
Computational Study of Crystallography, Defects, Ion Migration and Dopants in Almandine Garnet



Abstract
:
1. Introduction
2. Computational Methods
3. Results and Discussion
3.1. Crystal Structure of Almandine Garnet
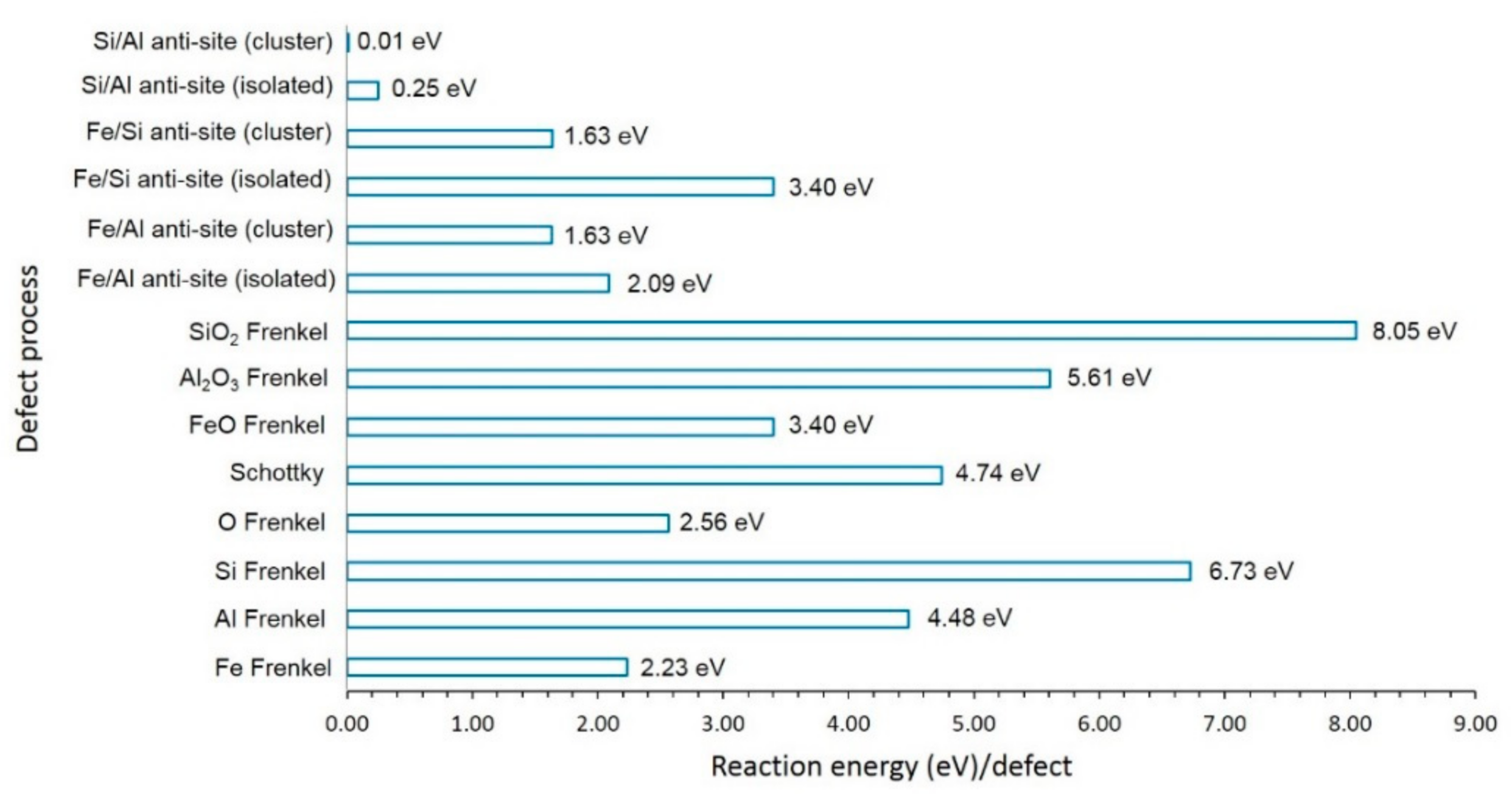
3.2. Intrinsic Defects
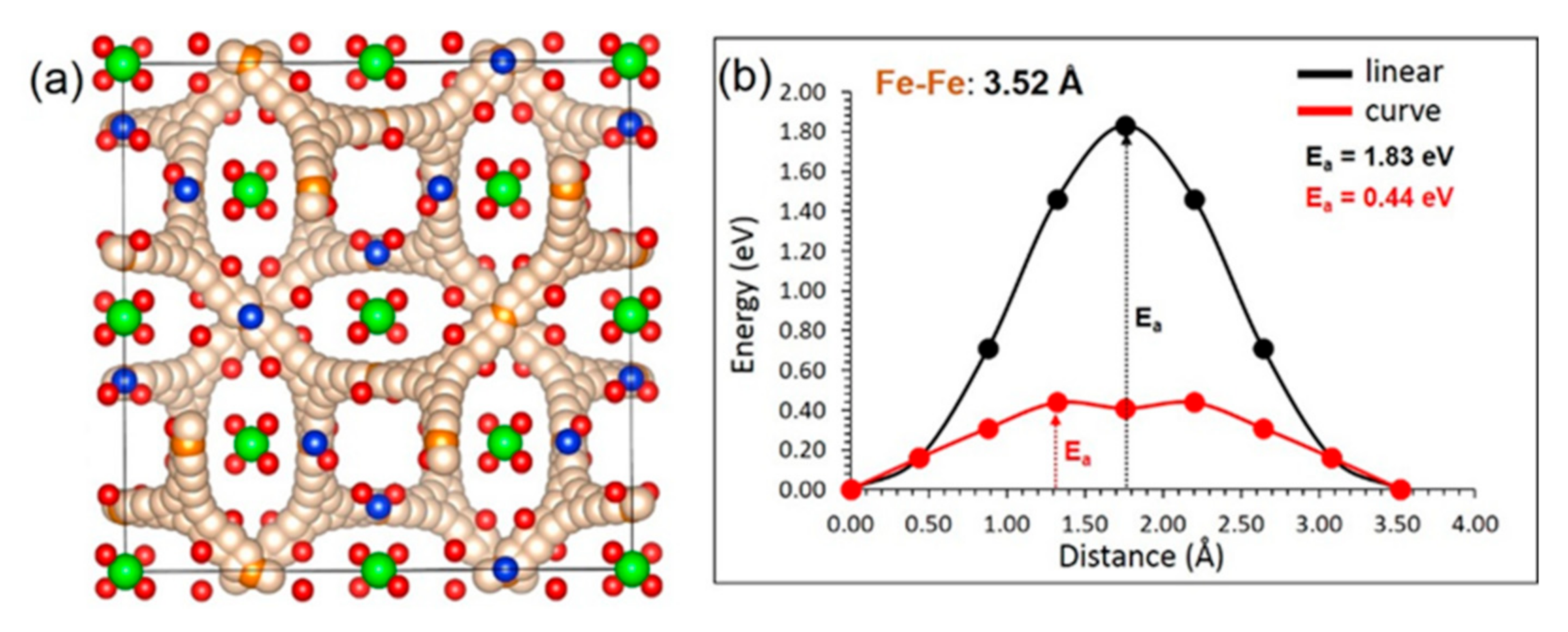
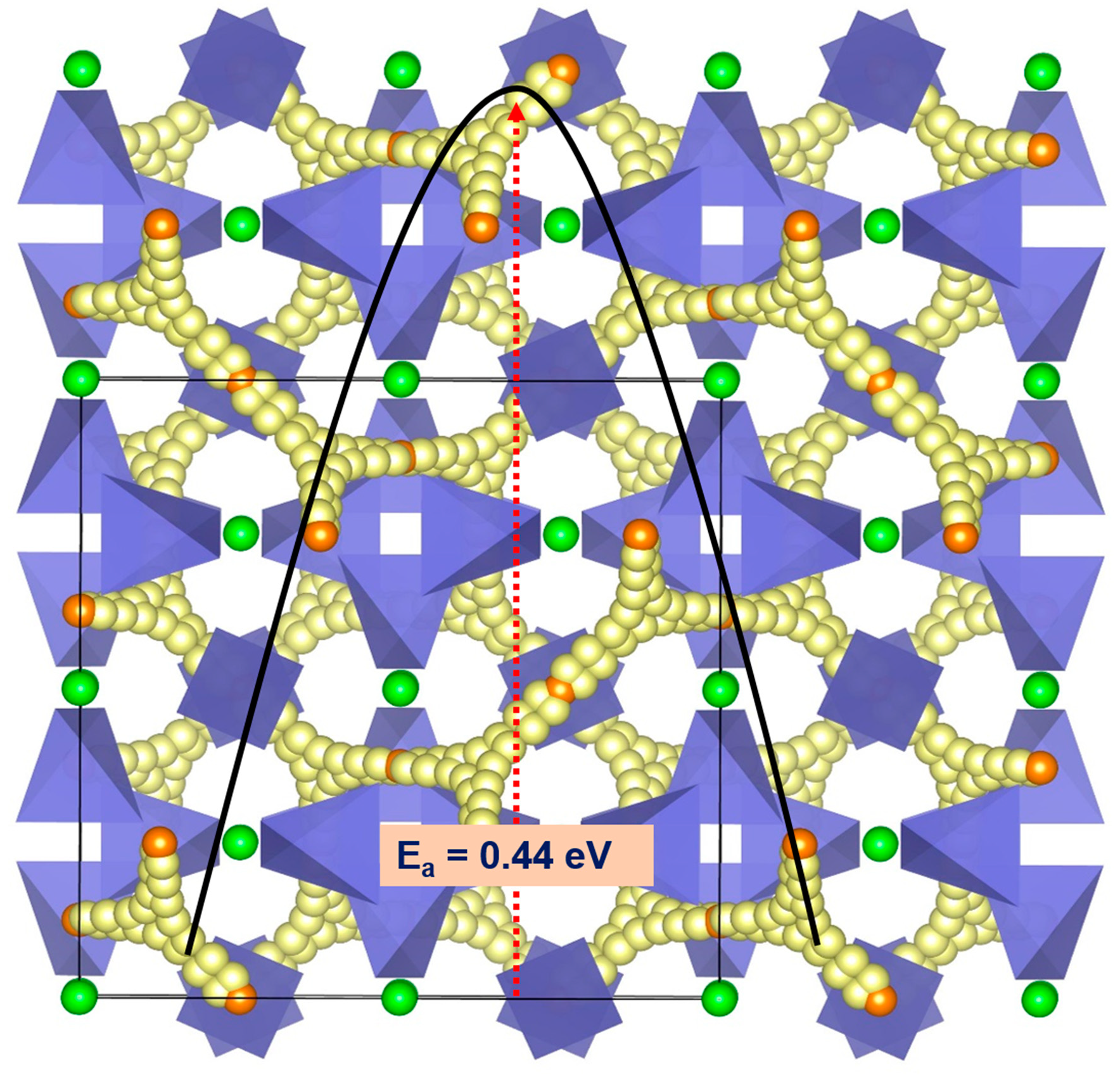
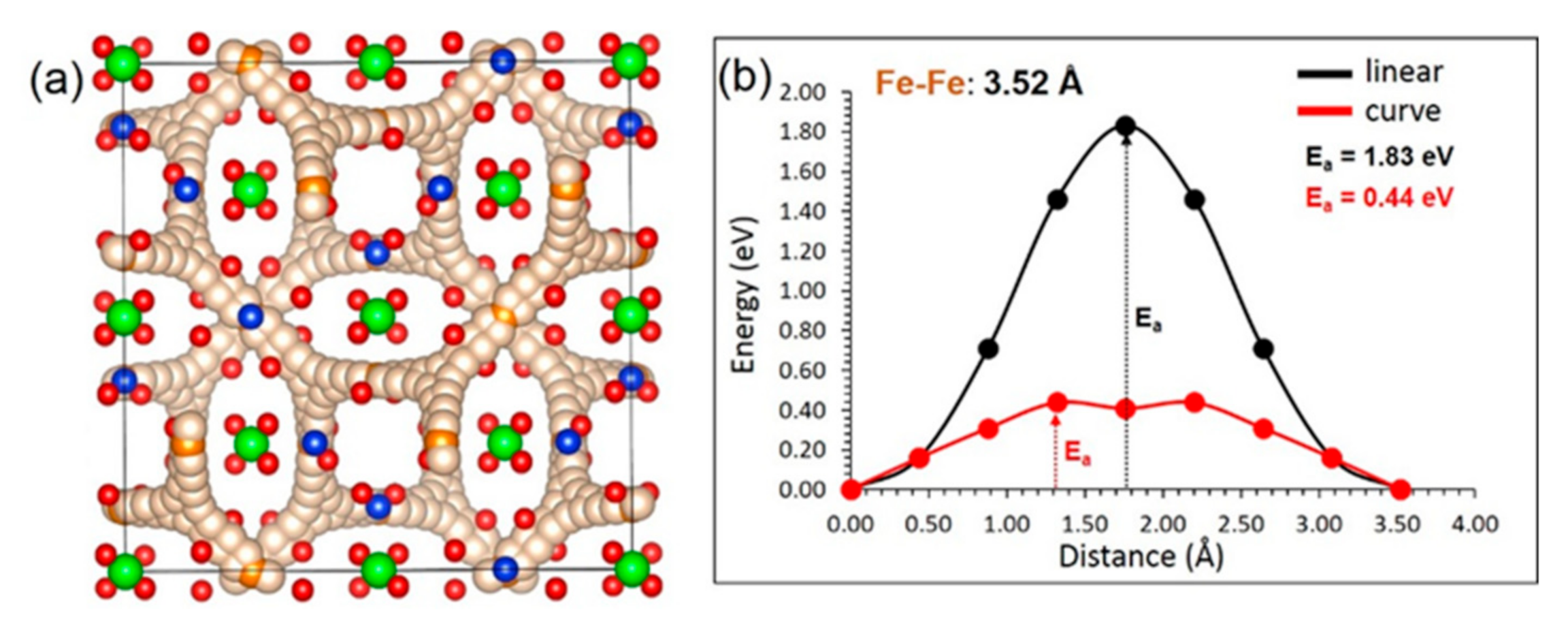
3.3. Fe Ion Migration
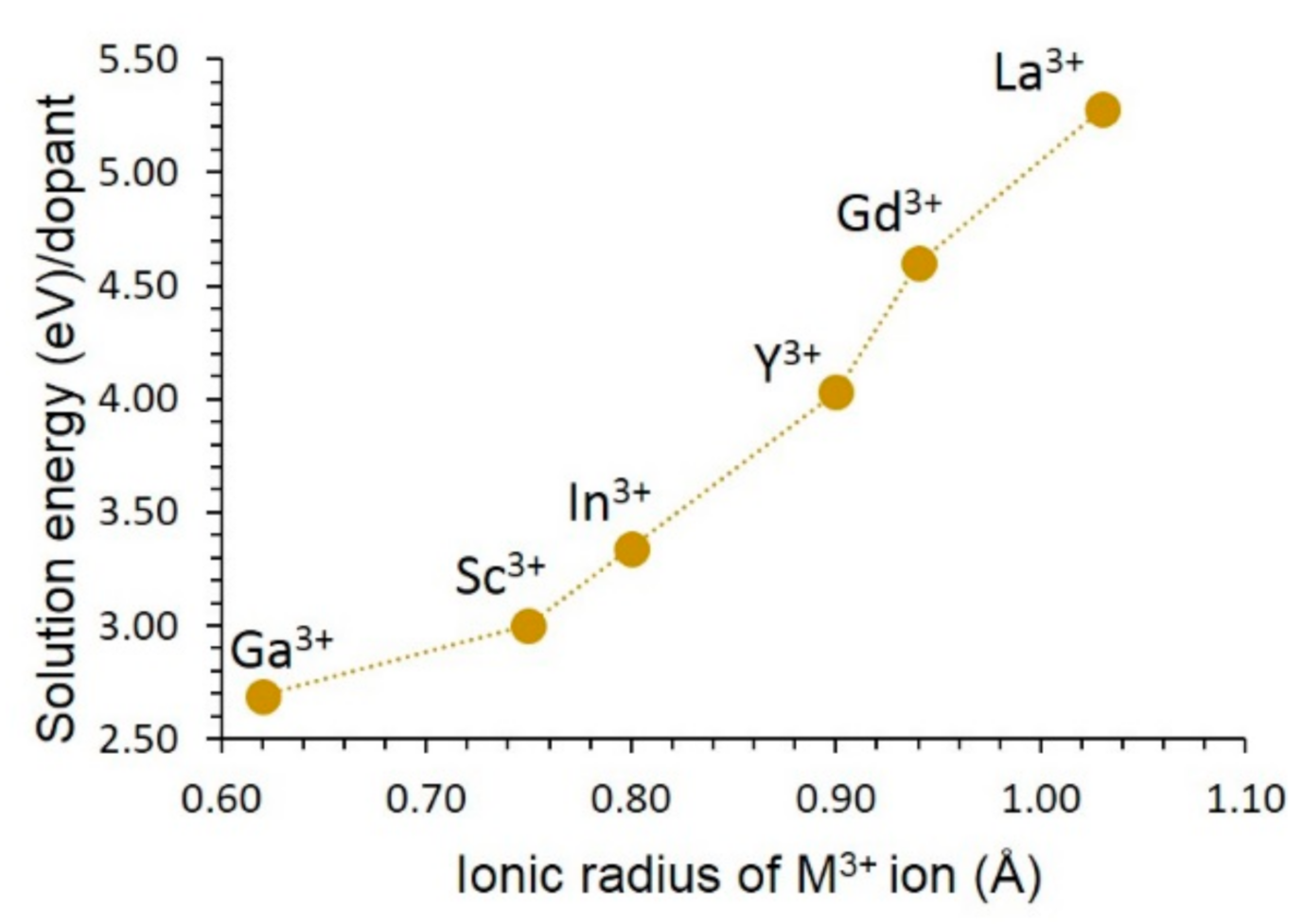
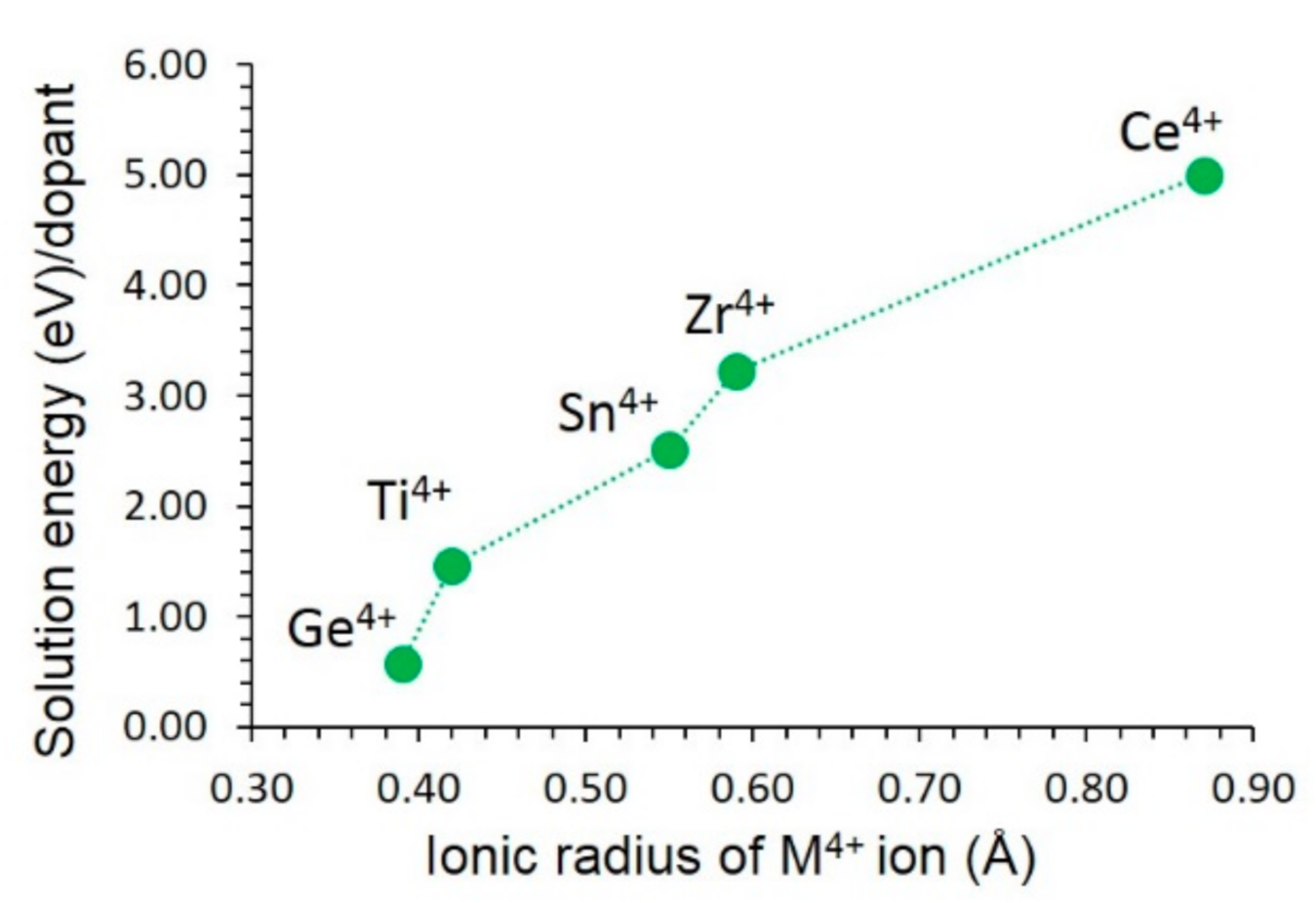
3.4. Solution of Dopants
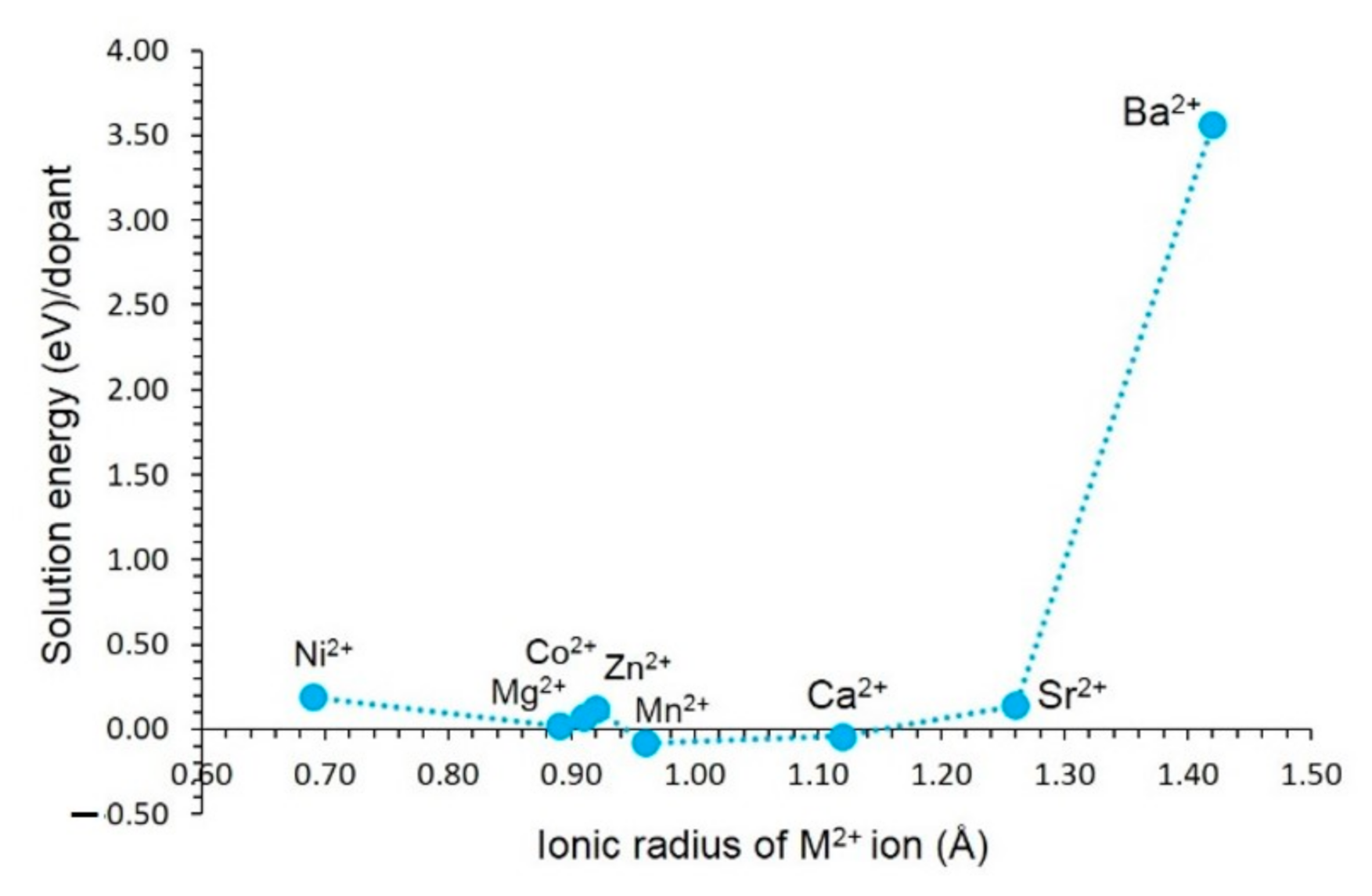
3.4.1. Divalent Dopants
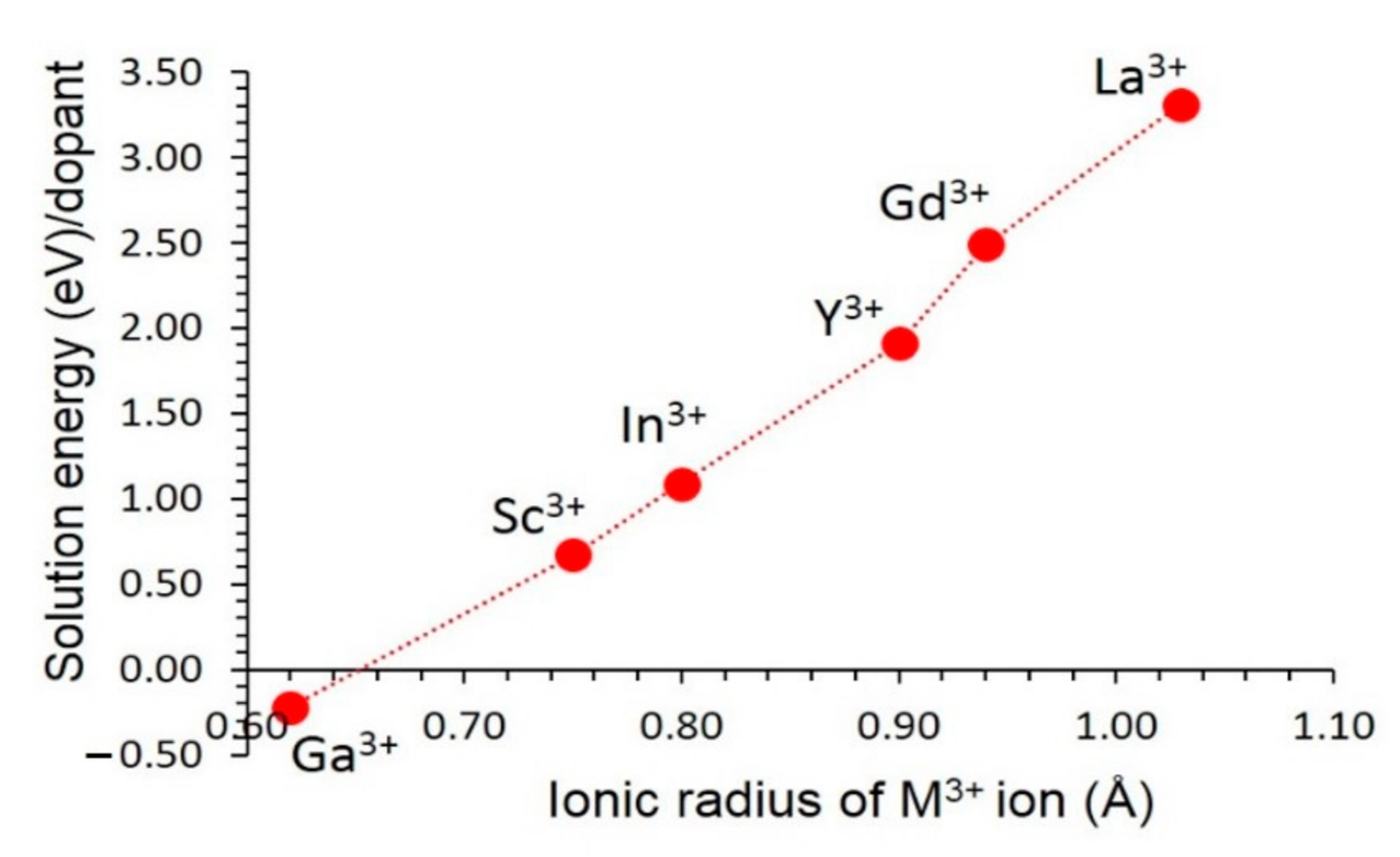
3.4.2. Trivalent Dopants
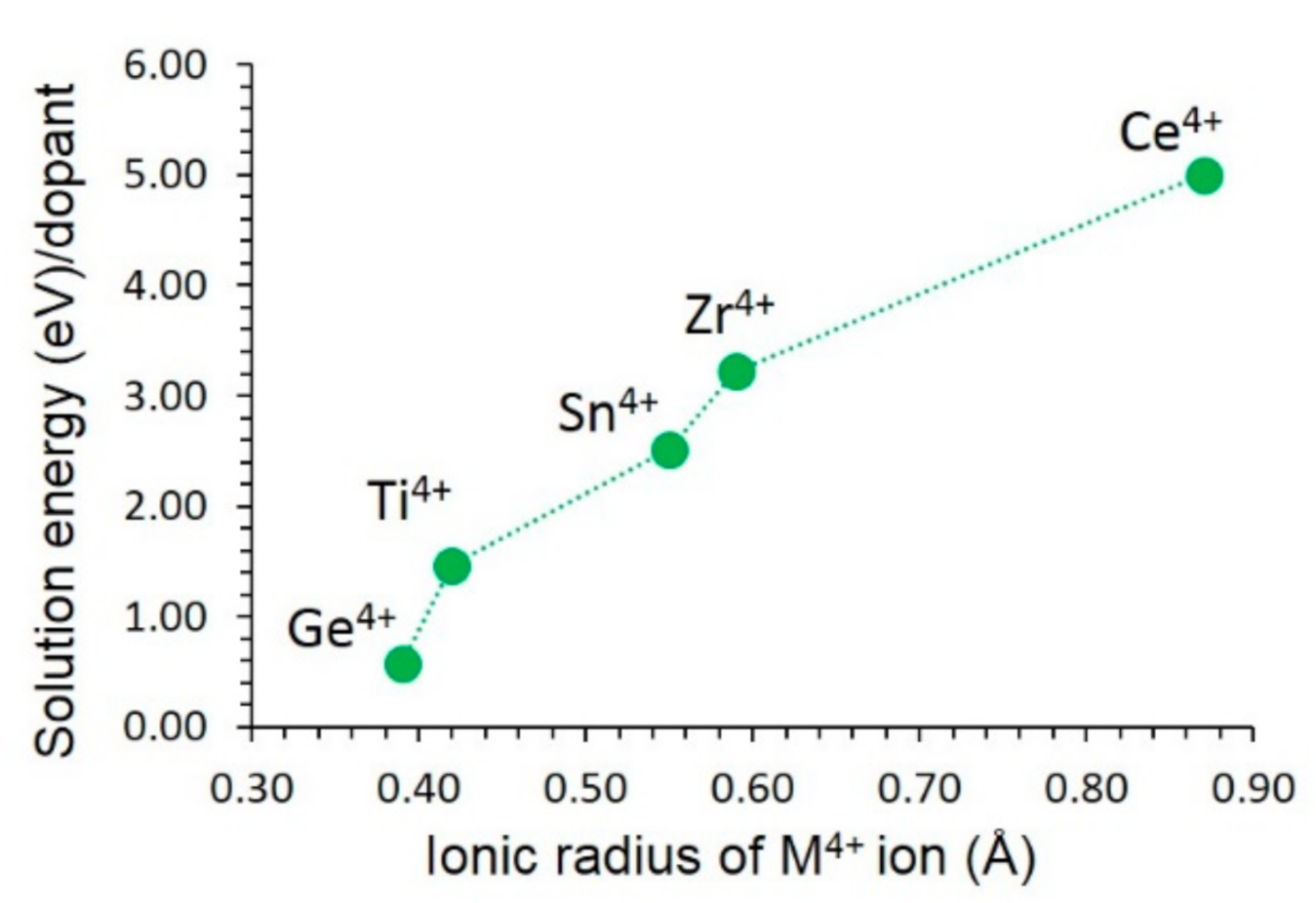
3.4.3. Tetravalent Dopants
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sultana, N. Trace Elements in Almandine Garnet, Vemireddipalle Area, Krishna District, Andhra Pradesh, India. Int. J. Sci. Res. Sci. Technol. 2019, 6, 184–187. [Google Scholar] [CrossRef]
- Yu, M.; Xia, Q.-X.; Zheng, Y.-F.; Zhao, Z.-F.; Chen, Y.-X.; Chen, R.-X.; Luo, X.; Li, W.-C.; Xu, H. The composition of garnet in granite and pegmatite from the Gangdese orogen in southeastern Tibet: Constraints on pegmatite petrogenesis. Am. Miner. 2021, 106, 265–281. [Google Scholar] [CrossRef]
- Sultana, N.; Podila, S.P. Almandine Gemstone—A review. Int. J. Recent Sci. Res. 2018, 9, 29204–29209. [Google Scholar]
- Geiger, C.A.; Feenstra, A. Molar volumes of mixing of almandine-pyrope and almandine-spessartine garnets and the crystal chemistry and thermodynamic-mixing properties of the aluminosilicate garnets. Am. Miner. 1997, 82, 571–581. [Google Scholar] [CrossRef]
- Čopjaková, R.; Sulovský, P.; Paterson, B.A. Major and trace elements in pyrope–almandine garnets as sediment provenance indicators of the Lower Carboniferous Culm sediments, Drahany Uplands, Bohemian Massif. Lithos 2005, 82, 51–70. [Google Scholar] [CrossRef]
- Sajeev, K.; Osanai, Y. Thermal gradients in the Sri Lankan granulite terrane: A garnet-biotite thermometric approach. J. Metamorph. Geol. 2005, 23, 383–397. [Google Scholar] [CrossRef]
- Usman, K.R.; Hainin, M.R.; Satar, M.K.I.M.; Warid, M.N.M.; Usman, A.; Al-Saffar, Z.H.; Bilema, M.A. A comparative assessment of the physical and microstructural properties of waste garnet generated from automated and manual blasting process. Case Stud. Constr. Mater. 2021, 14, e00474. [Google Scholar] [CrossRef]
- Poon, J.; Madden, D.C.; Wood, M.H.; van Tol, R.; Sonke, H.; Clarke, S.M. Surface Chemistry of Almandine Garnet. J. Phys. Chem. C 2020, 124, 5099–5117. [Google Scholar] [CrossRef]
- Stewart, H.A. Stock Removal Rates for Aluminium Oxide-Coated and Garnet-Coated Abrasive Belts. For. Prod. J. 1978, 28, 29–31. [Google Scholar]
- Jain, M.; Purushotham, G. Effect of Almandine on Microstructure and Corrosion properties of Nickel super alloy (K500) Composite for Aerospace Gas Turbine Application. IOP Conf. Ser. Mater. Sci. Eng. 2018, 376, 012051. [Google Scholar] [CrossRef]
- Chinner, G.A.; Clifford, T.N.; Nicolaysen, L.O.; Burger, A.J. Almandine in Thermal Aureoles. J. Pet. 1962, 3, 316–341. [Google Scholar] [CrossRef]
- Stone, M. The Significance of Almandine Garnets in the Lundy and Dartmoor Granites. Miner. Mag. 1988, 52, 651–658. [Google Scholar] [CrossRef]
- Guinel, M.J.-F.; Norton, M.G. The origin of asterism in almandine-pyrope garnets from Idaho. J. Mater. Sci. 2006, 41, 719–725. [Google Scholar] [CrossRef]
- Koziol, A.M.; Bohlen, S.R. Solution properties of almandine-pyrope garnet as determined by phase equilibrium experiments. Am. Mineral. 1992, 77, 765–773. [Google Scholar]
- Li, B.; Ge, J.; Zhang, B. Diffusion in garnet: A review. Acta Geochim. 2018, 37, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Woodland, A.B.; Wood, B.J. Electrochemical measurement of the free energy of almandine (Fe3Al2Si3O12) garnet. Geochim. Cosmochim. Acta 1989, 53, 2277–2282. [Google Scholar] [CrossRef]
- Freer, R. An experimental measurement of cation diffusion in almandine garnet. Nature 1979, 280, 220–222. [Google Scholar] [CrossRef]
- Price, J.; Bryan-Ricketts, D.S.; Anderson, D.; Velbel, M.A. Weathering of Almandine Garnet: Influence of Secondary Minerals on the Rate-Determining Step, and Implications for Regolith-Scale Al Mobilization. Clays Clay Miner. 2013, 61, 34–56. [Google Scholar] [CrossRef]
- Quartieri, S.; Antonioli, G.; Artioli, G.; Geiger, C.; Lottici, P. Temperature Dependence of Disorder and Correlation Effects in the Almandine X-Site. J. Phys. IV Fr. 1997, 7, C2-1157–C2-1158. [Google Scholar] [CrossRef]
- Romano, C.; Poe, B.T.; Kreidie, N.; McCammon, C.A. Electrical conductivities of pyrope-almandine garnets up to 19 GPa and 1700 °C. Am. Miner. 2006, 91, 1371–1377. [Google Scholar] [CrossRef]
- Hsiao, Y.-H.; La Plante, E.C.; Krishnan, N.M.A.; Dobbs, H.A.; Le Pape, Y.; Neithalath, N.; Bauchy, M.; Israelachvili, J.N.; Sant, G.N. Role of Electrochemical Surface Potential and Irradiation on Garnet-Type Almandine’s Dissolution Kinetics. J. Phys. Chem. C 2018, 122, 17268–17277. [Google Scholar] [CrossRef]
- Woodland, A.B.; Droop, G.; O’Neill, H.S.C. Almandine-rich garnet from near Collobrieres, southern France, and its petrological significance. Eur. J. Mineral. 1995, 7, 187–194. [Google Scholar] [CrossRef]
- Li, W.; Zheng, J.; Pei, J.; Xu, X.; Chen, T. Correlations between Garnet Species and Vibration Spectroscopy: Isomorphous Substitution Implications. Crystals 2022, 12, 104. [Google Scholar] [CrossRef]
- Borinski, S.A.; Hoppe, U.; Chakraborty, S.; Ganguly, J.; Bhowmik, S.K. Multicomponent diffusion in garnets I: General theoretical considerations and experimental data for Fe–Mg systems. Contrib. Miner. Pet. 2012, 164, 571–586. [Google Scholar] [CrossRef]
- Ferrari, A.M.; Valenzano, L.; Meyer, A.; Orlando, R.; Dovesi, R. Quantum-Mechanical ab Initio Simulation of the Raman and IR Spectra of Fe3Al2Si3O12 Almandine. J. Phys. Chem. A 2009, 113, 11289–11294. [Google Scholar] [CrossRef] [PubMed]
- Nobes, R.; Akhmatskaya, E.; Milman, V.; Winkler, B.; Pickard, C. Structure and properties of aluminosilicate garnets and katoite: An ab initio study. Comput. Mater. Sci. 2000, 17, 141–145. [Google Scholar] [CrossRef]
- Kuganathan, N.; Kordatos, A.; Anurakavan, S.; Iyngaran, P.; Chroneos, A. Li3SbO4 lithium-ion battery material: Defects, lithium ion diffusion and tetravalent dopants. Mater. Chem. Phys. 2019, 225, 34–41. [Google Scholar] [CrossRef]
- Kuganathan, N.; Ganeshalingam, S.; Chroneos, A. Defects, Dopants and Lithium Mobility in Li9V3(P2O7)3(PO4)2. Sci. Rep. 2018, 8, 8140. [Google Scholar] [CrossRef]
- Kuganathan, N.; Chroneos, A. Na3V(PO4)2 cathode material for Na ion batteries: Defects, dopants and Na diffusion. Solid State Ion. 2019, 336, 75–79. [Google Scholar] [CrossRef] [Green Version]
- Devanathan, R.; Weber, W.; Singhal, S.; Gale, J. Computer simulation of defects and oxygen transport in yttria-stabilized zirconia. Solid State Ion. 2006, 177, 1251–1258. [Google Scholar] [CrossRef] [Green Version]
- Pandey, R.; Gale, J.D.; Sampath, S.K.; Recio, J.M. Atomistic Simulation Study of Spinel Oxides: Zinc Aluminate and Zinc Gallate. J. Am. Ceram. Soc. 1999, 82, 3337–3341. [Google Scholar] [CrossRef]
- Williford, R.E.; Weber, W.J.; Devanathan, R.; Gale, J.D. Effects of Cation Disorder on Oxygen Vacancy Migration in Gd2Ti2O7. J. Electroceram. 1999, 3, 409–424. [Google Scholar] [CrossRef]
- Henson, N.J.; Cheetham, A.K.; Gale, J.D. Computational Studies of Aluminum Phosphate Polymorphs. Chem. Mater. 1996, 8, 664–670. [Google Scholar] [CrossRef]
- Gale, J.D.; Rohl, A.L. The General Utility Lattice Program (GULP). Mol. Simul. 2003, 29, 291–341. [Google Scholar] [CrossRef]
- Kuganathan, N.; Islam, M.S. Li2MnSiO4 Lithium Battery Material: Atomic-Scale Study of Defects, Lithium Mobility, and Trivalent Dopants. Chem. Mater. 2009, 21, 5196–5202. [Google Scholar] [CrossRef]
- Lewis, G.V.; Catlow, C.R.A. Potential models for ionic oxides. J. Phys. C Solid State Phys. 1985, 18, 1149–1161. [Google Scholar] [CrossRef]
- Gale, J.D. GULP: A computer program for the symmetry-adapted simulation of solids. J. Chem. Soc. Faraday Trans. 1997, 93, 629–637. [Google Scholar] [CrossRef]
- Mott, N.F.; Littleton, M.J. Conduction in polar crystals. I. Electrolytic conduction in solid salts. Trans. Faraday Soc. 1938, 34, 485–499. [Google Scholar] [CrossRef]
- Grimes, R.W.; Busker, G.; McCoy, M.A.; Chroneos, A.; Kilner, J.A.; Chen, S.-P. The Effect of Ion Size on Solution Mechanism and Defect Cluster Geometry. Ber. Bunsenges. Phys. Chem. 1997, 101, 1204–1210. [Google Scholar] [CrossRef]
- Fisher, C.A.J.; Prieto, V.M.H.; Islam, M.S. Lithium Battery Materials LiMPO4 (M = Mn, Fe, Co, and Ni): Insights into Defect Association, Transport Mechanisms, and Doping Behavior. Chem. Mater. 2008, 20, 5907–5915. [Google Scholar] [CrossRef]
- Nishimura, S.-I.; Kobayashi, G.; Ohoyama, K.; Kanno, R.; Yashima, M.; Yamada, A. Experimental visualization of lithium diffusion in LixFePO4. Nat. Mater. 2008, 7, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Wahid, F.A.; Thomson, G.B.; Graham, G.M.; Jackson, R.A. A computational study of the effect of doping divalent cations in barite. J. Mater. Chem. 2002, 12, 3799–3802. [Google Scholar] [CrossRef]
- Prandl, W. Die magnetische Struktur und die Atomparameter* des Almandins Al2Fe3(SiO4)3. Z. Krist.-Cryst. Mater. 1971, 134, 333–343. [Google Scholar] [CrossRef]
- Kröger, F.A.; Vink, H.J. Relations between the Concentrations of Imperfections in Crystalline Solids. In Solid State Physics; Seitz, F., Turnbull, D., Eds.; Academic Press: Cambridge, MA, USA, 1956; Volume 3, pp. 307–435. [Google Scholar]
- Devaraju, M.K.; Truong, Q.D.; Hyodo, H.; Sasaki, Y.; Honma, I. Synthesis, characterization and observation of antisite defects in LiNiPO4 nanomaterials. Sci. Rep. 2015, 5, 11041. [Google Scholar] [CrossRef] [Green Version]
- Politaev, V.V.; Petrenko, A.A.; Nalbandyan, V.B.; Medvedev, B.S.; Shvetsova, E.S. Crystal structure, phase relations and electrochemical properties of monoclinic Li2MnSiO4. J. Solid State Chem. 2007, 180, 1045–1050. [Google Scholar] [CrossRef]
- Nytén, A.; Abouimrane, A.; Armand, M.; Gustafsson, T.; Thomas, J.O. Electrochemical performance of Li2FeSiO4 as a new Li-battery cathode material. Electrochem. Commun. 2005, 7, 156–160. [Google Scholar] [CrossRef]
- Quinzeni, I.; Fujii, K.; Bini, M.; Yashima, M.; Tealdi, C. Na+ diffusion mechanism and transition metal substitution in tunnel-type manganese-based oxides for Na-ion rechargeable batteries. Mater. Adv. 2022, 3, 986–997. [Google Scholar] [CrossRef]
- Ward, R.E.; Freeman, C.L.; Dean, J.S.; Sinclair, D.C.; Harding, J.H. Using Metadynamics to Obtain the Free Energy Landscape for Cation Diffusion in Functional Ceramics: Dopant Distribution Control in Rare Earth-Doped BaTiO3. Adv. Funct. Mater. 2020, 30, 1905077. [Google Scholar] [CrossRef]
- Fisher, C.; Matsubara, H. Oxide ion diffusion along grain boundaries in zirconia: A molecular dynamics study. Solid State Ion. 1998, 113–115, 311–318. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interaction | A/eV | ρ/Å | C/eV•Å6 | Y/e | K/eV•Å–2 |
|---|---|---|---|---|---|
| Fe2+–O2− | 694.1 | 0.3399 | 0 | 2 | 10.92 |
| Al3+–O2− | 2409.5 | 0.2649 | 0 | 3 | 99,999.00 |
| Si4+–O2− | 1283.91 | 0.32052 | 10.66 | 4 | 99,999.00 |
| O2−–O2− | 149,734.35 | 0.1593 | 19.9 | –2.04 | 6.3 |
| Parameter | Calculated | Experiment [43] | |∆|(%) |
|---|---|---|---|
| a = b = c (Å) | 11.511 | 11.507 | 0.03 |
| α = β = γ (°) | 90 | 90 | 0 |
| V (Å3) | 1525.17 | 1523.65 | 0.10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subasinghe, J.L.; Ganeshalingam, S.; Kuganathan, N. Computational Study of Crystallography, Defects, Ion Migration and Dopants in Almandine Garnet. Physchem 2022, 2, 43-51. https://doi.org/10.3390/physchem2010004
Subasinghe JL, Ganeshalingam S, Kuganathan N. Computational Study of Crystallography, Defects, Ion Migration and Dopants in Almandine Garnet. Physchem. 2022; 2(1):43-51. https://doi.org/10.3390/physchem2010004
Chicago/Turabian StyleSubasinghe, Janya Lumbini, Sashikesh Ganeshalingam, and Navaratnarajah Kuganathan. 2022. "Computational Study of Crystallography, Defects, Ion Migration and Dopants in Almandine Garnet" Physchem 2, no. 1: 43-51. https://doi.org/10.3390/physchem2010004
APA StyleSubasinghe, J. L., Ganeshalingam, S., & Kuganathan, N. (2022). Computational Study of Crystallography, Defects, Ion Migration and Dopants in Almandine Garnet. Physchem, 2(1), 43-51. https://doi.org/10.3390/physchem2010004