
Reactions of Graphene Nano-Flakes in Materials Chemistry and Astrophysics

Division of Applied Chemistry, Faculty of Engineering, Hokkaido University, Sapporo 060-8628, Japan
*
Author to whom correspondence should be addressed.
Physchem 2022, 2(2), 145-162; https://doi.org/10.3390/physchem2020011
Submission received: 9 March 2022
/
Revised: 1 April 2022
/
Accepted: 27 April 2022
/
Published: 12 May 2022
Abstract
:The elucidation of the mechanism of the chemical evolution of the universe is one of the most important themes in astrophysics. Polycyclic aromatic hydrocarbons (PAHs) provide a two-dimensional reaction field in a three-dimensional interstellar space. Additionally, PAHs play an important role as a model of graphene nanoflake (GNF) in materials chemistry. In the present review, we introduce our recent theoretical studies on the reactions of PAH and GNF with several molecules (or radicals). Furthermore, a hydrogen storage mechanism for alkali-doped GNFs and the molecular design of a reversible hydrogen storage device based on GNF will be introduced. Elucidating these reactions is important in understanding the chemical evolution of the universe and gives deeper insight into materials chemistry.

1. Introduction
Understanding the mechanisms of the chemical evolution of the universe is one of the most important topics of recent research in astrophysics and astrochemistry [1,2,3] About 20% of the total carbon in the universe is widely distributed in space in the form of polycyclic aromatic hydrocarbons (PAHs) [4], although the actual distribution in space and structure of PAHs are still unclear [5]. PAH is thought to provide a two-dimensional reaction field in three-dimensional interstellar space [6,7,8]. The reaction probability of bimolecular collisions increases significantly with the adsorption of molecules to the surface of PAH. Therefore, understanding the interaction between PAH and molecules is important for understanding the chemical evolution of the universe.
On the other hand, in the field of materials science, PAH is used as a model compound of graphene nanoflake (GNF) [9,10,11,12]. GNF has a wide range of applications such as hydrogen storage and lithium-ion batteries. Other applications include solar cells, energy storage devices, bio-devices, thermal management and piezoelectric materials, antibacterial materials, and filtration materials. Thus, PAH and GNF play an important role in a wide range of fields. In this review article, we introduce our theoretical studies on the interaction between PAH (GNF) and molecules. For example, the reactions of GNF (PAH) with hydrogen atoms, methyl radicals, and organic radicals are introduced here. Chemical reactions occurring on the surface of GNF are also presented. A mechanism of hydrogen storage composed of alkali-doped GNF and the molecular design of a reversible hydrogen storage device based on GNFs are introduced. The interaction of GNFs with molecules and radicals will play an important role both in the early reactions in the chemical evolution of the universe and in materials chemistry.
2. Interaction of Hydrogen Atoms with GNF
Atomic and molecular hydrogen are most abundant in interstellar space [13] If atomic hydrogen is adsorbed onto a GNF surface, several reactions can take place with other small atoms and molecules, forming more complicated molecules. Hence, the interaction of GNFs with atomic hydrogen plays an important role in the initial reactions of chemical evolution in the universe. In the field of materials science, GNF is used as a model compound of graphene [14,15,16,17]. In this chapter, the mechanism of hydrogen addition to GNFs is introduced using the results from density functional theory (DFT) calculations.
2.1. Bonding Structure of Hydrogen Atoms to GNFs
In density functional theory (DFT) calculations of the interaction system composed of GNF and a hydrogen atom (H), H was added to the carbon atom near the center of GNF, and the geometry was optimized. The GNF and the hydrogen-added GNF are denoted as GNF(n) and H-GNF(n), respectively, where n is the number of benzene rings in the GNF. Figure 1 shows examples of the GNFs(n) (n = 19 and 37) used in the calculations. The DFT calculations were performed on GNFs consisting of 7–37 benzene rings (See Figure S1 in Supporting Information). The basis sets are 6-31G(d) and 6-311G(d,p) [18] and the functional Coulomb-attenuating method (CAM-B3LYP) [19] is used. Atomic charges and spin densities were determined by natural population analysis (NPA) and natural bond orbital (NBO) methods.
The optimized structure of H-GNF is shown in Figure 2. The planar structure around the H-bonded carbon atom (C0) of GNF is changed to a pyramidal one after the addition of H. The distance between the added hydrogen (H) and the bonded carbon atom (C0) is R1 = 1.114 Å. The atomic charges of C0 and H are −0.274 and +0.245, respectively, indicating that the C-H bond is locally polarized as (C0)−0.274–(H)+0.245. This suggests that the addition of H to the nonpolar GNF generates a dipole moment that facilitates the attraction of other molecules in interstellar space [20].
2.2. Potential Energy Curve
The potential energy curve (PEC) for the approach of H to the GNF surface is plotted in Figure 3A as a function of C0-H distance (R1). The calculations are carried out at the CAM-B3LYP/6-311G(d,p) level. All geometrical parameters except for the C0-H distance (R1) are optimized at each value of R1. When H approaches the GNF surface, the first energy minimum was found at R1 = 3.50 Å, corresponding to a van der Waals (vdW) complex composed of GNF and H. The binding energy of H to GNF is 0.2 kcal/mol. The H atom is bound by vdW force to the π-orbital of the GNF surface. Subsequently, the energy barrier corresponding to transition state (TS) is found at R1 = 1.71 Å, where the barrier height is 6.2 kcal/mol relative to the dissociation limit (GNF + H). The energy decreases significantly after TS and reaches the lowest energy point corresponding to the bound state of H-GNF, i.e., the product state (PD). The barrier is caused by the change in the electronic state in the carbon atom (C0) from sp2 to sp3 upon the addition of hydrogen.
The PEC for the addition of a proton (H+) is also given for comparison. The shape of PEC for the addition of H+ is significantly different from that of the addition of hydrogen. The H+ can bind directly to the graphene surface without an activation barrier. A large quantity of exothermic energy is generated in H+ because of the high proton affinity of GNF.
The spatial distribution of the spin density of H-GNF is shown in Figure 3B. In the vdW state, the spin density is localized to the H atom and does not flow into the PAH side. At TS, the spin density of H is 0.794, which indicates that about 20% of the unpaired electrons dissipated into the GNF side. The spin density on GNF is 0.988 in PD, indicating that the unpaired electron of the H atom is almost completely transferred to GNF.
2.3. Activation and Binding Energies
The size dependence of the activation barrier and binding energy of GNFs is shown in Figure 4. The activation energies are calculated to be 6.6 kcal/mol (n = 7), 5.6 kcal/mol (n = 14), 6.2 kcal/mol (n = 19), 5.2 kcal/mol (n = 29), and 7.0 kcal/mol (n = 37). These results indicate that the activation energy is independent of the GNF size and is nearly constant (~5–7 kcal/mol). In contrast, the binding energy was slightly dependent on the size of GNF. The binding energies for n = 14 and 29, specifically, are higher than the other binding energies.
2.4. Behavior of Hydrogen Atoms on GNF (PAH) Surfaces
In this section, the interactions and reactions between H atom and GNF surface ere described, which were obtained by the DFT method. A schematic energy diagram of the interaction between the hydrogen atom and the GNF surface is shown in Figure 5. The H atom from interstellar space is first weakly bound to the GNF surface by vdW interaction. The binding energy is about 0.1–0.2 kcal/mol, and the H atom is located at 2.8–3.5 Å from the surface. This binding energy is large enough to stay on the GNF surface in interstellar space because the temperature is lower than 10 K in interstellar space. Hydrogen atoms can diffuse freely on the surface at 10 K. The diffusion barrier is about 0.2 kcal/mol. The H atom trapped on the surface in the vdW state is an active species and can readily react with other molecules. If H atom gains excess energy higher than the activation barrier, hydrogenated GNF (H-GNF) is formed. H-GNF is reactive species due to a radical with dipole moment.
3. Interaction of GNFs with Methyl Radicals
The methyl radical (CH3) is the simplest organic radical observed in interstellar space and is the basis for the formation of more complex organic molecules. It was first discovered in space by Feuchtgruber et al. [21] in Sgr A* using the infrared space telescope of the European Space Agency. It was subsequently discovered by Knez et al. in NGC7538 [22]. Methyl radicals adsorbed on GNF surfaces can evolve by reacting with other atoms and molecules, as discussed by us [23].
In materials science, a methyl radical is an important intermediate generated from the catalytic reaction of alkane and its related compounds. In this chapter, the mechanism of the addition of a methyl radical to GNFs is introduced [22].
3.1. Binding Structure of Methyl Radical to GNF
The optimized binding structure of methyl radical added to GNF, CH3-GNF, is shown in Figure 6. The C-C bond distance of the binding site was calculated to be R1 = 1.574 Å. The molecular charges of CH3 and GNF parts are +0.084 and −0.084, respectively. Although CH3 shows a slight electron donor property, the magnitude of the charge transfer is very small. Therefore, CH3-GNF has no dipole moment.
3.2. Potential Energy Curve
Figure 7A shows potential energy along the intrinsic reaction coordinate (IRC) for the addition reaction of CH3 to the GNF surface. The barrier height of TS is 14.0 kcal/mol higher in energy than the initial vdW complex. The distance of the CH3 from GNF in TS is R1 = 2.131 Å. After TS, the energy decreases and reaches the PD of CH3-GNF. The activation energy associated with the binding of CH3 to GNF is 14.0 kcal/mol, which is larger than that of H-GNF (6.2 kcal/mol). The addition reaction of CH3 is a 3.0 kcal/mol exothermic reaction.
The spatial distribution of the spin density of CH3-GNF is shown in Figure 7B. In the vdW state, the spin density of CH3 is 0.994, which is almost localized to the CH3 radical. In TS, the spin density of the CH3 part is 0.661 and that of GNF is 0.339, which means that almost 30% of the unpaired electrons flow from CH3 to GNF. In PD, on the contrary, the spin density of GNF is 0.931, indicating that the unpaired electron of methyl radical is almost completely transferred to GNF.
3.3. Activation and Binding Energies
The addition of CH3 to GNF needs an activation barrier. The size dependence of the activation and binding energies of GNFs is shown in Figure S2 (in Supporting Information). The activation energies for n = 4, 7, 19, and 37 are 15.1, 14.7, 13.8, and 13.4 kcal/mol, respectively, at the CAM-B3LYP/6-311G(d,p) level. These results indicate that there is almost no size dependence of the activation energy of GNFs. In contrast, the binding energy was slightly dependent on the size of the GNF, and the binding energy increased slightly with size.
3.4. Behavior of Methyl Radicals on GNF Surface
In this section, the interaction of CH3 with the GNF surface was described. A schematic energy diagram of the interaction between CH3 and the GNF surface is shown in Figure 8. The methyl radical from interstellar space binds weakly to the GNF surface through the vdW interaction. The binding energy is about 0.1–0.2 kcal/mol. When the methyl radical receives a quantity of kinetic energy of about 14 kcal/mol, it reacts with carbon atoms on the surface to form CH3-GNF.
3.5. Activation Energies of Alkyl Radicals Addition to GNF
The activation energies of radical addition reaction, E(TS), reaction energies leading to PD, E(PD), and the binding energies of vdW complexes, E(vdW), are given in Table 1. The calculated values of methyl (CH3), ethyl (Et), n-propyl (n-Pr), iso-propyl (iso-Pr), n-butyl (n-Bu), secondary-butyl (sec-Bu), tertiary-butyl (tert-Bu), and iso-butyl (iso-Bu) radicals are given. The activation energy of CH3 is 13.8 kcal/mol, and the activation energy increases as the bulk of the molecule increases. The reaction energies are positive except for the CH3 radical, indicating that the addition of alkyl radicals is endothermic. In all radicals, vdW complexes composed of GNF and radical are exothermic formed.
4. Chemical Reaction on GNF Surface
When a molecule is adsorbed onto a GNF (PAH) surface, the electronic state of the molecule will be changed by the interaction with GNF. It is expected that a reaction mechanism will be affected by interaction from the surface. The effects of GNF on the reaction mechanism are considered in this chapter. The hydrogen addition reaction to acetylene (HCCH) on the GNF surface is presented as a sample reaction to elucidate the effect of the GNF surface on the reaction mechanism. The reaction is expressed as
HCCH + H → TS → H2CCH (radical)
The H2CCH radical is formed by the addition of hydrogen to HCCH as a product. The transition state (TS) structure for reaction (1) in the gas phase is illustrated in Figure 9. The hydrogen atom is located at 1.996 Å from the carbon atom of HCCH, and the linear structure of HCCH is changed to a slightly bent form in TS. The activation energy of the addition reaction is 1.3 kcal/mol in vacuum. Next, the structural parameters of TS on the GNF surface are given in Figure 9 (in parenthesis). Both lengths and angles on the GNF surface are close to those in vacuum. The structure on GNF is significantly similar to that in vacuum, indicating that the GNF surface effect on the electronic states of TS is quite small.
Potential energy along the IRC is illustrated on Figure 10A. First, HCCH and the H atom are adsorbed onto GNF due to the vdW interaction (vdW(1)). In the TS structure, the distance of the H atom from the carbon atom of HCCH is 1.998 Å, which is close to the structure in vacuum. After TS, the addition of a H atom to HCCH takes place and forms a radical product (H2CCH). This radical remains on the surface with vdW interaction (vdW(2)). The radical is then desorbed from the surface, yielding the same reaction product (H2CCH) as in vacuum.
The relative energies at each point along IRC are summarized in Table 2. In vacuum, the activation and reaction energies are 1.3 kcal/mol and −47.6 kcal/mol, respectively. In the reaction on GNF, the activation energy is 1.5 kcal/mol. There is almost no energy difference between the GNF surface and the vacuum is very small. This indicates that the GNF surface largely does not change the electronic state of the adsorbed molecules, but contributes only as a reaction field.
5. Mechanism of Hydrogen Storage in the Graphene Nanoflake Doped by Alkali-Metals
GNF acts as an important reaction field in interstellar space. On the other hand, GNFs have been studied in materials chemistry as a model molecule for graphene nanoflakes. Carbon materials, such as graphene nanoflakes and fullerene, can be used for hydrogen storage. Alkali doping these materials generally increases their H2-storage density. In this article, DFT studies on the mechanism of hydrogen storage using GNFs doped by alkali-metals are presented [24].
GNF (37) is used as a model of a graphene nanoflake (denoted GNF), and hydrogen added clusters, GNF-M-(H2)n and GNF-M+-(H2)n (M = Li and Na, n = 0–12), are employed as hydrogen storage systems.
First, the structure of GNF is optimized, and then alkali metal M (or M+) is placed in the central region of GNF. The structures of GNF-M is optimized, where all GNF-M+/M atoms are fully optimized. The binding energy of M to GNF is defined as follows:
where E(X) is the total energy of X. If Ebind(M) is positive, M can bind exothermally to GNF. The binding energy of H2 to GNF-M (per one H2 molecule) is defined as defined as follows:
If Ebind(n) is positive, the H2 molecule binds exothermally to GNF-M.
5.1. Structures of the Li Doped-Graphene Nanoflake
First, the structures of GNF-Li and GNF-Li+ are optimized, and the binding energies are summarized in Table 3. Both Li and Li+ are bound to the hexagonal site of the GNF surface. The similar heights, i.e., the distance of Li (or Li+) from the GNF surface, are obtained (1.736 Å for Li and 1.771 Å for Li+). The binding energies are 17.1 kcal/mol (Li) and 52.8 kcal/mol (Li+). These results are in good agreement with previous calculations of the coronene-Li (or Li+) system [25]. The NPA atomic charges on Li and Li+ were +0.93 and +0.94, respectively, suggesting that the net charge of Li is very similar to that of Li+. This implies that significant electron transfer takes place from Li to GNF after binding (0.93 e).
5.2. Binding Structures of the Hydrogen Molecules to GNF-Li
Figure 11 shows the binding structures of the H2 molecules to GNF-Li. In case of first H2 addition (n = 1), both distances of hydrogen atoms from Li are equivalent with 2.027 Å, indicating that H2 binds to Li with a side-on structure. For n = 2–3, the binding structures are side-on, similar to that of the n = 1 case.
For n = 4, the fourth hydrogen molecule cannot bind directly to Li, and instead is weakly bound to the hydrogen molecules already binding to Li. The outer hydrogen molecules (n = 5 and 6) are also bound to H2 in the inner shell. The similar binding structures are obtained for the GNF-Li+ (ion)-H2 system because the electronic state of GNF-Li+ (ion) is very similar to that of GNF-Li (atom).
5.3. Binding Energy of H2 to GNF-Li
The binding energy of H2 to GNF-Li plotted as a function of n (per H2 molecule) are in Figure 12. The binding energy for the first addition of H2 (n = 1) is 3.83 kcal/mol, and it decreases with increasing n. The binding energies are 2.85 (n = 3) and 1.43 kcal/mol (n = 7). The energy is almost saturated at n = 12. The large energy strongly indicates that GNF-Li can be used as a H2 storage material. Similar features were observed in the GNF-Li+-(H2)n system.
To elucidate the effect of GNF on the binding energy, the binding energy of H2 to bare Li was calculated without GNF and the results are plotted in Figure S3 (open squares). The calculated energies for n = 4, 7, and 12 are 1.35, 0.82, and 0.50 kcal/mol, respectively. The binding energy in Li-(H2)n without GNF is significantly lower than that with GNF, suggesting that GNF activates Li via electron capture. This electron transfer activates the Li atom on the GNF.
5.4. Binding Structures of H2 to GNF-Na
Establishing an appropriate means of transportation is an important element in achieving a hydrogen energy society [27,28]. Carbon materials can safely store hydrogen [29,30] and lithium (Li)-doped graphene is a particularly effective storage material [31,32]. Unfortunately, lithium is high cost metal because it must travel long distances to be commercialized. In this regard, sodium (Na) is an inexpensive metal with chemical properties similar to those of Li [26].
The geometry optimizations of GNF-Na and GNF-Na+ indicate that both Na and Na+ bind to the hexagonal sites of GNF. The heights of Na (or Na+) from GNF, binding energies of Na to GNF, and NPA atomic charge of Na are given in Table 3. The heights are 2.247 Å (Na) and 2.288 Å (Na+), and both Na and Na+ are located at a similar distance from the GNF surface, although the binding energies are different from each other: 4.4 (Na) and 37.5 kcal/mol (Na+).
The atomic charges are +0.978 (Na) and +0.979 (Na+), indicating that the net charge of Na on GNF is very similar to that of Na+ on GNF, which suggests that significant electron transfer occurs from Na to GNF (0.98e). These features in GNF-Na are in good agreement with the GNF-Li system, as summarized in Table 3, where the atomic charges on Li and Li+ adsorbed on GNF are +0.929 and +0.937, respectively.
Next, the binding of H2 to GNF-Na is examined to elucidate its hydrogen storage ability. The binding structures of H2 to GNF-Na (n = 1–6) are illustrated in Figure 13. In n = 1, H2 is bound to Na with a side-on structure, where the two hydrogen atoms of H2 are equivalently bound to Na. Similar binding structures are obtained for n = 1–4. The fifth H2 molecule binds to Na in a manner orthogonal to the surface, with a distance of 2.833 Å, which is larger than those of n = 1–4 (2.45 Å). These results indicate that the first coordination shell is saturated at n = 4. The sixth H2 molecule interacts with a H2 molecule in the first coordination shell and is not directly bound to Na (the distances between the sixth H2 molecule and Na and the nearest H2 molecule are 5.037 and 3.625 Å, respectively). Side-on coordination structures were observed in all clusters (n = 7–12).
5.5. Binding Energies of H2 to GNF-Na
The binding energies of H2 to GNF-Na or GNF-Na+ plotted as a function of n (per H2 molecule) are in Figure 12. The binding energy for n = 1 is 2.72 kcal/mol, and it decreases with increasing n. The binding energies are 2.67 (n = 2), 2.50 (n = 3), 2.34 (n = 4), and 2.01 kcal/mol (n = 5).
In the case of a GNF-Li-H2 system, the binding energies are 3.83 (n = 1), 3.29 (n = 2), 2.85 (n = 3), 2.20 (n = 4), and 1.83 kcal/mol (n = 5). These trend indicates that GNF-Li interacts more strongly with H2 than GNF-Na for n = 1–3, while the interactions are comparable for both Li and Na at n = 4. GNF-Na interacts slightly more strongly with H2 in larger systems (n = 5–12). Thus, GNF-Na appears to have a higher H2-storage ability than GNF-Li. The GNF-Na+-(H2)n (Na+ ionic system) exhibits similar features (Figure 12). Since the Na–(H2)n binding energy is close to zero in the absence of GNF, GNF clearly enhances binding through electron transfer from Na to GNF: GNF + Na → (GNF)−-Na+.
These results strongly suggest that GNF-Na is a suitable candidate for the efficient storage of hydrogen gas for various applications in the hydrogen economy, and that sodium is an alternative to lithium for that purpose.
For comparison, the hydrogen adsorption capacity of K+ was examined. The binding energy of H2 to GNF-K+ is plotted in Figure 12. The binding energies in GNF-K+ were lower than those of GNF-Li+ and GNF-Na+, suggesting that GNF-K+ is poor as a H2 storage.
6. Molecular Design of Reversible Hydrogen Storage Device
In previous sections, the hydrogen storages of Li- and Na-doped GNFs were introduced. In this section, the molecular design of a reversible hydrogen storage device based on GNFs is presented. Magnesium has three valence states: neutral, mono-, and divalent, expressed as Mg, Mg+, and Mg2+. Here, the GNF-Mg system is examined as a reversible hydrogen storage device [33].
6.1. Structures of Mg-Doped GNF
First, the structures of Mg-doped GNF, GNF-Mgm+ (m = 2, 1, and 0), were fully optimized. Figure 14A shows the structures of GNF-Mgm+ (m = 2), and those for m = 1 and 2 are illustrated in Figures S4 and S5, respectively. Neutral and ionic species of Mg are bound to the hexagonal site of GNF. The binding energies of Mgm+ to GNF are 0.3 kcal/mol (m = 0), 39.9 kcal/mol (m = 1), and 159.8 kcal/mol (m = 2), indicating that the binding energy is strongly dependent on the charge of Mg. The height of Mg from the GNF surface (h) is also dependent on the atomic charge of Mg: h = 4.339 Å (m = 0), 2.214 Å (m = 1), and 1.806 Å (m = 2). Mg2+ is shorter than Mg+, and a neutral Mg atom cannot bind to GNF. The atomic charges of the Mg moiety of GNF-Mgm+ for m = 0, 1, and 3 are −0.001, +0.960, and +1.882, respectively, indicating that the electron transfer from GNF to Mg species is small in the GNF-Mg system.
6.2. Binding Structures and Energies of H2 to GNF-Mgm+ (m = 1 and 2)
Figures S6 and S7 show the binding structures of H2 to GNF-Mg2+ (m = 2). The H2 molecule binds to Mg2+ with a side-on structure. In both cases (m = 1 and 2), the first coordination shells were closed up to n = 4. The second shell consisted of one H2 molecule (5-th H2). Figure 14B shows the binding energy of H2 to GNF-Mgm+ (per H2 molecule) plotted as a function of n. The first addition of H2 to GNF-Mg2+ causes a binding energy of 13.22 kcal/mol, which gradually decreases as a function of n: the binding energies are 9.99 kcal/mol (n = 3), 6.79 kcal/mol (n = 5), and 5.03 kcal/mol (n = 7).
In contrast, the binding energies of GNF-Mg+-(H2)n (monovalent state, m = 1) are significantly lower than those for Mg2+: 0.31 kcal/mol (n = 1), 0.28 kcal/mol (n = 3), 0.26 kcal/mol (n = 5), and 0.24 kcal/mol (n = 7). These trends strongly indicate that GNF-Mg2+ can be used as a H2 storage material, whereas the ability of GNF-Mg+ is quite low. Additionally, it was found that the ability for H2 storage in GNF-Mgm+ is changed by the charge of GNF-Mg. Adsorption–desorption is controlled by the molecular charge in the GNF-Mgm+-(H2)n system (m).
6.3. Electron Capture Dynamics of GNF-Mg2+-H2
In previous section, it was found that the structure of GNF-Mg-H2 and the binding energy of H2 to GNF-Mg are largely dependent on the atomic charge of Mg. This specific property in the Mg system is much different from the Li and Na-GNF systems. Additionally, this makes GNF-Mg-H2 suitable for use as a H2 storage device with reversible adsorption–desorption properties. In this section, direct ab initio molecular dynamics (AIMD) calculations [34,35,36] for the electron and hole capture processes of the GNF-Mg-H2 system are described.
The result of the electron capture dynamics of GNF-Mgm+-(H2)4 (m = 2), calculated by means of the direct AIMD method, is given in Figure 15, where snapshots of GNF-Mg+-(H2)4 are shown. The initial structure at time zero was set to the optimized geometry of GNF-Mg2+-(H2)4 and then a trajectory of GNF-Mg+-(H2)4 following the electron capture of GNF-Mg2+-(H2)4 was started without excess energy. The electron capture makes a change to the charge on Mg (Mg2+ to Mg+). The intermolecular distance between the Mg and H2 molecules (denoted as <R>, average distance) and the height of Mg+ from the GNF surface (h) were drastically varied as a function of time. The H2 molecules and Mg+ were located at <R> = 2.253 Å and h = 1.908 Å at time = 0 fs (vertical electron capture point), respectively. After the electron capture, the H2 molecules gradually left the Mg+: <R> = 2.327 Å (51.7 fs) and 3.034 Å (85.9 fs). In the final stage of electron capture reaction (100 fs), the H2 molecules were fully dissociated as <R> = 4.014 Å. The position of Mg+ was elongated to h = 2.526 Å at 100.0 fs. The reaction is expressed as follows:
GNF-Mg2+-(H2)n + e− → GNF-Mg+-(H2)n (H2 in the gas phase)
Thus, H2 molecules were released into the gas phase after the electron capture.
6.4. Hole Capture Dynamics of GNF-Mg+-H2
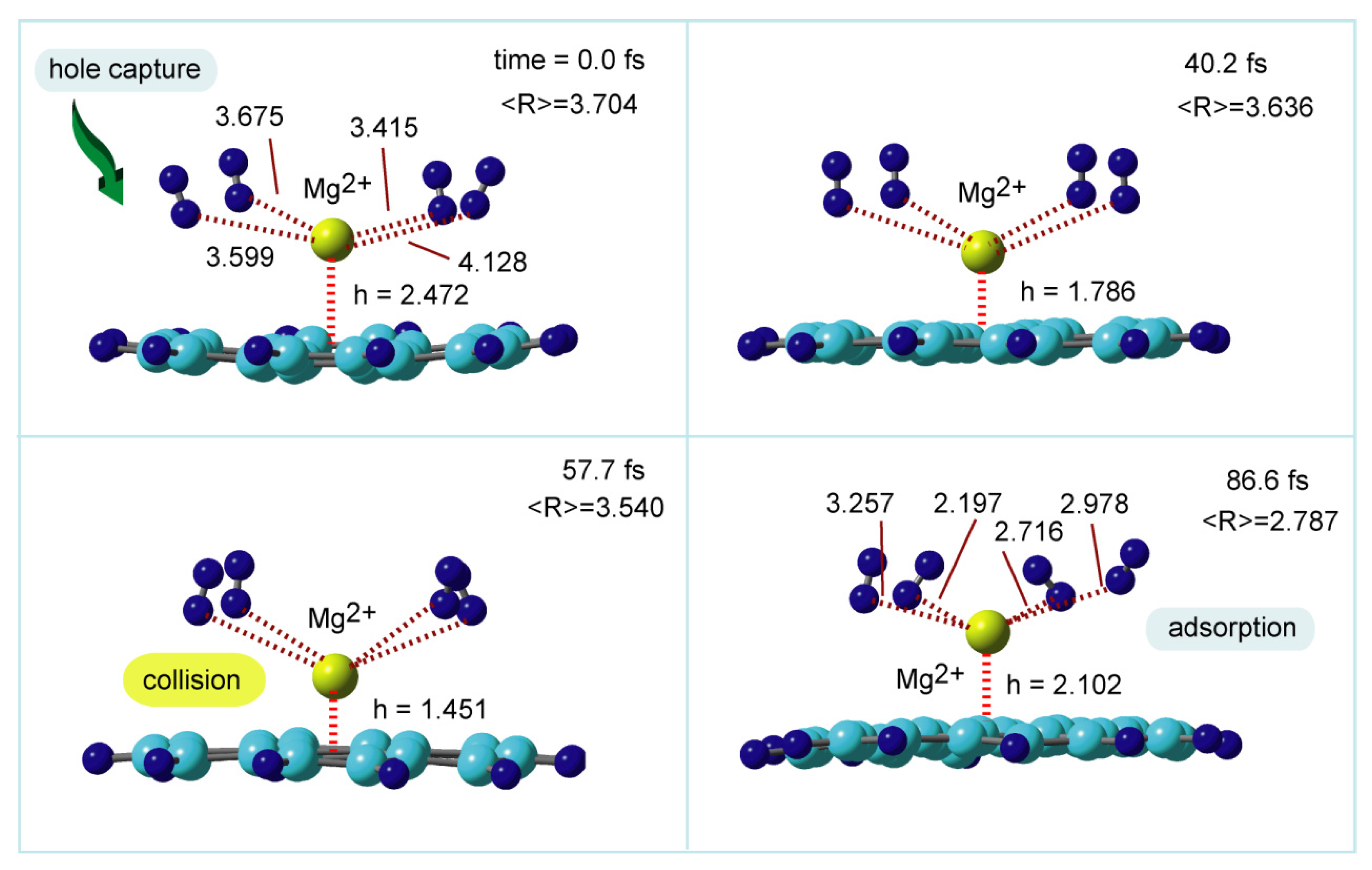
In contrast, the dissociated hydrogen molecules return from gas phase to GNF-Mg2+ after a hole capture of GNF-Mg+. The hole capture dynamics of GNF-Mgm+-(H2)4 (m = 1) are given in Figure 16. The molecular charge of GNF-Mg is changed from GNF-Mg+ to GNF-Mg2+ by a hole capture, and the electronic state is thus drastically changed. The initial geometry of GNF-Mg2+-(H2)4 was chosen as one of the dissociation structures in the gas phase. At the vertical hole capture point (time zero), the distance between GNF-Mg and H2 molecules was <R> = 3.704 Å (average distance), while the height of Mg+ from the GNF surface was h = 2.472 Å. At 40.2 fs, the distance of H2 was shortened to be <R> = 3.636 Å, while the height of Mg2+ became very low (1.786 Å). Mg2+ collided with the GNF surface at 57.7 fs (h = 1.451 Å). The hole capture reaction took 86.6 fs become placid. The distance of H2 and Mg from the GNF surface was <R> = 2.787 and h = 2.787 Å, respectively. These geometrical changes indicate that the H2 molecules can be fully rebound to GNF-Mg2+ after hole capture, and the structure was recovered by the addition of H2 to GNF-Mg2+. The reverse process is finished at ~90 fs. The reaction is expressed as follows:
GNF-Mg+ + (H2)n (gas phase) + hole → GNF-Mg2+–(H2)n (adsorption)
These results strongly indicate that the reversible adsorption–desorption reaction of H2 is easily controlled by the charge switching of GNF-Mg.
7. Conclusions
In the present review article, the reactions of hydrogen atom and methyl radicals with graphene nanoflakes (GNFs) are first studied from a quantum-chemical point of view. The reactions of alkyl radicals with GNF are also presented. These species can react with GNFs, and radical-added GNFs are formed as products. The addition reactions need activation energies corresponding to sp2-sp3 hybridization. In contrast, the addition of a halogen atom (F and Cl) to the GNF surface proceeds without an activation barrier due to the large exothermic energy involved. The dipole moment is generated by the addition of a H atom to the GNF, and spin density is widely distributed over H-GNF, indicating that the non-reactive GNF is activated by the addition of H in interstellar space.
Second, the effects of the GNF surface on the chemical reaction are presented. The reaction associated with the addition of a hydrogen atom to acetylene was examined as a sample reaction. The GNF surface largely did not affect the electronic states of the adsorbed molecule. This indicates that the GNF surface does not much change the electronic state of the adsorbed molecules, but contributes as a two-dimensional reaction field for the reaction to proceed in three-dimensional interstellar space.
Next, the hydrogen storage mechanism of alkali doped GNFs is presented. Lithium and sodium were examined as an alkali atom and ion. In both atoms and ions, hydrogen molecules are efficiently stored in doped GNFs, indicating that the GNF-Li and GNF-Na systems can be used as hydrogen storage devices.
Lastly, the molecular design of a reversible hydrogen storage device based on GNFs was introduced. Magnesium takes three valence states: neutral, mono-, and divalent, expressed as Mg, Mg+, and Mg2+. A molecular device composed of GNF and Mg, GNF-Mg, was examined as a reversible hydrogen storage device. GNF-Mg2+ can efficiently store hydrogen molecules, whereas GNF-Mg+ releases H2 molecules to the gas phase. Direct AIMD calculations showed that the reversible processes take place within 100 fs. These results strongly suggest that the GNF-Mg system has the potential to be used as a reversible H2 adsorption–desorption device.
Thus, GNFs can take on a wide variety of reactivity and electronic states. In interstellar space, it provides an important reaction field for space molecules. In addition, GNFs show many characteristic properties in material chemistry. The development of new GNF materials is expected to continue in the future.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/physchem2020011/s1. Figure S1. Models of graphene nanoflakes used in the present calculations. GNF(n) means graphene nanoflake composed of n benzene rings. GNF(7) is coronene; Figure S2. Binding energies (in kcal/mol) and activation energies (in kcal/mol) of CH3 addition to GNF(n). The calculations were carried out at the CAM-B3LYP/6-31G(d) and 6-311G(d, p) levels of theory; Figure S3. Binding energies of H2 to GNF-Li and GNF-Li+ (per H2 molecule). The open squares represent the binding energies of H2 to the Li atom without GNF, Li(H2)n; Figure S4. Optimized structures of GNF-Mg (atom). The calculations were performed at the CAM-B3LYP/6-311G(d,p) level. Value indicates height of Mg from GNF surface in Å; Figure S5. Optimized structures of GNF-Mg+ (ion). The calculations were performed at the CAM-B3LYP/6-311G(d,p) level. Value indicates height of Mg from GNF surface in Å; Figure S6. Optimized structures of GNF-Mg+-(H2)n with stable form (n = 1–6). The calculations were performed at the CAM-B3LYP/6-311G(d,p) level.
Author Contributions
H.T.; writing—original draft preparation, T.I.; review, H.T.; project administration, H.T.; funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by Japan Society for the Promotion of Science (JSPS): Grant Numbers 21K04973 and 21H05415.
Data Availability Statement
Not applicable.
Acknowledgments
H.T. acknowledges partial support from JSPS (Grant Numbers 21K04973 and 21H05415).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pandey, G. Chemical elements in the Universe: Origin and evolution. J. Astrophys. Astr. 2020, 41, 31. [Google Scholar] [CrossRef]
- Phua, Y.Y.; Sakakibara, N.; Ito, T.; Terashima, K. Low Temperature Plasma for Astrochemistry: Toward a Future Understanding with Continuous and Precise Temperature Control. Plasma Fusion Res. 2020, 15, 1506041. [Google Scholar] [CrossRef]
- Kauffman, S.; Jelenfi, D.P.; Vattay, G. Theory of chemical evolution of molecule compositions in the universe, in the Miller-Urey experiment and the mass distribution of interstellar and intergalactic molecules. J. Theor. Biol. 2020, 486, 110097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwek, E.; Arendt, R.G.; Fixsen, D.J.; Sodroski, T.J.; Odegard, N.; Weiland, J.L.; Reach, W.T.; Hauser, M.G.; Kelsall, T.; Moseley, S.H.; et al. Detection and Characterization of Cold Interstellar Dust and Polycyclic Aromatic hydrocarbon Emission, from COBE Observations. Astrophys. J. 1997, 475, 565–579. [Google Scholar] [CrossRef] [Green Version]
- Mori, T.I.; Onaka, K.; Sakon, I.; Ishihara, D.; Shimonishi, T.; Ohsawa, R.; Bell, A.C. Observational studies on the Near-infrared Unidentified Emission Bands in Galactic H II regions. Astrophys. J. 2014, 784, 53. [Google Scholar] [CrossRef] [Green Version]
- Vats, A.; Pathak, A.; Onaka, T.; Buragohain, M.; Sakon, I.; Endo, I. Theoretical study of infrared spectra of interstellar PAH molecules with N, NH, and NH2 incorporation. Publ. Astron. Soc. Jpn. 2022, 74, 161–174. [Google Scholar] [CrossRef]
- Hu, X.; Yang, Y.; Zhang, C.; Chen, Y.; Zhen, J.; Qin, L. Gas-phase laboratory formation of large, astronomically relevant PAH-organic molecule clusters. Astron. Astrophys. 2021, 656, A80. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, C.; Hu, X.; Zhang, D.; Chen, Y.; Zhen, J.; Qin, L. Gas-phase laboratory study on PAH/amino-acid cluster cations. MNRAS 2021, 508, 3009–3022. [Google Scholar] [CrossRef]
- Qu, J.; Dai, X.-X.; Cui, J.-S.; Chen, R.-X.; Wang, X.; Lin, Y.-H.; Verduzco, R.; Wang, H.-L. Hierarchical polyaromatic hydrocarbons (PAH) with superior sodium storage properties. J. Mater. Chem. A 2021, 9, 16554–16564. [Google Scholar] [CrossRef]
- Hjertenaes, E.; Andersson, S.; Koch, H. Potential Energy Surfaces and Charge Transfer of PAH-Sodium-PAH Complexes. Chemphyschem 2016, 17, 2908–2915. [Google Scholar] [CrossRef]
- Munoz-Castro, A.; Gomez, T.; MacLeod Carey, D.; Miranda-Rojas, S.; Mendizabal, F.; Zagal, J.H.; Arratia-Perez, R. Surface on Surface. Survey of the Monolayer Gold-Graphene Interaction from Au12 and PAH via Relativistic DFT Calculations. J. Phys. Chem. C 2016, 120, 7358–7364. [Google Scholar] [CrossRef]
- Gloriozov, I.P.; Demyanov, P.I.; Zhulyaev, N.S.; Nechaev, M.S.; Oprunenko, Y.F.; Gam, F.; Saillard, J.-Y.; Kuznetsov, A.E. DFT Investigation of the eta(6) reversible arrow eta(6)-Inter-ring Haptotropic Rearrangement of the Group 8 Metals Complexes [(graphene)MCp](+) (M = Fe, Ru, Os). J. Phys. Chem. A 2021, 125, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, G.R. Atomic and Molecular Hydrogen in Interstellar Space. Space Sci. Rev. 1970, 10, 459–482. [Google Scholar] [CrossRef]
- Sancheza, M.; Ruette, F. Hydrogen atom chemisorption and diffusion on neutral and charged polycyclic aromatic hydrocarbon (PAH) flakes in the interstellar media. Chem. Phys. Lett. 2015, 640, 11–15. [Google Scholar] [CrossRef]
- Wang, Y.; Qian, H.-J.; Morokuma, K.; Irle, S. Coupled Cluster and Density Functional Theory Calculations of Atomic Hydrogen Chemisorption on Pyrene and Coronene as Model Systems for Graphene Hydrogenation. J. Phys. Chem. A 2012, 116, 7154–7160. [Google Scholar] [CrossRef]
- Beitollahi, A.; Sheikhaleslami, M.A.S. A novel approach for development of graphene structure in mesoporous carbon of high specific surface area. Carbon 2016, 107, 440–447. [Google Scholar] [CrossRef]
- Mutoh, M.; Abe, S.; Kusaka, T.; Nakamura, M.; Yoshida, Y.; Iida, J.; Hiroto Tachikawa, H. Density Functional Theory (DFT) Study on the Ternary Interaction System of the Fluorinated Ethylene Carbonate, Li+ and Graphene Model. Atoms 2016, 4, 4. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian-basis sets for molecular calculations. 1. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.; Handy, N. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Tachikawa, H. Hydrogen atom addition to the surface of graphene nanoflakes: A density functional theory study. Appl. Surf. Sci. 2017, 396, 1335–1342. [Google Scholar] [CrossRef]
- Feuchtgruber, H.; Helmich, F.P.; van Dishoeck, E.F.; Wright, C.M. Detection of Interstellar CH3. Astrophys. J. 2000, 535, L111–L114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knez, C.; Lacy, J.H.; Evans, N.J., II; van Dishoeck, E.F.; Richter, M.J. High-Resolution Mid-Infrared Spectroscopy of NGC 7538 IRS 1: Probing Chemistry in a Massive Young Stellar Object. Astrophys. J. 2009, 696, 471–483. [Google Scholar] [CrossRef] [Green Version]
- Tachikawa, H. Methyl radical addition to the surface of graphene nanoflakes: A density functional theory study. Surf. Sci. 2019, 679, 196–201. [Google Scholar] [CrossRef]
- Tachikawa, H.; Iyama, T. Mechanism of Hydrogen Storage in the Graphene Nanoflake-Lithium-H2 System. J. Phys. Chem. C 2019, 123, 8709–8716. [Google Scholar] [CrossRef]
- Umadevi, D.; Sastry, G.N. Molecular and Ionic Interaction with Graphene Nanoflakes: A Computational Investigation of CO2, H2O, Li, Mg, Li+, and Mg2+ Interaction with Polycyclic Aromatic Hydrocarbons. J. Phys. Chem. C 2011, 115, 9656–9667. [Google Scholar] [CrossRef]
- Tachikawa, H.; Yi, H.; Iyama, T.; Yamasaki, S.; Azumi, K. Hydrogen Storage Mechanism in Sodium-Based Graphene Nanoflakes: A Density Functional Theory Study. Hydrogen 2022, 3, 43–52. [Google Scholar] [CrossRef]
- Guo, Z.; Wei, W.; Chen, L.; Zhang, X.; Mei, S. Equilibrium Model of a Regional Hydrogen Market with Renewable Energy Based Suppliers and Transportation Costs. Energy 2021, 220, 119608. [Google Scholar] [CrossRef]
- Vijayakumar, V.; Jenn, A.; Fulton, L. Low Carbon Scenario Analysis of a Hydrogen-Based Energy Transition for On-Road Transportation in California. Energies 2021, 14, 7163. [Google Scholar] [CrossRef]
- Lobo, R.; Alvarez, N.; Shanov, V. Hydrogen Nanometrology in Advanced Carbon Nanomaterial Electrodes. Nanomaterials 2021, 11, 1079. [Google Scholar] [CrossRef]
- Lobo, R.; Ribeiro, J.H.F.; Inok, F. Hydrogen Uptake and Release in Carbon Nanotube Electrocatalysts. Nanomaterials 2021, 11, 975. [Google Scholar] [CrossRef]
- Wu, R.; Zhang, X.; Liu, Y.; Zhang, L.; Hu, J.; Gao, M.; Pan, H. A Unique Double-Layered Carbon Nanobowl-Confined Lithium Borohydride for Highly Reversible Hydrogen Storage. Small 2020, 16, 202001963. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Tam, J.; Singh, C.V. A First Principles Study of Hydrogen Storage on Lithium Decorated Two Dimensional Carbon Allotropes. Int. J. Hydrogen Energy 2015, 40, 6128–6136. [Google Scholar] [CrossRef]
- Tachikawa, H.; Izumi, Y.; Iyama, T.; Azumi, K. Molecular Design of a Reversible Hydrogen Storage Device Composed of the Graphene Nanoflake-Magnesium-H2 System. ACS Omega 2021, 6, 7778–7785. [Google Scholar] [CrossRef] [PubMed]
- Tachikawa, H. Reaction mechanism of an intracluster SN2 reaction induced by electron capture. Phys. Chem. Chem. Phys. 2022, 24, 3941–3950. [Google Scholar] [CrossRef]
- Tachikawa, H. Reactions of Photoionization-Induced CO-H2O Cluster: Direct Ab Initio Molecular Dynamics Study. ACS Omega 2021, 6, 16688–16695. [Google Scholar] [CrossRef]
- Tachikawa, H. Formation of Hydrogen Peroxide from O-(H2O)n Clusters. J. Phys. Chem. A 2021, 125, 4598–4605. [Google Scholar] [CrossRef]
Figure 1.
Models of graphene nanoflakes used in the present calculations. GNF(n) means graphene nanoflake composed of n benzene rings. GNF(19) and GNF(37) are circumcoronene (C54H18) and circumcircumcoronene (C96H24), respectively.
Figure 1.
Models of graphene nanoflakes used in the present calculations. GNF(n) means graphene nanoflake composed of n benzene rings. GNF(19) and GNF(37) are circumcoronene (C54H18) and circumcircumcoronene (C96H24), respectively.

Figure 2.
Binding structure of hydrogen atom to graphene nanoflake, H-GNF(19) calculated at the CAM-B3LYP/6-311G(d, p) level. Reprinted with permission from [20] (Tachikawa, Appl. Surf. Sci. 2017, 396, 1335–1342). Copyright 2017 Elsevier.
Figure 2.
Binding structure of hydrogen atom to graphene nanoflake, H-GNF(19) calculated at the CAM-B3LYP/6-311G(d, p) level. Reprinted with permission from [20] (Tachikawa, Appl. Surf. Sci. 2017, 396, 1335–1342). Copyright 2017 Elsevier.
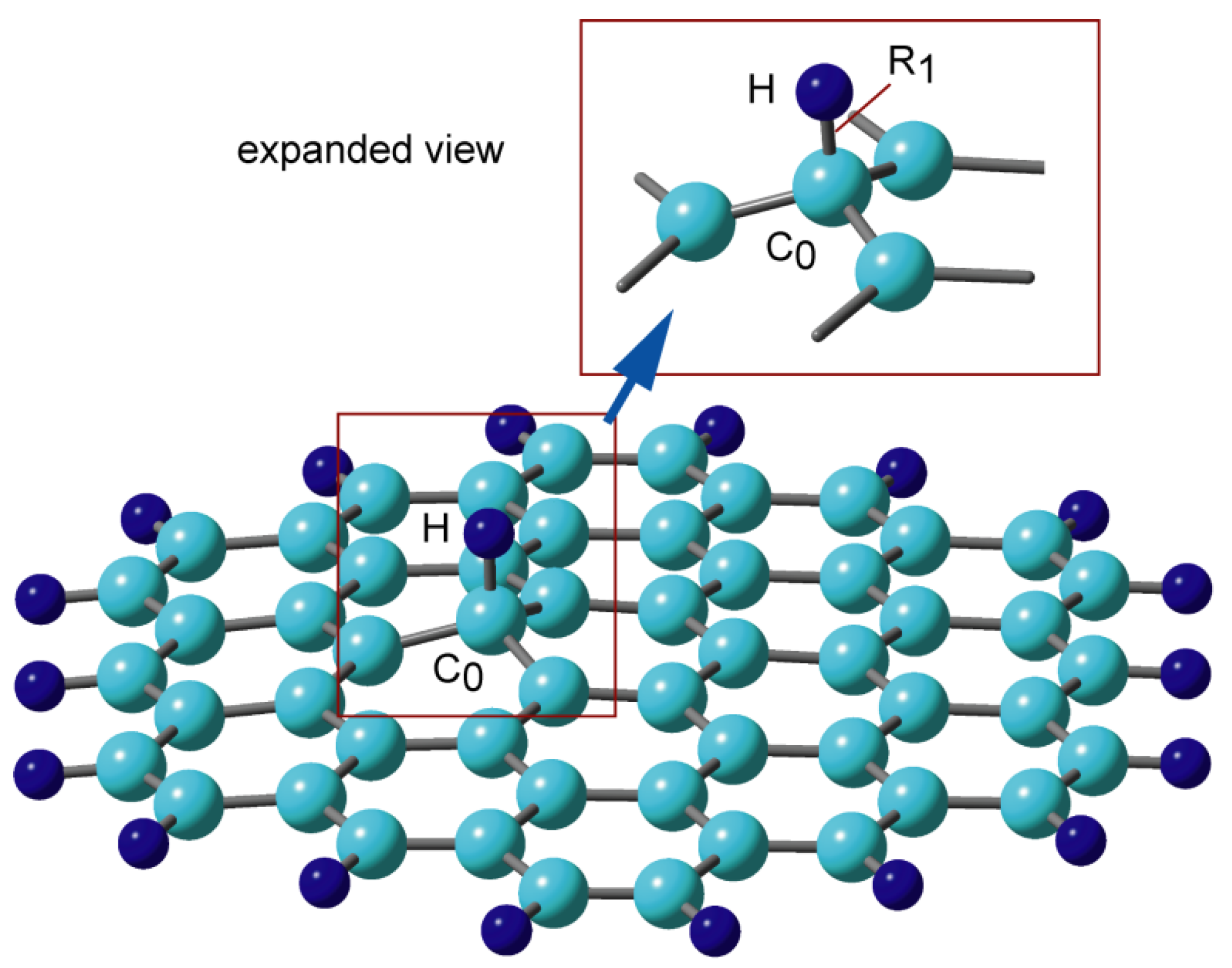
Figure 3.
(A) Potential energy curves for the hydrogen atom (H) or proton (H+) addition to GNF(19), and (B) spatial distributions of spin density in H-GNF(19) along the reaction coordinate (vdW, TS, and PD states). The calculations were carried out at the CAM-B3LYP/6-311G(d,p) level. Reprinted with permission from [20] (Tachikawa, Appl. Surf. Sci. 2017, 396, 1335–1342). Copyright 2017 Elsevier.
Figure 3.
(A) Potential energy curves for the hydrogen atom (H) or proton (H+) addition to GNF(19), and (B) spatial distributions of spin density in H-GNF(19) along the reaction coordinate (vdW, TS, and PD states). The calculations were carried out at the CAM-B3LYP/6-311G(d,p) level. Reprinted with permission from [20] (Tachikawa, Appl. Surf. Sci. 2017, 396, 1335–1342). Copyright 2017 Elsevier.
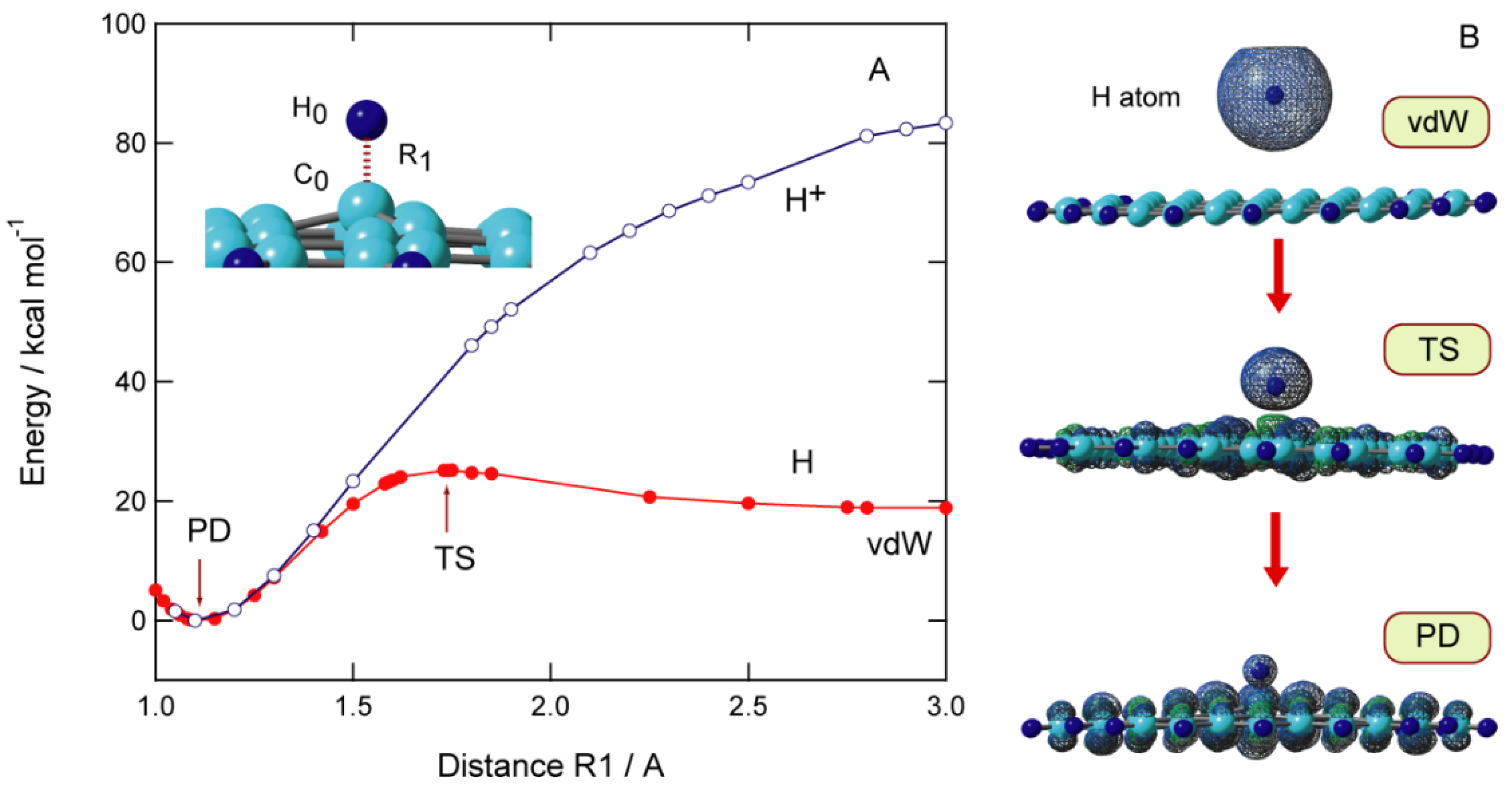
Figure 4.
Binding energies (in kcal/mol) and activation energies (in kcal/mol) of hydrogen atom addition to GNF(n). The calculations were carried out at the CAM-B3LYP/6-31G(d) and 6-311G(d, p) levels of theory. Reprinted with permission from [20] (Tachikawa, Appl. Surf. Sci. 2017, 396, 1335–1342). Copyright 2017 Elsevier.
Figure 4.
Binding energies (in kcal/mol) and activation energies (in kcal/mol) of hydrogen atom addition to GNF(n). The calculations were carried out at the CAM-B3LYP/6-31G(d) and 6-311G(d, p) levels of theory. Reprinted with permission from [20] (Tachikawa, Appl. Surf. Sci. 2017, 396, 1335–1342). Copyright 2017 Elsevier.
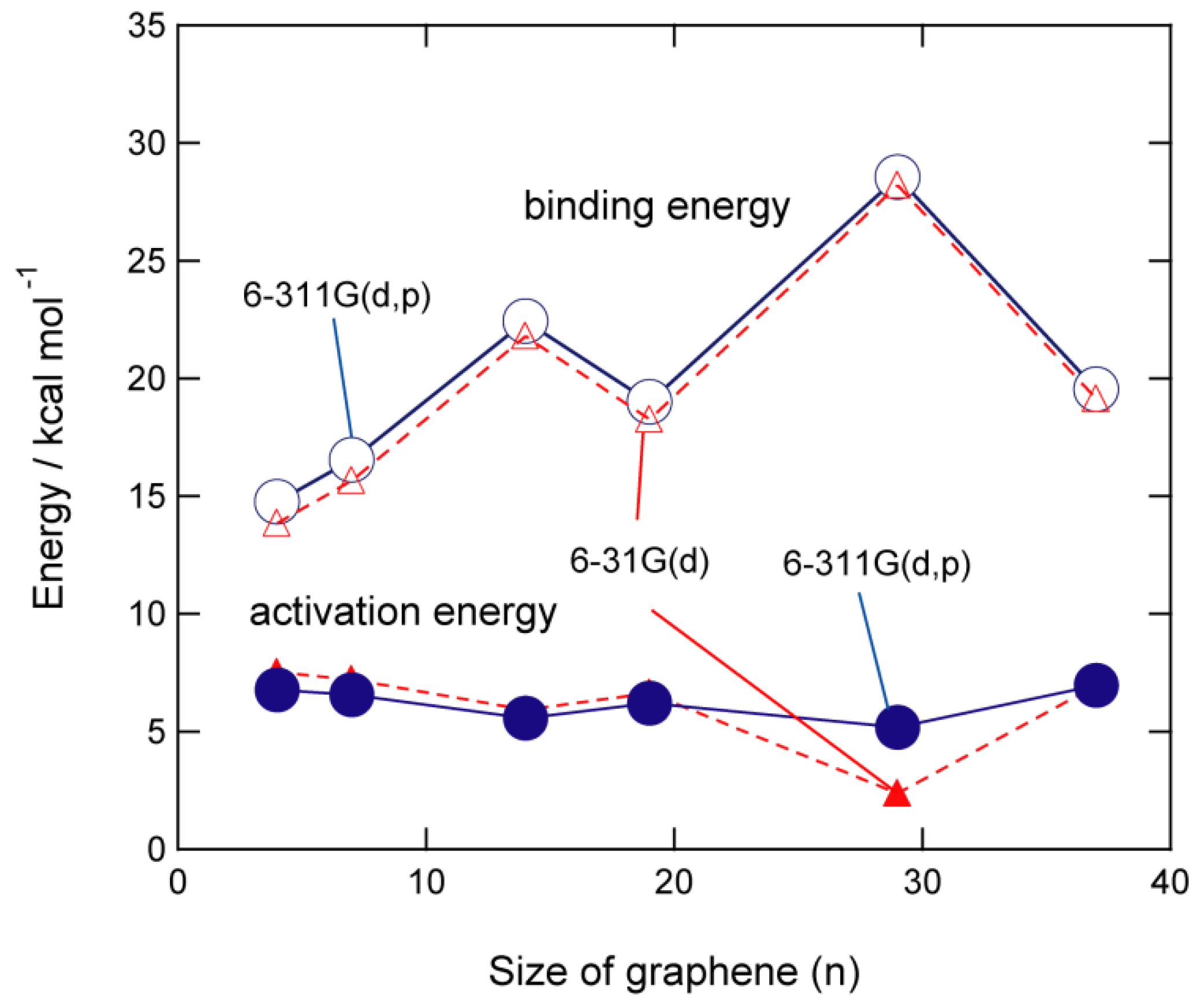
Figure 5.
Schematic illustration of the interaction of hydrogen atom with PAH (graphene) surface. Reprinted with permission from [20] (Tachikawa, Appl. Surf. Sci. 2017, 396, 1335–1342). Copyright 2017 Elsevier.
Figure 5.
Schematic illustration of the interaction of hydrogen atom with PAH (graphene) surface. Reprinted with permission from [20] (Tachikawa, Appl. Surf. Sci. 2017, 396, 1335–1342). Copyright 2017 Elsevier.
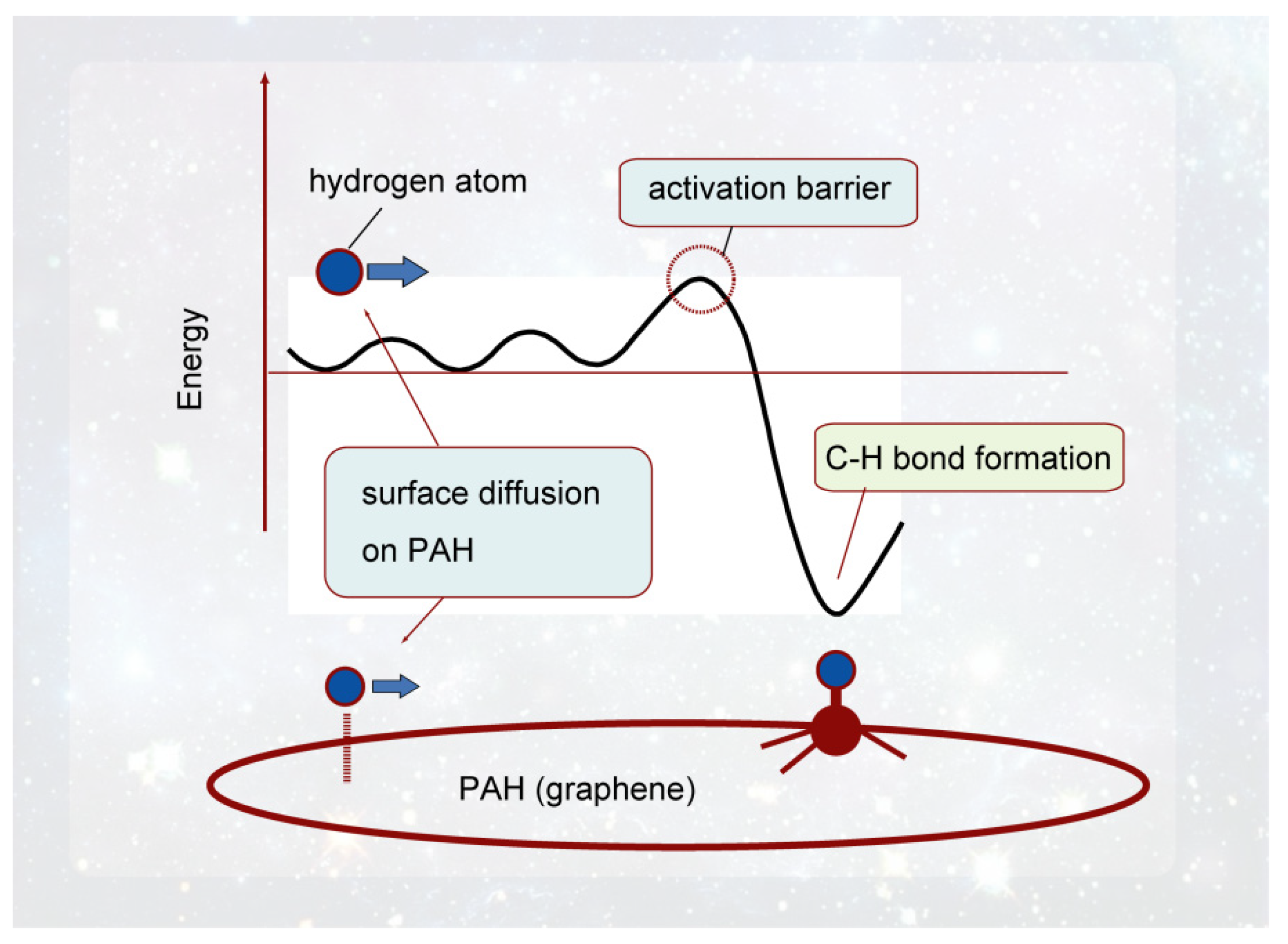
Figure 6.
Binding structure of methyl radical to graphene nanoflake, CH3-GNF(19) calculated at the CAM-B3LYP/6-311G(d, p) level. Reprinted with permission from [23] (Tachikawa, Surf. Sci. 2019, 679, 196–201). Copyright 2019 Elsevier.
Figure 6.
Binding structure of methyl radical to graphene nanoflake, CH3-GNF(19) calculated at the CAM-B3LYP/6-311G(d, p) level. Reprinted with permission from [23] (Tachikawa, Surf. Sci. 2019, 679, 196–201). Copyright 2019 Elsevier.

Figure 7.
(A) Intrinsic reaction coordinate (IRC) for the CH3 addition reaction to GNF(19), and (B) spatial distributions of spin density in CH3-GNF(19) along the reaction coordinate (vdW, TS, and PD states). The calculations were carried out at the CAM-B3LYP/6-311G(d,p) level. The values are (A) relative energies in kcal/mol and intermolecular distance (R1) in Å. Reprinted with permission from [23] (Tachikawa, Surf. Sci. 2019, 679, 196–201). Copyright 2019 Elsevier.
Figure 7.
(A) Intrinsic reaction coordinate (IRC) for the CH3 addition reaction to GNF(19), and (B) spatial distributions of spin density in CH3-GNF(19) along the reaction coordinate (vdW, TS, and PD states). The calculations were carried out at the CAM-B3LYP/6-311G(d,p) level. The values are (A) relative energies in kcal/mol and intermolecular distance (R1) in Å. Reprinted with permission from [23] (Tachikawa, Surf. Sci. 2019, 679, 196–201). Copyright 2019 Elsevier.
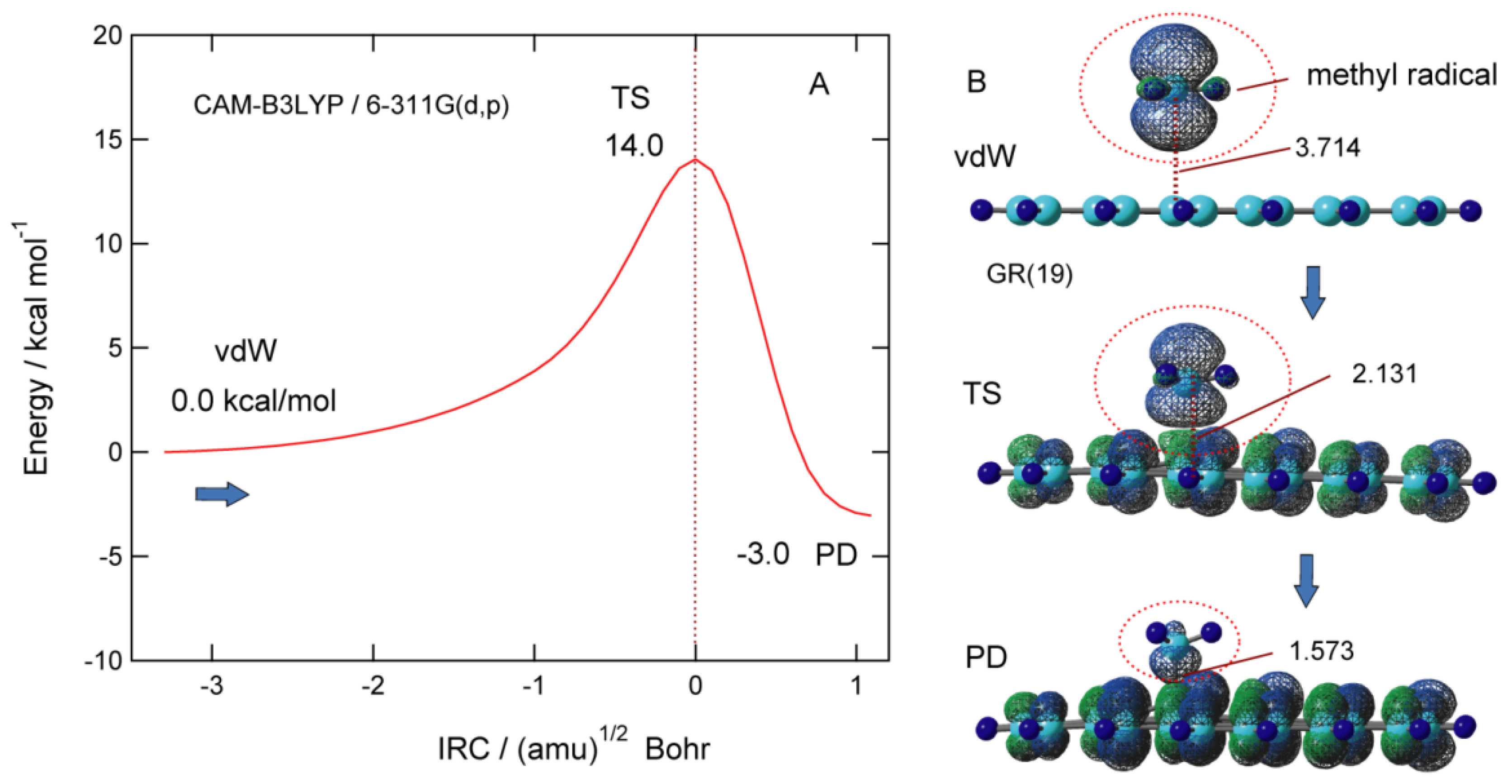
Figure 8.
Schematic illustration of the interaction of CH3 with PAH (graphene) surface. Reprinted with permission from [23] (Tachikawa, Surf. Sci. 2019, 679, 196–201). Copyright 2019 Elsevier.
Figure 8.
Schematic illustration of the interaction of CH3 with PAH (graphene) surface. Reprinted with permission from [23] (Tachikawa, Surf. Sci. 2019, 679, 196–201). Copyright 2019 Elsevier.

Figure 9.
Structure of transition state (TS) in hydrogen addition reaction: H + HCCH → TS → H2CCH calculated at the CAM-B3LYP/6-311G(d,p) level. Bond lengths and angles are in Å and degrees, respectively. The values for the GNF surface reaction are given in parenthesis.
Figure 9.
Structure of transition state (TS) in hydrogen addition reaction: H + HCCH → TS → H2CCH calculated at the CAM-B3LYP/6-311G(d,p) level. Bond lengths and angles are in Å and degrees, respectively. The values for the GNF surface reaction are given in parenthesis.
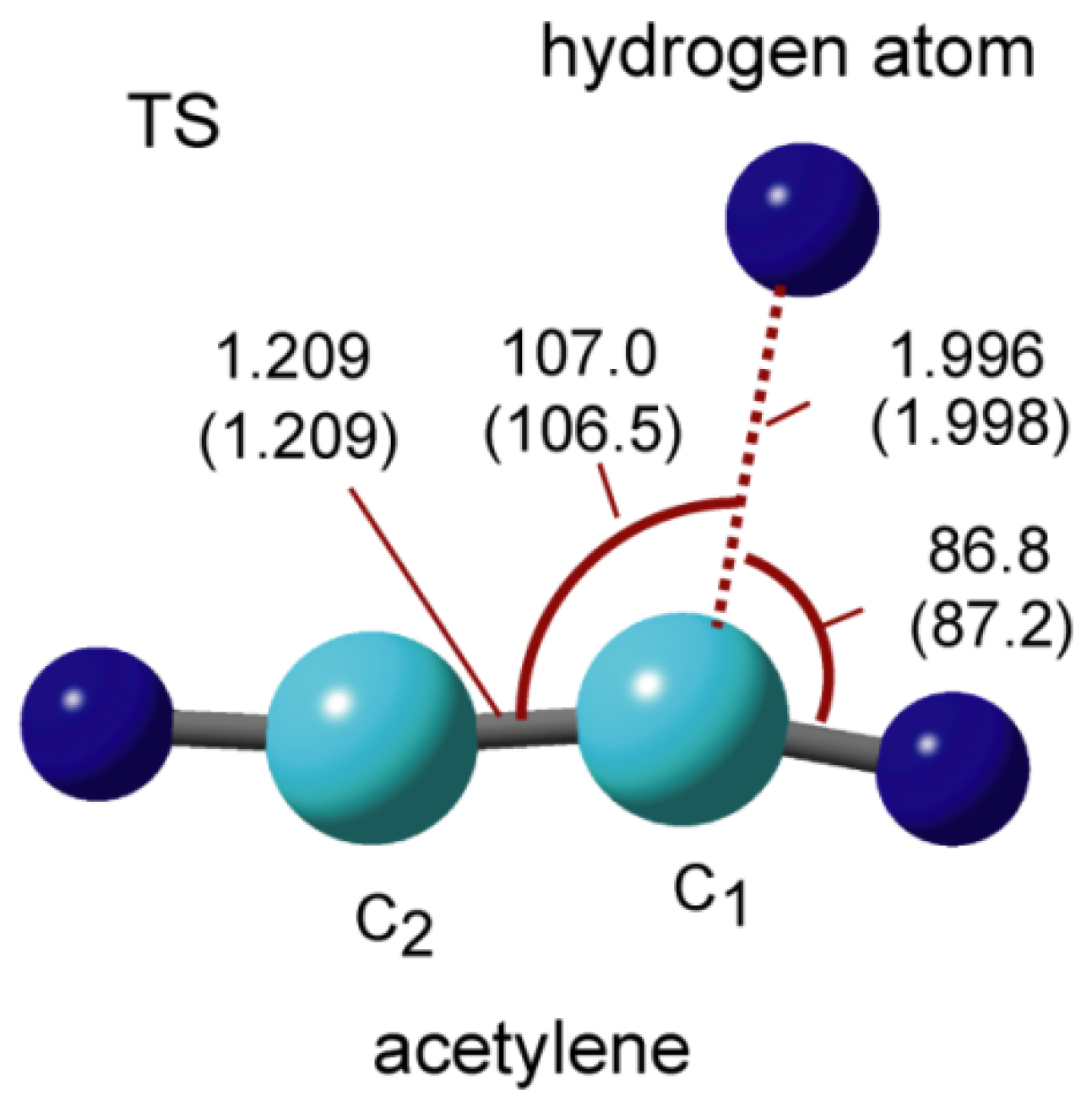
Figure 10.
(A) Intrinsic reaction coordinate (IRC) for hydrogen addition reaction on GNF surface: H + HCCH → TS → H2CCH, and (B) structures along IRC. The calculations were carried out at the CAM-B3LYP/6-311G(d,p) level. Bond lengths are in Å.
Figure 10.
(A) Intrinsic reaction coordinate (IRC) for hydrogen addition reaction on GNF surface: H + HCCH → TS → H2CCH, and (B) structures along IRC. The calculations were carried out at the CAM-B3LYP/6-311G(d,p) level. Bond lengths are in Å.

Figure 11.
Optimized structures of GNF-Li(atom)-(H2)n (n = 1, 2, 3, 4, 7, and 11). The calculations were performed at the CAM-B3LYP/6-311G(d,p) level. Reprinted with permission from [24] (Tachikawa et al. J. Phys. Chem. C 2019, 123, 14, 8709–8716). Copyright 2019 American Chemical Society.
Figure 11.
Optimized structures of GNF-Li(atom)-(H2)n (n = 1, 2, 3, 4, 7, and 11). The calculations were performed at the CAM-B3LYP/6-311G(d,p) level. Reprinted with permission from [24] (Tachikawa et al. J. Phys. Chem. C 2019, 123, 14, 8709–8716). Copyright 2019 American Chemical Society.

Figure 12.
Binding energies of H2 to GNF-M (per H2 molecule) plotted as functions of n (M = Li, Li+, Na, Na+, and K+). Calculations for M = Li, Li+, Na, Na+ were carried out at the CAM-B3LYP/6-311G(d,p) level and for M = K+ at the CAM-B3LYP/6-31G(d) level. Reprinted with permission from [26] (Tachikawa et al. Hydrogen 2022, 3, 43–52.)
Figure 12.
Binding energies of H2 to GNF-M (per H2 molecule) plotted as functions of n (M = Li, Li+, Na, Na+, and K+). Calculations for M = Li, Li+, Na, Na+ were carried out at the CAM-B3LYP/6-311G(d,p) level and for M = K+ at the CAM-B3LYP/6-31G(d) level. Reprinted with permission from [26] (Tachikawa et al. Hydrogen 2022, 3, 43–52.)

Figure 13.
Optimized structures of GNF-Na (atom)-(H2)n (n = 1–6). The calculations were performed at the CAM-B3LYP/6-311G(d,p) level. Reprinted with permission from [26] (Tachikawa et al. Hydrogen 2022, 3, 43–52).
Figure 13.
Optimized structures of GNF-Na (atom)-(H2)n (n = 1–6). The calculations were performed at the CAM-B3LYP/6-311G(d,p) level. Reprinted with permission from [26] (Tachikawa et al. Hydrogen 2022, 3, 43–52).

Figure 14.
(A) Optimized structure of the GNF-Mg2+ calculated at the CAM-B3LYP/6-311G(d,p) level with a GNF composed of seven benzene rings. The notation h means height of Mg species from GNF surface (in Å), and (B) Binding energies of H2 to GNF-Mg2+ and GNF-Mg+ (per H2 molecule) plotted as a function of the number of H2 molecules (n). Dashed lines indicate the binding energies of H2 to GNF37-Mgm+ (m = 1 and 2). Reprinted with permission from [34] (Tachikawa et al. ACS Omega 2021, 6, 11, 7778–7785). Copyright 2021 American Chemical Society.
Figure 14.
(A) Optimized structure of the GNF-Mg2+ calculated at the CAM-B3LYP/6-311G(d,p) level with a GNF composed of seven benzene rings. The notation h means height of Mg species from GNF surface (in Å), and (B) Binding energies of H2 to GNF-Mg2+ and GNF-Mg+ (per H2 molecule) plotted as a function of the number of H2 molecules (n). Dashed lines indicate the binding energies of H2 to GNF37-Mgm+ (m = 1 and 2). Reprinted with permission from [34] (Tachikawa et al. ACS Omega 2021, 6, 11, 7778–7785). Copyright 2021 American Chemical Society.
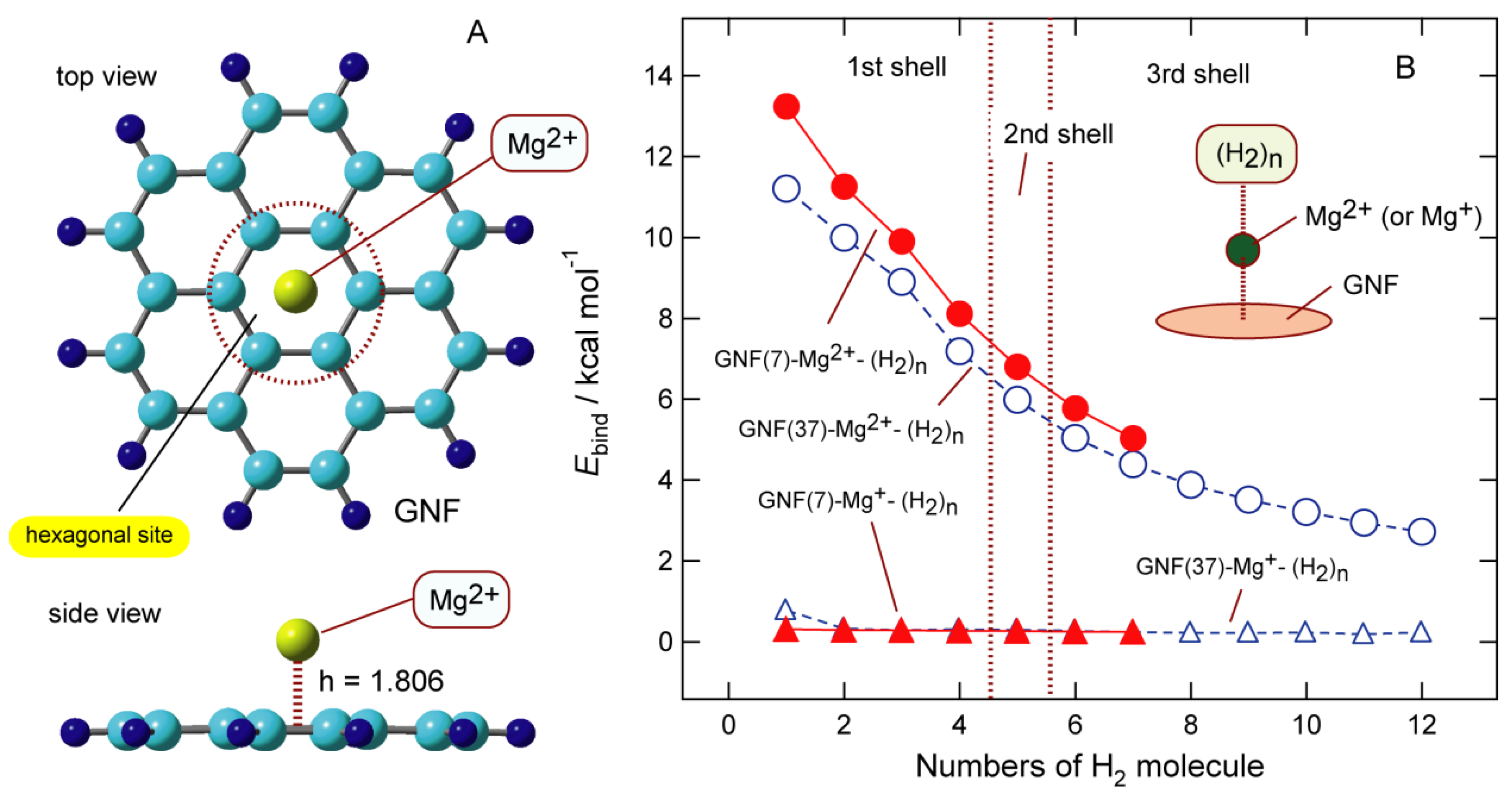
Figure 15.
Snapshots of GNF-Mg+-(H2)4 following the electron capture of GNF-Mg2+-(H2)4. The initial geometry was taken from the optimized structure of GNF-Mg2+-(H2)4 (CAM-B3LYP/6-31G(d) level). <R> and h mean average distance of H2 molecules from Mg+ and height of Mg from GNF surface (in Å). Direct AIMD calculations were carried out at the CAM-B3LYP/6-31G(d) level. Reprinted with permission from [33] (Tachikawa et al. ACS Omega 2021, 6, 11, 7778–7785). Copyright 2021 American Chemical Society.
Figure 15.
Snapshots of GNF-Mg+-(H2)4 following the electron capture of GNF-Mg2+-(H2)4. The initial geometry was taken from the optimized structure of GNF-Mg2+-(H2)4 (CAM-B3LYP/6-31G(d) level). <R> and h mean average distance of H2 molecules from Mg+ and height of Mg from GNF surface (in Å). Direct AIMD calculations were carried out at the CAM-B3LYP/6-31G(d) level. Reprinted with permission from [33] (Tachikawa et al. ACS Omega 2021, 6, 11, 7778–7785). Copyright 2021 American Chemical Society.

Figure 16.
Snapshots of GNF- Mg2+-(H2)4 following the hole capture of GNF-Mg+-(H2)4. Direct AIMD calculations were carried out at the CAM-B3LYP/6-31G(d) level. The initial geometry was taken from one of the structures in the simulation of GNF-Mg+-(H2)4. The distances and heights are in Å. Reprinted with permission from [33] (Tachikawa et al. ACS Omega 2021, 6, 11, 7778–7785). Copyright 2021 American Chemical Society.
Figure 16.
Snapshots of GNF- Mg2+-(H2)4 following the hole capture of GNF-Mg+-(H2)4. Direct AIMD calculations were carried out at the CAM-B3LYP/6-31G(d) level. The initial geometry was taken from one of the structures in the simulation of GNF-Mg+-(H2)4. The distances and heights are in Å. Reprinted with permission from [33] (Tachikawa et al. ACS Omega 2021, 6, 11, 7778–7785). Copyright 2021 American Chemical Society.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Activation energies of radical addition reaction to graphene nanoflake, E(TS), reaction energies leading to PD, E(PD), and binding energies of vdW complexes, E(vdW) (in kcal/mol). Notations are CH3, Et, n-Pr, iso-Pr, n-Bu, sec-Bu, tert-Bu, iso-Bu mean methyl, ethyl, iso-propyl, normal-butyl, secondary butyl, tertiary-butyl radicals, respectively. Zero level of relative energy corresponds to dissociation limit (radical + GNF). The calculations were carried out at the CAM-B3LYP/6-311G(d,p) level. Halogen atoms (F and Cl) can bind to GNF without activation barrier (i.e., E(TS) = 0.0 kcal/mol).
Table 1.
Activation energies of radical addition reaction to graphene nanoflake, E(TS), reaction energies leading to PD, E(PD), and binding energies of vdW complexes, E(vdW) (in kcal/mol). Notations are CH3, Et, n-Pr, iso-Pr, n-Bu, sec-Bu, tert-Bu, iso-Bu mean methyl, ethyl, iso-propyl, normal-butyl, secondary butyl, tertiary-butyl radicals, respectively. Zero level of relative energy corresponds to dissociation limit (radical + GNF). The calculations were carried out at the CAM-B3LYP/6-311G(d,p) level. Halogen atoms (F and Cl) can bind to GNF without activation barrier (i.e., E(TS) = 0.0 kcal/mol).
| Radical | E(TS) | E(PD) | E(vdW) |
|---|---|---|---|
| CH3 | 13.8 | −3.6 | −0.3 |
| Et | 14.7 | 1.1 | −0.6 |
| n-Pr | 15.2 | 1.3 | −0.2 |
| iso-Pr | 17.6 | 8.1 | −0.8 |
| n-Bu | 15.2 | 1.4 | −0.2 |
| sec-Bu | 18.7 | 9.7 | −0.4 |
| tert-Bu | 22.1 | 16.6 | −0.9 |
| iso-Bu | 20.1 | 9.0 | −0.7 |
| F | 0.0 | −29.4 | -- |
| Cl | 0.0 | −6.5 | -- |
Table 2.
Effects of GNF surface on energetics for hydrogen addition reaction to acetylene: H(atom) + HCCH → H2CCH (radical). Relative energies with respect to vdW state are given in parenthesis. The calculations were carried out at the CAM-B3LYP/6-311G(d,p) level.
Table 2.
Effects of GNF surface on energetics for hydrogen addition reaction to acetylene: H(atom) + HCCH → H2CCH (radical). Relative energies with respect to vdW state are given in parenthesis. The calculations were carried out at the CAM-B3LYP/6-311G(d,p) level.
| State | Vacuo | on GNF |
|---|---|---|
| RC (H + HCCH) | 0.0 | 0.0 (1.3) |
| vdW(1) | -- | −1.3 (0.0) |
| TS | 1.3 | 0.2 (1.5) |
| vdW(2) | -- | −48.7 (−47.4) |
| PD (H2CCH) | −47.6 | −47.6 (−46.3) |
Table 3.
Binding energies of M–GNF and M+–GNF (Ebind in kcal/mol, M = Li and Na), M–GNF-surface distances (h in Å), and NPA-determined atomic charges for M on GNF, calculated at the CAM-B3LYP/6-311G(d,p) level.
Table 3.
Binding energies of M–GNF and M+–GNF (Ebind in kcal/mol, M = Li and Na), M–GNF-surface distances (h in Å), and NPA-determined atomic charges for M on GNF, calculated at the CAM-B3LYP/6-311G(d,p) level.
| Ebind | Height (h) | NPA | |
|---|---|---|---|
| GNF-Li | 17.1 | 1.736 | +0.929 |
| GNF-Li+ | 52.8 | 1.771 | +0.937 |
| GNF-Na | 4.4 | 2.247 | +0.978 |
| GNF-Na+ | 37.5 | 2.288 | +0.979 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tachikawa, H.; Iyama, T. Reactions of Graphene Nano-Flakes in Materials Chemistry and Astrophysics. Physchem 2022, 2, 145-162. https://doi.org/10.3390/physchem2020011
AMA Style
Tachikawa H, Iyama T. Reactions of Graphene Nano-Flakes in Materials Chemistry and Astrophysics. Physchem. 2022; 2(2):145-162. https://doi.org/10.3390/physchem2020011
Chicago/Turabian StyleTachikawa, Hiroto, and Tetsuji Iyama. 2022. "Reactions of Graphene Nano-Flakes in Materials Chemistry and Astrophysics" Physchem 2, no. 2: 145-162. https://doi.org/10.3390/physchem2020011