1. Introduction
The increasing importance of the conservation and protection of clean water supplies [
1] presents a pressing opportunity for the use and development of computational physical chemistry methods and modelling over the next decade. Technological advances continue to impact our waterways in ways that derive acute and chronic risk to human health. In this perspective, I pose a few modest areas where computational chemistry modelling should prove beneficial in understanding these impacts. For obvious reasons, quantum chemistry has historically only been able to be applied to the simplest
model systems. The complexity needed to accurately model “real” systems is often treated approximately—or not at all—in deference to feasible and efficient models. To provide the best computational predictions, the models must move toward more realism. This includes a full accounting of stereochemistry, conformation, and solvation effects in physicochemical property prediction, realistic and dynamic amorphous models, and treatment of the interfacial chemistry of perfluorocarbon molecules and polymers. Each of these areas requires an additional level of computational expense that simpler models generally do not consider.
Computing power, however, is catching up with the imagination of the computational physical chemist to the point that further advances require new software paradigms [
2]. A new age of computing is on the horizon, with the prospect of quantum computing edging ever closer [
3]. Recent decades have seen remarkable advances in computer infrastructure, which have made possible the execution of ever-larger molecular simulations, and ever-more-accurate electronic structure calculations. Over the past decade, graphic processing unit (GPU) processors, in particular, have provided platforms to massively parallelize computational efforts. Even before the widespread use of GPUs, parallelization alone allowed decreases in real-world execution times to the point where jobs that would likely never be attempted if executed serially were feasible. Cloud computing infrastructure facilitates access to such high-performance computing for almost any researcher [
4]. With an eye toward environmental water quality applications, the physical chemistry community will be able to reap the rewards of these current and future speed-ups over the next decade to increase the computational accuracy in simulations of realistic systems, which will translate to further understanding of field-level water quality issues involving acute chemical spill incident responses, and to understanding and potential remediation of persistent environmental pollutants such as microplastics and perfluorinated alkyl substances (PFAS).
2. When Horses Are Actually Zebras: Physicochemical Properties of Novel Molecules
An oft-cited adage from medical schools goes something like “When in Tennessee, if you hear hoofbeats, think horses, not zebras”. The idea is to train doctors to focus on the most common/most probable explanation of patient symptoms, and not on the exotic. An analogy can be made to chemicals in commerce and how we deal with spill incidents. An overwhelming majority of spilled chemicals into waterways are things such as petroleum products (i.e., diesel, gasoline), natural gas, and sewage [
5,
6]; none of these are things we
want in our water, but we know how to deal with their cleanup and removal. Due to their common and heavy usage, comprehensive Safety Data Sheets are available with important information such as toxicity, chemical compatibility, and cleanup and disposal procedures; much of this information exists because it is mandated by the Toxic Substance Control Act (TSCA) in the United States, and by other such legislation around the developed world [
7,
8]. What happens, however, when a novel chemical is spilled into a waterway? A vast number of compounds in commerce are not used in amounts large enough to be considered “high volume”, are not used in food or cosmetics, and, in general, are never intended for human exposure. Producers of these low volume industrial chemicals are, therefore, not bound by the TSCA and other guiding rules [
5,
7]. When these types of substances are involved in spill incidents, the Safety Data Sheets are often sparsely populated, best practices are not established, and detection methods may not exist. Zebras have arrived, but we only know how to round up the horses.
Computational physical chemistry can play an important role in responding to chemical spill incidents involving novel or poorly characterized materials. High-level models have been developed that can provide responders with guidance through predicted information about such things as environmental fate and transport, bioaccumulation, and acute toxicity; however, the high-level models all need regular physicochemical information as input. When these properties are unknown, they can in turn be predicted with various chemistry-based methods. Such programs as the US Environmental Protection Agency’s (USEPA) EPISuite uses group additivity methods applied to 2D stick figure representations of molecules [
9]. These models are built on training sets of existing properties, and thus, they work well for molecules that “look like” the training data. However, it cannot be expected for such empirically based models to be able to describe all conceivable molecules [
5]. Additionally, the 2D static nature of such models fails to incorporate the important effects of stereochemical isomers, conformational diversity, or solvation effects [
10,
11,
12]. All three of these aspects can largely affect the behavior of species in waterways.
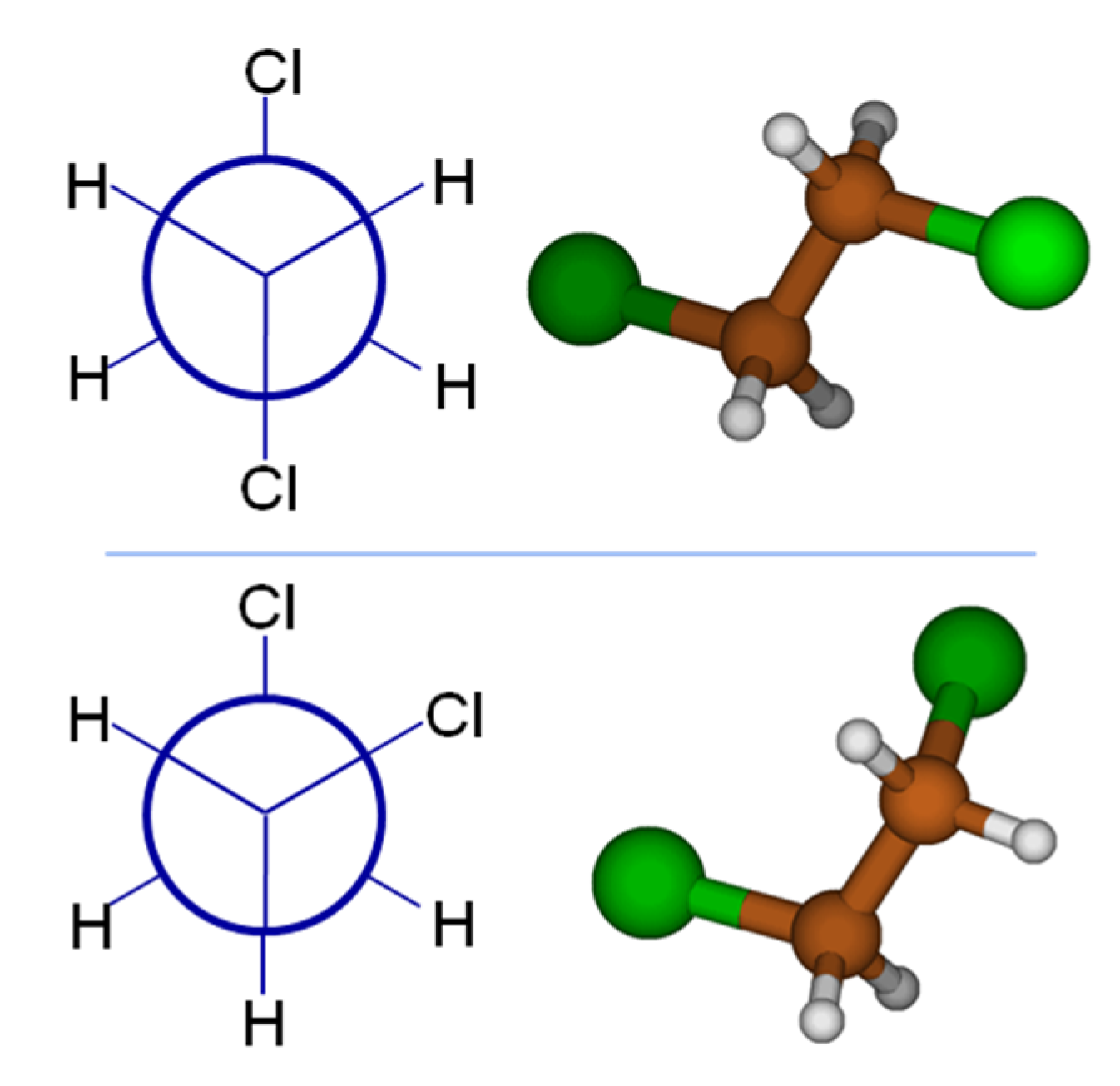
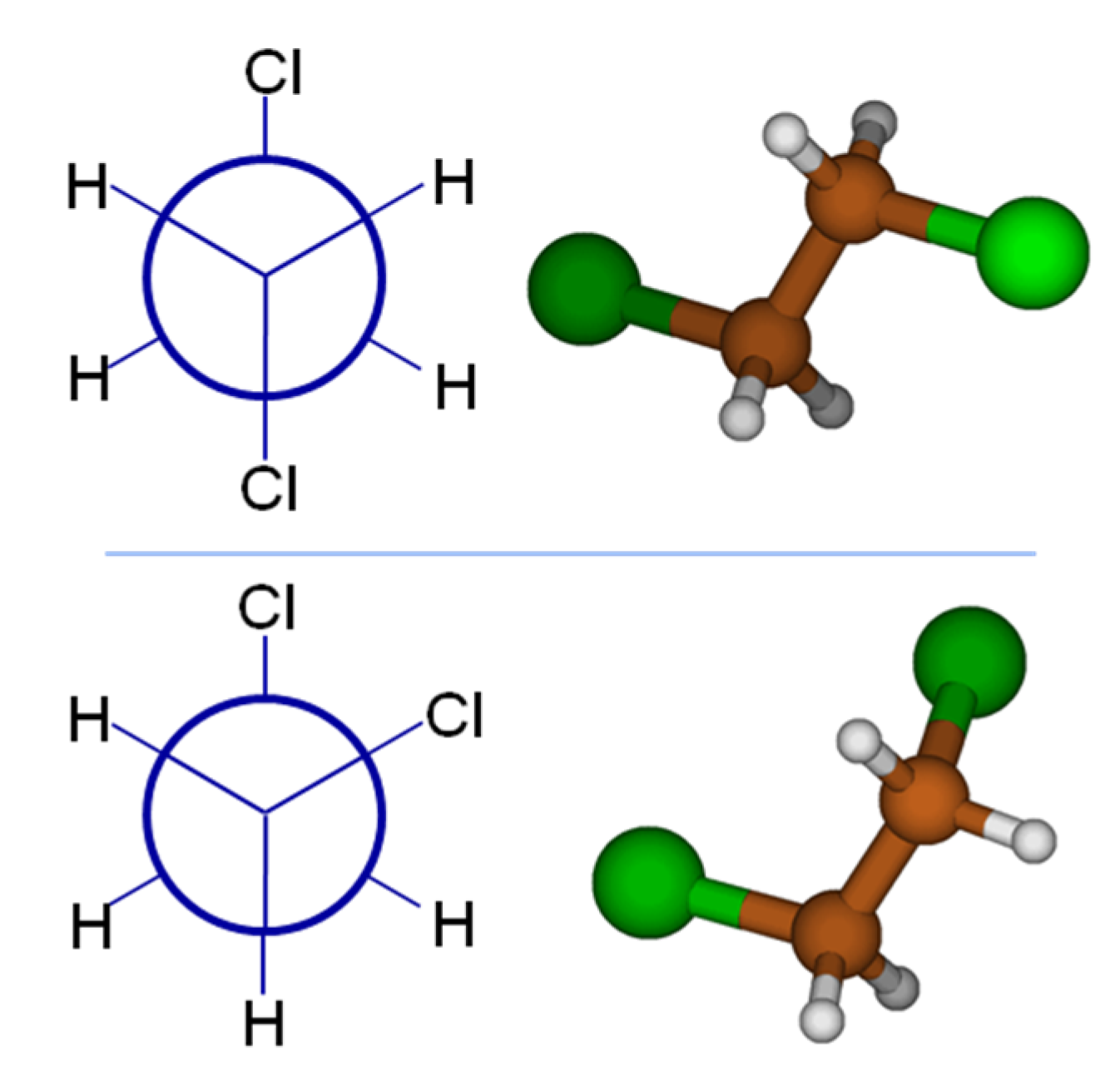
To provide a simple example of how a simple statistical mechanical treatment can qualitatively affect results, consider 1,2-dichloroethane (DCE), a classic example from organic chemistry class. One could desire to use the dipole moment, a very good measure of a molecule’s polarity, to aid predictions of aqueous solubility. If we are to draw 1,2-dichloroethane using concepts from organic chemistry class, our chemistry training and intuition tell us that the global minimum energy conformation will position the bulky chlorine atoms farthest apart; the top panel of
Figure 1 shows the Newman projection and 3D structure for DCE. It is straightforward to argue based on the vector addition of identical bond dipoles that this global minimum (
anti form) of DCE has a dipole moment of 0 Debye (D). At this point, perhaps full credit has been earned on an organic chemistry quiz, but only considering the global minimum structure will provide a poor comparison to the tabulated, experimentally known dipole value for DCE of 1.8 D! Examining the Newman projection in the bottom panel of
Figure 1, the Cl atom in the foreground has rotated 120° from its original position, to be in a
gauche conformation. This skewed conformation will have a substantial molecular dipole moment. Density Functional Theory computations (B3LYP/6-31G*) predict 2.91 D for the dipole moment of the
gauche conformer, with an energy 1.7 kcal mol
−1 higher than the
anti conformer. (All calculations mentioned in this section were performed with Gaussian 09 [
13].) With this energy difference, Boltzmann analysis shows that the
gauche conformer will not contribute significantly to the overall average dipole. At the MP2 level of theory with a larger basis set (MP2/aug-cc-pwCVDZ), the
gauche DCE conformer is only 1.4 kcal mol
−1 higher in energy, which translates to 9% relative population and still does not rationalize the experimental dipole moment. Adding solvation effects (MP2/aug-cc-pwCVDZ + SMD model) to the MP2 calculation brings the energy difference between the conformers down to 0.17 kcal mol
−1. This corresponds to a 57/43% population ratio between the
anti and
gauche conformers, resulting in a Boltzmann-averaged dipole moment of 1.88 D. Clearly, accounting for the effects of both conformation and implicit solvation are required to properly determine molecular dipoles.
It was found that a full accounting of conformational, stereochemical, and solvation effects was required to properly predict the aqueous behavior of MCHM (methyl cyclohexane methanol), the principal contaminant involved in the 2014 Elk River chemical spill [
10,
11,
12]; conformational averaging and high-level electronic structure calculations were also applied in other systems to good effect [
14]. However, even for the smallest of molecules, properly accounting for conformational, stereochemical, and solvation effects requires roughly an order-of-magnitude increase in raw computational power, and this scales as the size and “floppiness” of the molecule increases. Because of this, it has historically been common to only interrogate properties of the global minimum structure for compounds. With the ever-increasing computational power available, the community should more fully embrace the realistic picture of how properties arise from dynamic, rather than static, molecular structures.
3. Will It Stay or Will It Go? Contaminant Binding Affinities on Water-Adjacent Surfaces
Once a contaminant is in the water system, where will it go? Will it be deposited into oils and clays in the riverbed? Is it able to be removed with carbon or sand filters at the treatment plant? Will it absorb into the polymer piping and linings in premise plumbing? The fate and transport behavior of such contaminants can be predicted based on partitioning behavior at surfaces throughout the system. Binding energies calculated at different infrastructure and environmental surfaces/matrices can aid in the selection of filter materials and or predict where the molecules will “stick” and concentrate in the water system infrastructure. If information about surface-binding properties can be computed quickly—for instance, the time it would take for the flow of a spill plume down a waterway to a facility intake—this information can help make decisions on remediation, and ultimately (and most importantly), whether or not to even allow the plume into the facility.
One problem with computing binding properties to the relevant interfaces is that they mostly comprise amorphous surfaces. While one may only require a single or a few “poses” of a contaminant molecule on a crystalline surface to obtain a good representative binding energy, accurately computing binding behavior to amorphous surfaces is more complicated. To gain a full picture requires averaging over multiple absorption sites on a simulated surface, and this simulated surface should be one of many sampled from the overall equilibrium ensemble to provide a sufficient site- and time-averaged binding estimate. A realistic simulation of an amorphous surface requires generally many thousands of atoms, and the dynamic interface configuration should be allowed to evolve at equilibrium in time for as long as needed to fully capture the fluctuational behavior. This timescale is generally not known
a priori, but it is safe to say that, in real-world minutes, this requires “a long time” relative to the travel time mentioned of a plume drifting toward a water intake. Efforts are underway to build libraries of amorphous surface “snapshots” to alleviate this long simulation time [
15]. With banked surface trajectory snapshots at the ready, it becomes a much quicker prospect to obtain binding estimates. The contaminant can be immediately posed at multiple sites on each snapshot and the needed computations for each of these poses can be executed simultaneously in a “ridiculously parallel” scheme. In this way, the time required to obtain accurate binding estimates on the amorphous surface is now analogous (or better) than that to obtain the few poses needed for similar information on a crystal surface.
Conformational complexity in the contaminant molecule should be accounted for in the initial posing (i.e., the overall ensemble includes multiple conformers posed at the same site). The optimized structure and binding energy of the adsorbed contaminant can be calculated and refined in turn with increasing levels of accurate theory (i.e., general molecular mechanics forcefield → HF → DFT and/or MP2; basis set sizes can also be increased). The higher levels of theory cannot be applied to full surface slab models, so these are achieved through QM/MM approaches. A simple example of this can be seen in
Figure 2, which depicts an MCHM molecule adsorbed on an amorphous carbon model [
12].
Finally, the true adsorption behavior of contaminants on water infrastructure surfaces is mediated/altered by whatever species are already adsorbed to the surface before the new molecule comes along. For instance, a contaminant molecule may interact much differently with a pristine model PVC surface than it would on the surface of a bio-film coated pipe in premise plumbing. To describe these matrix effects, more sophisticated and detailed models may require the inclusion of co-adsorbates, bio-films, and surface aging considerations, to name a few. It is not currently clear how to best account for these effects, and this is a frontier where increased research is needed to build realistic and accurate computational models to aid our understanding, and ultimately prediction, of the contaminant behavior at surfaces within the water system infrastructure.
4. Cheap and Disposable (and in Fourteen Years Decomposable?): Aging of Microplastics
The heading of this section references a song lyric by sometimes controversial country musician Toby Keith [
16]. Keith’s non-scientific prediction about the breakdown of plastic party cups within 14 years is unfortunately far from accurate. Instead, the usual polystyrene party cups require hundreds of years to fully degrade in the environment [
17]. As such, the vast majority of all the plastic humans have ever produced still exists. What does happen more quickly is that through physical and partial degradation processes, plastic goods are broken down to microplastics (particles smaller than 5 mm); these microplastics may enter our waterways and pose a variety of threats to freshwater and marine wildlife, and can significantly impact drinking water and sewage treatment processing [
18,
19,
20,
21]. Due to their size, their collective relative active surface areas are large, and these surfaces may act in ways that are important for the environmental fate and transport of chemicals in our waters. An array of processes can be conceived that may be impacted by these ubiquitous microplastics. For instance, bisphenol A, a hormone-active compound, has been a frequent additive to plastics, and the leeching of bisphenol A from microplastics into waterways may have important health implications [
21,
22]. Microplastics may also be thought of as a vast distributed “catalytic surface” for environmentally relevant surface chemistry. Aged plastics, with even higher surface areas, may participate in chemistry at rates much different than for bulky plastics. Heavy metal adsorption and uptake, for instance, is significantly different on new and aged polymer surfaces [
23,
24]. Computational modelling of these microparticulate systems is needed to understand the surface chemistry of these small particles. Improving our knowledge of microplastic processes can be of great importance to understand and potentially remediate these microplastic pollutants in the environment. Accurate polymer models (as mentioned earlier) plus reaction modelling on such surfaces could provide insights for timescales that experiments may not allow. Just as mentioned in the previous section, environmental matrix effects may alter the physical and chemical processes involved on these plastic surfaces.
5. I’ll Be Here ‘til the End of Time: Dealing New Fates to Forever Chemicals
If it seems as though hydrocarbon-based plastics will never degrade, let us now turn to “forever chemicals”—a moniker given to perfluorinated chemical species (PFAS; per- and polyfluorinated alkyl substances). PFAS have likely environmental degradation lifetimes in the
thousands of years. They have come under increasing scrutiny and are of rising concern in the environmental, economic, social equity, and health sectors [
25,
26]. Species such as C8 (PFOA, perfluorooctanoic acid) and GenX (a six-carbon perfluorinated ether-containing carboxylic acid) gain much of the attention, but many different species are of concern. The USEPA recently listed 160 PFAS as reportable to the Toxics Release Inventory (TRI) under EPCRA (Emergency Planning and Community Right-to-know Act) guidance [
27]. Due to the highly persistent and bioaccumulative nature of PFAS species, it is imperative to understand these species’ behavior in the water system. The conversion or removal of such species may be some of the only ways to keep such chemicals from causing deleterious health effects in peoples around the world. High-throughput reaction modelling can aid in the identification of potentially useful chemistries to try for these species. Perfluorocarbon species have been the subject of markedly fewer quantum chemical computational studies than their hydrocarbon counterparts, as applying quantum chemical methods to fluorocarbons may have been prohibitively expensive. The net increase of eight electrons for each H→F substitution requires large increases in computational power (e.g., it is ~70× more expensive to compute energies for PFOA than the hydrocarbon octanoic acid counterpart at the Hartree-Fock (HF) level; including basic electron correlation with MP2 theory raises this scaling factor to ~200×). However, advances in computational algorithms, such as linear scaling approaches [
28], and the continual increases in raw computational power should ameliorate these difficulties. As new information emerges on new endemic PFAS species, computational modelling can aid in a complete understanding of their behaviors.



{kind=link}
{kind=link}
{kind=link}