |
| CHARMM | 1.100 ± 0.003 |
| ρ (gm/cc) | Experiment [37] | OPLS-AMBER | CHARMM | Coarse-Grain (CG) |
|---|---|---|---|---|
| T = 303.15 K | 1.09866 | 1.104 ± 0.003 (+0.5%) | 1.100 ± 0.003 (+0.4%) | 1.162 ± 0.004 (+5.8%) |
| T = 343.15 K | 1.07277 | 1.077 ± 0.003 (+0.4%) | 1.072 ± 0.003 (−0.3%) | 1.118 ± 0.002 (+4.1%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konar, S.; Elahi, A.; Chaudhuri, S. Deciphering the Coarse-Grained Model of Ionic Liquid by Tunning the Interaction Level and Bead Types of Martini 3 Force Field. Physchem 2024, 4, 420-430. https://doi.org/10.3390/physchem4040029
Konar S, Elahi A, Chaudhuri S. Deciphering the Coarse-Grained Model of Ionic Liquid by Tunning the Interaction Level and Bead Types of Martini 3 Force Field. Physchem. 2024; 4(4):420-430. https://doi.org/10.3390/physchem4040029
Chicago/Turabian StyleKonar, Sukanya, Arash Elahi, and Santanu Chaudhuri. 2024. "Deciphering the Coarse-Grained Model of Ionic Liquid by Tunning the Interaction Level and Bead Types of Martini 3 Force Field" Physchem 4, no. 4: 420-430. https://doi.org/10.3390/physchem4040029
APA StyleKonar, S., Elahi, A., & Chaudhuri, S. (2024). Deciphering the Coarse-Grained Model of Ionic Liquid by Tunning the Interaction Level and Bead Types of Martini 3 Force Field. Physchem, 4(4), 420-430. https://doi.org/10.3390/physchem4040029