
Oxidative Stress in the Pathogenesis of Neurodegenerative Diseases


Abstract
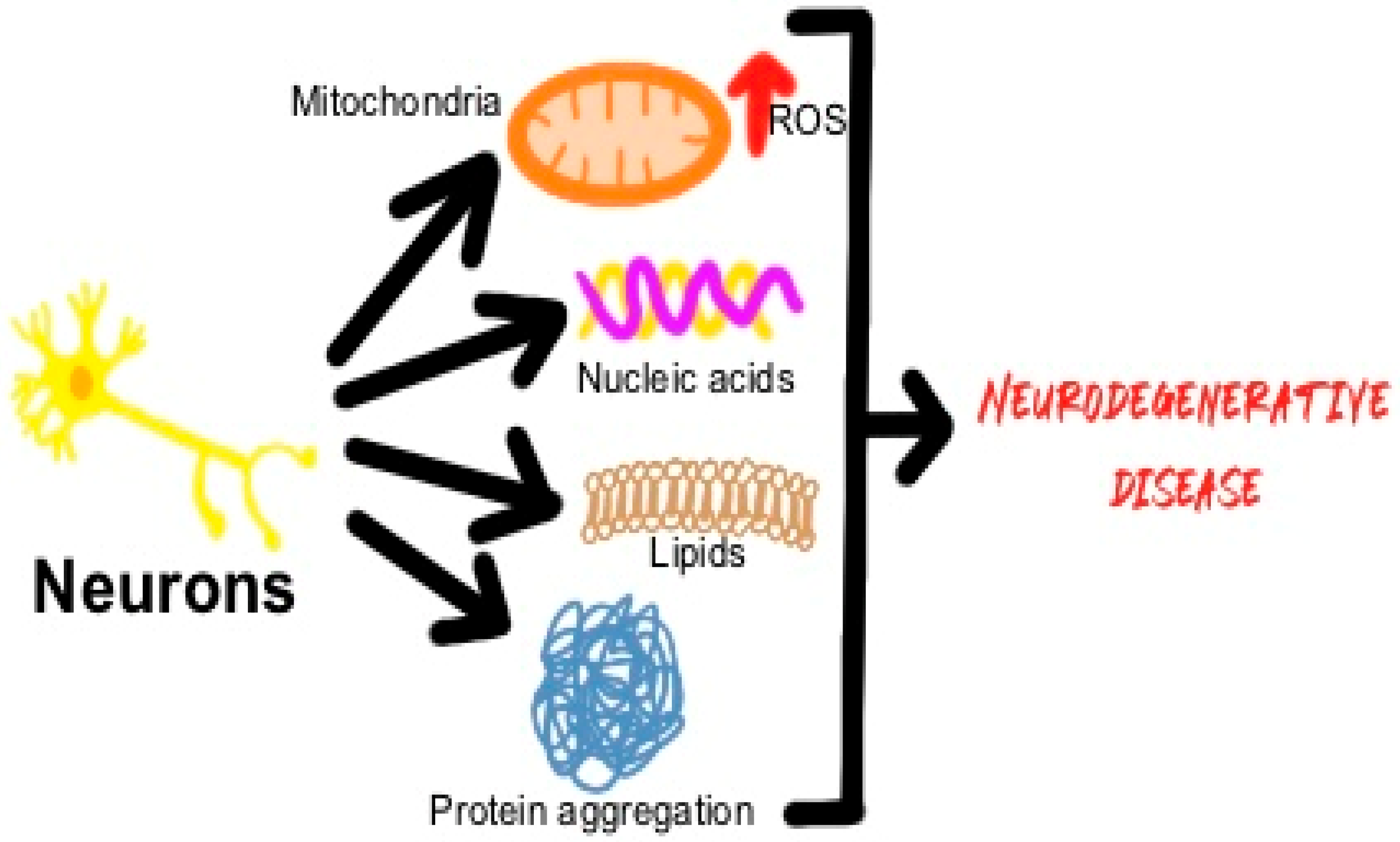
1. Introduction
2. Oxidative Stress (OS)
2.1. Definition
2.2. ROS Generation
- -
- The main generator appears to be the mitochondrial activity. Around 1% of a healthy brain cell’s mitochondrial electron flow produces O2−, preponderantly via complex I (NADH dehydrogenase) and complex III (ubiquinone cytochrome c reductase) [21,22]. Superoxide is neutralized by superoxide dismutases (SOD1, SOD2, SOD3), thus resulting in H2O2 molecules. Hydrogen peroxide is less toxic than superoxide, but its danger lies in its potential to create even more harmful byproducts, e.g., hydroxyl radicals (–OH) by reacting with Fenton’s reagent or peroxynitrite anions (ONOO–) by reacting with NO [23]. Various metabolic factors can influence mitochondrial ROS production, such as a shift of the NADH/NAD+ balance toward reduction of NADH [24], increases in succinate levels [25], or alterations in the mitochondrial membrane potential, as occurs in conditions of hypoxia [26].
- -
- Monoamine oxidases (MAOs), enzymes located on the outer mitochondrial membrane, metabolize serotonin, epinephrine, and dopamine [27]. MAO-A is expressed in neurons, and glial cells express both MAO-A and -B [28]. They use flavin adenine dinucleotide (FAD) for metabolizing monoamines, and hydrogen peroxide results from the FAD-FADH2 cycle [29].
- -
- Several other mitochondrial enzymes can produce significant amounts of ROS, such as α-ketoglutarate dehydrogenase, glycerol phosphate dehydrogenase, and p66shc [30].
- -
- Peroxisomes participate in the beta-oxidation of fatty acids, a process leading to the generation of H2O2. However, other enzymes, such as xanthine oxidase, acyl CoA oxidases, D-amino acid oxidase, D-aspartate oxidase, and L-α-hydroxy oxidase, may contribute to the generation of superoxide, hydroxyl radicals, nitric oxide, and hydrogen peroxide [31].
- -
- Several nicotinamide adenine dinucleotide phosphate oxidases (NADPH oxidases) are expressed in brain cells and microglia, and they are involved in the regulation of cell survival/apoptosis, neuroinflammation, migration, differentiation, the proliferation of brain cells, and synaptic plasticity [32]. NOX isoforms localize to the mitochondria, nucleus, endoplasmic reticulum, and plasma membrane. NOX4 mainly produces hydrogen peroxide, while NOX2 activity generates superoxide [33]. Moreover, NOX-generated ROS can lead to mitochondrial dysfunction or even depolarize the mitochondrial membrane, open the mitochondrial permeability transition pore, and ignite apoptosis [34].
2.3. Antioxidant Defenses
2.3.1. Superoxide Dismutases (SODs)
- SOD1 is active in cytosol and organelles.
- SOD2 is active in mitochondria.
- SOD3 is an extracellular enzyme with a comparatively restricted expression in only a few types of cells [36].
2.3.2. Catalase
2.3.3. Glutathione Peroxidase (GPx)
2.3.4. Glutathione (GSH)
2.3.5. Vitamins C and E
2.3.6. Trace Elements
- Action potentials propagated in the CNS cause calcium influx, with raised intracellular calcium leading to the activation of neuronal nitric oxide synthase (nNOS) and the production of nitric oxide [51].
- Mitochondria attempt to buffer the excess intracellular calcium, but mitochondrial calcium overload leads to dysfunction of the organelles and impairs energy homeostasis [52].
- Activated microglia produce high amounts of ROS, mainly superoxide, and increase the transcription of SOD2 that converts superoxide into H2O2 [53].
- ROS are also generated via the metabolization or auto-oxidation of neurotransmitters, such as dopamine, serotonin, or adrenaline [54].
- The relatively high contents of redox-active transition metals of the brain, such as Fe2+ and Cu+, act as catalyzers in the Fenton reaction and promote the generation of ROS [55].
- The cellular membranes are rich in polyunsaturated fatty acids and very susceptible to lipid peroxidation. The high membrane surface/cytoplasmic volume ratio of the brain cells creates the premises for the chain propagation of peroxidation reactions following ROS attack [57].
3. Pathways Through Which ROS Promote Neurodegeneration
3.1. Oxidation of Proteins
3.2. Lipid Peroxidation
3.3. DNA Oxidative Damage
3.4. RNA Oxidative Damage
4. Oxidative Stress Is Intricately Linked to Other Pathogenic Cascades in Neurodegeneration
5. Oxidative Stress in Specific Neurodegenerative Diseases
5.1. Alzheimer’s Disease
5.2. Parkinson’s Disease
5.3. Amyotrophic Lateral Sclerosis
6. Antioxidant Therapeutic Strategies in Neurodegenerative Diseases
6.1. Antioxidant Therapeutic Strategies in Alzheimer’s Disease
6.2. Antioxidant Therapeutic Strategies in Parkinson’s Disease
6.3. Antioxidant Therapeutic Strategies in Amyotrophic Lateral Sclerosis
6.4. The Road to Finding Efficient Therapies for Neurodegenerative Diseases Is Paved with Many Trial Failures
- -
- Insufficient dose of the chosen antioxidant.
- -
- Inappropriate timing and insufficient duration of the treatment.
- -
- Poor solubility and blood–brain penetration of the antioxidant. Antioxidant drugs are usually polar molecules with high molecular weights and poor absorption, which, together with their quick metabolism, limit their bioavailability [200].
- -
- Antioxidant supplementation might affect the natural redox equilibrium between pro-oxidant and antioxidant species and reduce the natural antioxidant response, further increasing the redox homeostasis failure in neurodegenerative diseases [201].
- -
- It may be that we have a poor understanding of the antioxidant effect exerted by a particular antioxidant compound. For example, glutathione can act via post-translational modifications to mediate protective effects, which may be confused with an antioxidant effect [200]
- -
- The animal models used are mainly transgenic animals, which do not recapitulate the complexity of the human brain or the complex pathogenic cascades of human disease. These limitations could be overcome by studies performed on brain organoids [202].
7. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Herculano-Houzel, S. The human brain in numbers: A linearly scaled-up primate brain. Front. Hum. Neurosci. 2009, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Pakkenberg, B.; Pelvig, D.; Marner, L.; Bundgaard, M.J.; Gundersen, H.J.G.; Nyengaard, J.R.; Regeur, L. Aging and the human neocortex. Exp. Gerontol. 2003, 38, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Aranda-Anzaldo, A. The post-mitotic state in neurons correlates with a stable nuclear higher-order structure. Commun. Integr. Biol. 2012, 5, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef]
- Plascencia-Villa, G.; Perry, G. Roles of Oxidative Stress in Synaptic Dysfunction and Neuronal Cell Death in Alzheimer’s Disease. Antioxidants 2023, 12, 1628. [Google Scholar] [CrossRef]
- Lee, J.; Kim, H.-J. Normal Aging Induces Changes in the Brain and Neurodegeneration Progress: Review of the Structural, Biochemical, Metabolic, Cellular, and Molecular Changes. Front. Aging Neurosci. 2022, 14, 931536. [Google Scholar] [CrossRef]
- Temple, S. Advancing Cell Therapy for Neurodegenerative Diseases. Cell Stem Cell 2023, 30, 512–529. [Google Scholar] [CrossRef]
- GBD 2019 Dementia Forecasting Collaborators. Estimation of the Global Prevalence of Dementia in 2019 and Forecasted Prevalence in 2050: An Analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef]
- Gomberg, M. An Instance of Trivalent Carbon: Triphenylmethyl. J. Am. Chem. Soc. 1900, 22, 757–771. [Google Scholar] [CrossRef]
- Commoner, B.; Townsend, J.; Pake, G.E. Free Radicals in Biological Materials. Nature 1954, 174, 689–691. [Google Scholar] [CrossRef]
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Gerschman, R.; Gilbert, D.L.; Nye, S.W.; Dwyer, P.; Fenn, W.O. Oxygen Poisoning and X-Irradiation: A Mechanism in Common. Science 1954, 119, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I.; Storey, K.B. Oxidative stress concept updated: Definitions, classifications, and regulatory pathways implicated. EXCLI J. 2021, 20, 956–967. [Google Scholar] [CrossRef] [PubMed]
- Augusto, O.; Miyamoto, S. Oxygen radicals and related species. In Principles of Free Radical Biomedicine; Pantopoulos, K., Schipper, H.M., Eds.; Nova Science Publishers Inc.: Hauppauge, NY, USA, 2011. [Google Scholar]
- Egea, J.; Fabregat, I.; Frapart, Y.M.; Ghezzi, P.; Görlach, A.; Kietzmann, T.; Kubaichuk, K.; Knaus, U.G.; Lopez, M.G.; Olaso-Gonzalez, G.; et al. European Contribution to the Study of ROS: A Summary of the Findings and Prospects for the Future from the COST Action BM1203 (EU-ROS). Redox Biol. 2017, 13, 94–162. [Google Scholar] [CrossRef]
- Jurcău, M.C.; Andronie-Cioara, F.L.; Jurcău, A.; Marcu, F.; Tit, D.M.; Pașcalău, N.; Nistor-Cseppentö, D.C. The Link between Oxidative Stress, Mitochondrial Dysfunction and Neuroinflammation in the Pathophysiology of Alzheimer’s Disease: Therapeutic Implications and Future Perspectives. Antioxidants 2022, 11, 2167. [Google Scholar] [CrossRef]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef]
- Frijhoff, J.; Winyard, P.G.; Zarkovic, N.; Davies, S.S.; Stocker, R.; Cheng, D.; Knight, A.R.; Taylor, E.L.; Oettrich, J.; Ruskovska, T.; et al. Clinical Relevance of Biomarkers of Oxidative Stress. Antioxid. Redox Signal. 2015, 23, 1144–1170. [Google Scholar] [CrossRef]
- Mani, S. Production of Reactive Oxygen Species and Its Implication in Human Diseases. In Free Radicals in Human Health and Disease; Springer: Berlin/Heidelberg, Germany, 2014; pp. 3–15. [Google Scholar] [CrossRef]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef]
- Kumar, V.; Bishayee, K.; Park, S.; Lee, U.; Kim, J. Oxidative stress in cerebrovascular disease and associated diseases. Front. Endocrinol. 2023, 14, 1124419. [Google Scholar] [CrossRef]
- Klein, J.A.; Ackerman, S.L. Oxidative stress, cell cycle, and neurodegeneration. J. Clin. Investig. 2003, 111, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Reinstadler, B.; Engelstad, K.; Skinner, O.S.; Stackowitz, E.; Haller, R.G.; Clish, C.B.; Pierce, K.; Walker, M.A.; Fryer, R.; et al. Circulating markers of NADH-reductive stress correlate with mitochondrial disease severity. J. Clin. Investig. 2021, 131, e136055. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, M.; Zhu, W.; Yu, J.; Wang, Q.; Zhang, J.; Cui, Y.; Pan, X.; Gao, X.; Sun, H. Succinate accumulation induces mitochondrial reactive oxygen species generation and promotes status epilepticus in the kainic acid rat model. Redox Biol. 2020, 28, 101365. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A.; Ardelean, I.A. Molecular pathophysiological mechanisms of ischemia/reperfusion injuries after recanalization therapy for acute ischemic stroke. J. Integr. Neurosci. 2021, 20, 727–744. [Google Scholar] [CrossRef]
- Jones, D.N.; Raghanti, M.A. The role of monoamine oxidase enzymes in the pathophysiology of neurological disorders. J. Chem. Neuroanat. 2021, 114, 101957. [Google Scholar] [CrossRef]
- Rahman, M.S.; Uddin, M.S.; Rahman, M.A.; Samsuzzaman, M.; Behl, T.; Hafeez, A.; Perveen, A.; Barreto, G.E.; Ashraf, G.M. Exploring the Role of Monoamine Oxidase Activity in Aging and Alzheimer’s Disease. Curr. Pharm. Des. 2021, 27, 4017–4029. [Google Scholar] [CrossRef]
- Bettendorf, L. Reduced Nucleotides, Thiols and O2 in Cellular Redox Balance: A Biochemist’s View. Antioxidants 2022, 11, 1877. [Google Scholar] [CrossRef]
- Starkov, A.A. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann. N. Y. Acad. Sci. 2006, 1147, 37–52. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Baes, M.; Ribeiro, D.; Ferdinandusse, S.; Waterham, H.R. The physiological functions of human peroxisomes. Physiol. Rev. 2023, 103, 957–1024. [Google Scholar]
- Fang, J.; Sheng, R.; Qin, Z.H. NADPH Oxidases in the Central Nervous System: Regional and Cellular Localization and the Possible Link to Brain Diseases. Antioxid. Redox Signal. 2021, 35, 951–973. [Google Scholar] [CrossRef]
- Jurcau, A. Insights into the Pathogenesis of Neurodegenerative Diseases: Focus on Mitochondrial Dysfunction and Oxidative Stress. Int. J. Mol. Sci. 2021, 22, 11847. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J. Neurosci. 2004, 24, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Al-Mamary, A.M.; Moussa, Z. Antioxidant Activity: The Presence and Impact of Hydroxyl Groups in Small Molecules of Natural and Synthetic Origin. In Antioxidants—Benefits, Sources, Mechanisms of Action; Waisundara, V.Y., Ed.; IntechOpen: London, UK, 2021; pp. 253–280. [Google Scholar] [CrossRef]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide Dismutase Multigene Family: A Comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) Gene Structures, Evolution, and Expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Dasuri, K.; Zhang, L.; Keller, J.N. Oxidative Stress, Neurodegeneration, and the Balance of Protein Degradation and Protein Synthesis. Free Radic. Biol. Med. 2013, 62, 170–185. [Google Scholar] [CrossRef]
- Gandhi, S.; Abramov, A.Y. Mechanism of Oxidative Stress in Neurodegeneration. Oxidative Med. Cell. Longev. 2012, 2012, 428010. [Google Scholar] [CrossRef]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione Peroxidases. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Power, J.H.T.; Blumbergs, P.C. Cellular Glutathione Peroxidase in Human Brain: Cellular Distribution, and Its Potential Role in the Degradation of Lewy Bodies in Parkinson’s Disease and Dementia with Lewy Bodies. Acta Neuropathol. 2008, 117, 63–73. [Google Scholar] [CrossRef]
- Sabetta, W.; Paradiso, A.; Paciolla, C.; Concetta, M. Chemistry, Biosynthesis, and Antioxidative Function of Glutathione in Plants. In Glutathione in Plant Growth, Development, and Stress Tolerance; Springer: Berlin/Heidelberg, Germany, 2017; pp. 1–27. [Google Scholar] [CrossRef]
- Dringen, R. Metabolism and Functions of Glutathione in Brain. Prog. Neurobiol. 2000, 62, 649–671. [Google Scholar] [CrossRef]
- Dringen, R.; Hirrlinger, J. Glutathione Pathways in the Brain. Biol. Chem. 2003, 384, 505–516. [Google Scholar] [CrossRef]
- Presnell, C.E.; Bhatti, G.; Numan, L.S.; Lerche, M.; Alkhateeb, S.K.; Ghalib, M.; Shammaa, M.; Kavdia, M. Computational Insights into the Role of Glutathione in Oxidative Stress. Curr. Neurovascular Res. 2013, 10, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Kang, S.M.; Lee, W.T.; Park, K.A.; Lee, K.M.; Lee, J.E. Glutathione Protects Brain Endothelial Cells from Hydrogen Peroxide-Induced Oxidative Stress by Increasing Nrf2 Expression. Exp. Neurobiol. 2014, 23, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Zou, M.-H. Roles of Reactive Oxygen Species in Physiology and Pathology. Atherosclerosis 2015, 27, 379–392. [Google Scholar] [CrossRef]
- Bjørklund, G.; Shanaida, M.; Lysiuk, R.; Antonyak, H.; Klishch, I.; Shanaida, V.; Peana, M. Selenium: An Antioxidant with a Critical Role in Anti-Aging. Molecules 2022, 27, 6613. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.R.; Akash, S.; Jony, M.H.; Alam, M.N.; Nowrin, F.T.; Rahman, M.M.; Rauf, A.; Thiruvengadam, M. Exploring the potential function of trace elements in human health: A therapeutic perspective. Mol. Cell. Biochem. 2023, 478, 2141–2171. [Google Scholar] [CrossRef] [PubMed]
- Olechno, E.; Puścion-Jakubik, A.; Socha, K.; Zujko, M.E. Coffee Infusions: Can They Be a Source of Microelements with Antioxidant Properties? Antioxidants 2021, 10, 1709. [Google Scholar] [CrossRef]
- Araki, S.; Osuka, K.; Takata, T.; Tsuchiya, Y.; Watanabe, Y. Coordination between Calcium/Calmodulin-Dependent Protein Kinase II and Neuronal Nitric Oxide Synthase in Neurons. Int. J. Mol. Sci. 2020, 21, 7997. [Google Scholar] [CrossRef]
- Faria-Pereira, A.; Morais, V.A. Synapses: The Brain’s Energy-Demanding Sites. Int. J. Mol. Sci. 2022, 23, 3627. [Google Scholar] [CrossRef]
- Ishihara, Y.; Itoh, K. Microglial inflammatory reactions regulated by oxidative stress. J. Clin. Biochem. Nutr. 2023, 72, 23–27. [Google Scholar] [CrossRef]
- Olufunmilayo, E.O.; Gerke-Duncan, M.B.; Holsinger, R.M.D. Oxidative Stress and Antioxidants in Neurodegenerative Disorders. Antioxidants 2023, 12, 517. [Google Scholar] [CrossRef]
- Que, E.L.; Domaille, D.W.; Chang, C.J. Metals in neurobiology: Probing their chemistry and biology with molecular imaging. Chem. Rev. 2008, 39, 1517–1549. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K. Glutathione in the Brain. Int. J. Mol. Sci. 2021, 22, 5010. [Google Scholar] [CrossRef] [PubMed]
- Cobley, N.J.; Fiorello, M.L.; Bailey, D.M. 13 reasons the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–513. [Google Scholar] [CrossRef] [PubMed]
- Aliperti, V.; Skonieczna, J.; Cerase, A. Long Non-Coding RNA (lncRNA) Roles in Cell Biology, Neurodevelopment and Neurological Disorders. Non-Coding RNA 2021, 7, 36. [Google Scholar] [CrossRef]
- Giorgio, M.; Dellino, G.I.; Gambino, V.; Roda, N.; Pelicci, P.G. On the epigenetic role of guanosine oxidation. Redox Biol. 2020, 29, 101398. [Google Scholar] [CrossRef]
- McQuillen, P.S.; Ferriero, D.M. Selective Vulnerability in the Developing Central Nervous System. Pediatr. Neurol. 2004, 30, 227–235. [Google Scholar] [CrossRef]
- Fujii, J.; Homma, T.; Osaki, T. Superoxide Radicals in the Execution of Cell Death. Antioxidants 2022, 11, 501. [Google Scholar] [CrossRef]
- Luo, J.; Mills, K.; le Cessie, S.; Noordam, R.; van Heemst, D. Ageing, age-related diseases and oxidative stress: What to do next? Ageing Res. Rev. 2020, 57, 100982. [Google Scholar] [CrossRef]
- Yun, H.R.; Jo, Y.H.; Kim, J.; Shin, Y.; Kim, S.S.; Choi, T.G. Roles of Autophagy in Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 3289. [Google Scholar] [CrossRef]
- Wesch, N.; Kirkin, V.; Rogov, V.V. Atg8-Family Proteins—Structural Features and Molecular Interactions in Autophagy and Beyond. Cells 2020, 9, 2008. [Google Scholar] [CrossRef]
- Bernardo, V.S.; Torres, F.F.; da Silva, D.G.H. FoxO3 and oxidative stress: A multifaceted role in cellular adaptation. J. Mol. Med. 2023, 101, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Hu, Z.W.; Mao, C.Y.; Shi, C.H.; Xu, Y.M. CHIP as a therapeutic target for neurological diseases. Cell Death Dis. 2020, 11, 727. [Google Scholar] [CrossRef] [PubMed]
- Valgimigli, L. Lipid Peroxidation and Antioxidant Protection. Biomolecules 2023, 13, 1291. [Google Scholar] [CrossRef] [PubMed]
- Zgorzynska, E.; Dziedzic, B.; Walczewska, A. An Overview of the Nrf2/ARE Pathway and Its Role in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 9592. [Google Scholar] [CrossRef]
- Capece, D.; Verzella, D.; Flati, I.; Arboretto, P.; Cornice, J.; Franzoso, G. NF-κB: Blending metabolism, immunity, and inflammation. Trends Immunol. 2022, 43, 757–775. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, T.; Li, J.; Xia, M.; Li, Y.; Wang, X.; Liu, C.; Zheng, T.; Chen, R.; Kan, D.; et al. Oxidative Stress and 4-hydroxy-2-nonenal (4-HNE): Implications in the Pathogenesis and Treatment of Aging-related Diseases. J. Immunol. Res. 2022, 2022, 2233906. [Google Scholar] [CrossRef]
- Montecillo-Aguado, M.; Tirado-Rodriguez, B.; Huerta-Yepez, S. The Involvement of Polyunsaturated Fatty Acids in Apoptosis Mechanisms and Their Implications in Cancer. Int. J. Mol. Sci. 2023, 24, 11691. [Google Scholar] [CrossRef]
- Shadfar, S.; Parakh, S.; Jamali, M.S.; Atkin, J.D. Redox dysregulation as a driver for DNA damage and its relationship to neurodegenerative diseases. Transl. Neurodegener. 2023, 12, 18. [Google Scholar] [CrossRef]
- Jurcau, M.C.; Jurcau, A.; Cristian, A.; Hogea, V.O.; Diaconu, R.G.; Nunkoo, V.S. Inflammaging and Brain Aging. Int. J. Mol. Sci. 2024, 25, 10535. [Google Scholar] [CrossRef]
- Wong, G.C.; Chow, K.H. DNA Damage Response-Associated Cell Cycle Re-Entry and Neuronal Senescence in Brain Aging and Alzheimer’s Disease. J. Alzheimers Dis. 2023, 94, S429–S451. [Google Scholar] [CrossRef]
- Rong, Z.; Tu, P.; Xu, P.; Sun, Y.; Yu, F.; Tu, N.; Guo, L.; Yang, Y. The Mitochondrial Response to DNA Damage. Front. Cell Dev. Biol. 2021, 9, 669379. [Google Scholar] [CrossRef]
- Basu, S.; Song, M.; Adams, L.; Jeong, I.; Je, G.; Guhathakurta, S.; Jiang, J.; Boparai, N.; Dai, W.; Cardozo-Pelaez, F.; et al. Transcriptional mutagenesis of α-synuclein caused by DNA oxidation in Parkinson’s disease pathogenesis. Acta Neuropathol. 2023, 146, 685–705. [Google Scholar] [CrossRef]
- Bazzani, V.; Equisoain Redin, M.; McHale, J.; Perrone, L.; Vascotto, C. Mitochondrial DNA Repair in Neurodegenerative Diseases and Ageing. Int. J. Mol. Sci. 2022, 23, 11391. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, X.; Li, Z.; Ye, W.; Ding, H.; Li, P.; Aung, L.H.H. Role of RNA Oxidation in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 5022. [Google Scholar] [CrossRef]
- Akiyama, Y.; Ivanov, P. Oxidative Stress, Transfer RNA Metabolism, and Protein Synthesis. Antioxid. Redox Signal. 2024, 40, 715–735. [Google Scholar] [CrossRef]
- Eshraghi, M.; Adlimoghaddam, A.; Mahmoodzadeh, A.; Sharifzad, F.; Yasavoli-Sharahi, H.; Lorzadeh, S.; Albensi, B.C.; Ghavami, S. Alzheimer’s disease pathogenesis: Role of autophagy and mitophagy focusing in microglia. Int. J. Mol. Sci. 2021, 22, 3330. [Google Scholar] [CrossRef]
- Perluigi, M.; Di Domenico, F.; Barone, E.; Butterfield, D.A. mTOR in Alzheimer disease and its earlier stages: Links to oxidative damage in the progression of this dementing disorder. Free Radic. Biol. Med. 2021, 169, 382–396. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell. 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Kharitonova, E.K.; Snyder, A.C.; Hou, S.S.; Sanchez-Mico, M.V.; Das, S.; Fan, Z.; Shirani, H.; Nilsson, K.P.R.; Serrano-Pozo, A.; et al. Real-time imaging of mitochondrial redox reveals increased mitochondrial oxidative stress associated with amyloid β aggregates in vivo in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2024, 19, 6. [Google Scholar] [CrossRef]
- Tamagno, E.; Guglielmotto, M.; Vasciaveo, V.; Tabaton, M. Oxidative Stress and Beta Amyloid in Alzheimer’s Disease. Which Comes First: The Chicken or the Egg? Antioxidants 2021, 10, 1479. [Google Scholar] [CrossRef]
- Alqahtani, T.; Deore, S.L.; Kide, A.A.; Shende, B.A.; Sharma, R.; Dadarao Chakole, R.; Nemade, L.S.; Kishor Kale, N.; Borah, S.; Shrikant Deokar, S.; et al. Mitochondrial dysfunction and oxidative stress in Alzheimer’s disease, and Parkinson’s disease, Huntington’s disease and Amyotrophic Lateral Sclerosis—An updated review. Mitochondrion 2023, 71, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Thorne, N.J.; Tumbarello, D.A. The relationship of alpha-synuclein to mitochondrial dynamics and quality control. Front. Mol. Neurosci. 2022, 15, 947191. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Balachandran, Y.L.; Chong, W.P.; Chan, K.W.Y. Roles of Cytokines in Alzheimer’s Disease. Int. J. Mol. Sci. 2024, 25, 5803. [Google Scholar] [CrossRef] [PubMed]
- Brahadeeswaran, S.; Sivagurunathan, N.; Calivarathan, L. Inflammasome Signaling in the Aging Brain and Age-Related Neurodegenerative Diseases. Mol. Neurobiol. 2022, 59, 2288–2304. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A.; Andronie-Cioara, F.L.; Nistor-Cseppento, D.C.; Pascalau, N.; Rus, M.; Vasca, E.; Jurcau, M.C. The Involvement of Neuroinflammation in the Onset and Progression of Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 14582. [Google Scholar] [CrossRef]
- Mattson, M.P. Pathways towards and Away from Alzheimer’s Disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef]
- Houldsworth, A. Role of Oxidative Stress in Neurodegenerative Disorders: A Review of Reactive Oxygen Species and Prevention by Antioxidants. Brain Commun. 2024, 6, fcad356. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.; Zhu, X. Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef]
- Ebanks, B.; Chakrabarti, L. Mitochondrial ATP Synthase is a Target of Oxidative Stress in Neurodegenerative Diseases. Front. Mol. Biosci. 2022, 9, 854321. [Google Scholar] [CrossRef]
- Patro, S.; Ratna, S.; Yamamoto, H.A.; Ebenezer, A.T.; Ferguson, D.S.; Kaur, A.; McIntyre, B.C.; Snow, R.; Solesio, M.E. ATP Synthase and Mitochondrial Bioenergetics Dysfunction in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 11185. [Google Scholar] [CrossRef]
- Maruthiyodan, S.; Mumbrekar, K.D.; Guruprasad, K.P. Involvement of mitochondria in Alzheimer’s disease pathogenesis and their potential as targets for phytotherapeutics. Mitochondrion 2024, 76, 101868. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Oliver, D.M. Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer’s disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [PubMed]
- Nabi, S.U.; Khan, A.; Siddiqui, E.M.; Rehman, M.U.; Alshahrani, S.; Arafah, A.; Mehan, S.; Alsaffar, R.M.; Alexiou, A.; Shen, B. Mechanisms of Mitochondrial Malfunction in Alzheimer’s Disease: New Therapeutic Hope. Oxidative Med. Cell. Longev. 2022, 2022, 4759963. [Google Scholar] [CrossRef] [PubMed]
- Tobore, T.O. On the central role of mitochondria dysfunction and oxidative stress in Alzheimer’s disease. Neurol. Sci. 2019, 40, 1527–1540. [Google Scholar] [CrossRef]
- Fermaintt, C.S.; Wacker, S.A. Malate dehydrogenase as a multi-purpose target for drug discovery. Essays Biochem. 2024, 68, 147–160. [Google Scholar]
- Chen, F.; Bai, J.; Zhong, S.; Zhang, R.; Zhang, X.; Xu, Y.; Zhao, M.; Zhao, C.; Zhou, Z. Molecular Signatures of Mitochondrial Complexes Involved in Alzheimer’s Disease via Oxidative Phosphorylation and Retrograde Endocannabinoid Signaling Pathways. Oxidative Med. Cell. Longev. 2022, 2022, 9565545. [Google Scholar] [CrossRef]
- Cores, Á.; Carmona-Zafra, N.; Clerigué, J.; Villacampa, M.; Menéndez, J.C. Quinones as Neuroprotective Agents. Antioxidants 2023, 12, 1464. [Google Scholar] [CrossRef]
- Di Domenico, F.; Tramutola, A.; Butterfield, D.A. Role of 4-hydroxynonenal (HNE) in the pathogenesis of Alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic. Biol. Med. 2017, 111, 253–261. [Google Scholar] [CrossRef]
- Ebanks, B.; Ingram, T.L.; Chakrabarti, L. ATP synthase and Alzheimer’s disease: Putting a spin on the mitochondrial hypothesis. Aging 2020, 12, 16647–16662. [Google Scholar] [CrossRef]
- Campos-Peña, V.; Pichardo-Rojas, P.; Sánchez-Barbosa, T.; Ortíz-Islas, E.; Rodríguez-Pérez, C.E.; Montes, P.; Ramos-Palacios, G.; Silva-Adaya, D.; Valencia-Quintana, R.; Cerna-Cortes, J.F.; et al. Amyloid β, Lipid Metabolism, Basal Cholinergic System, and Therapeutics in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 12092. [Google Scholar] [CrossRef]
- Lin, X.; Kapoor, A.; Gu, Y.; Chow, M.J.; Peng, J.; Zhao, K.; Tang, D. Contributions of DNA Damage to Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1666. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Guo, J.; Ye, X.Y.; Xie, Y.; Xie, T. Oxidative stress: The core pathogenesis and mechanism of Alzheimer’s disease. Ageing Res. Rev. 2022, 77, 101619. [Google Scholar] [CrossRef] [PubMed]
- Paß, T.; Wiesner, R.J.; Pla-Martin, D. Selective neuron vulnerability in common and rare diseases–mitochondria in the focus. Front. Mol. Biosci. 2021, 8, 676187. [Google Scholar] [CrossRef] [PubMed]
- Khanam, H.; Ali, A.; Asif, M. Shamsuzzaman Neurodegenerative diseases linked to misfolded proteins and their therapeutic approaches: A review. Eur. J. Med. Chem. 2016, 124, 1121–1141. [Google Scholar] [CrossRef]
- Hevner, R.F.; Wong-Riley, M.T. Entorhinal cortex of the human, monkey, and rat: Metabolic map as revealed by cytochrome oxidase. J. Comp. Neurol. 1992, 326, 451–469. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of glutamate and NMDA receptors in Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef]
- Hof, P.R.; Nimchinsky, E.A.; Celio, M.R.; Bouras, C.; Morrison, J.H. Calretinin-immunoreactive neocortical interneurons are unaffected in Alzheimer’s disease. Neurosci. Lett. 1993, 152, 145–148. [Google Scholar] [CrossRef]
- Roussarie, J.-P.; Yao, V.; Rodriguez-Rodriguez, P.; Oughtred, R.; Rust, J.; Plautz, Z.; Kasturia, S.; Albornoz, C.; Wang, W.; Schmidt, E.F.; et al. Selective neuronal vulnerability in Alzheimer’s disease: A network-based analysis. Neuron 2020, 107, 821–835. [Google Scholar] [CrossRef]
- Kaufman, S.K.; Del Tredici, K.; Thomas, T.L.; Braak, H.; Diamond, M.I. Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer’s disease and PART. Acta Neuropathol. 2018, 136, 57–67. [Google Scholar] [CrossRef]
- Zhu, J.; Cui, Y.; Zhang, J.; Yan, R.; Su, D.; Zhao, D.; Wang, A.; Feng, T. Temporal Trends in the Prevalence of Parkinson’s Disease from 1980 to 2023: A Systematic Review and Meta-Analysis. Lancet Healthy Longev. 2024, 5, e464–e479. [Google Scholar] [CrossRef]
- Pathania, A.; Garg, P.; Sandhir, R. Impaired mitochondrial functions and energy metabolism in MPTP-induced Parkinson’s disease: Comparison of mice strains and dose regimens. Metab. Brain Dis. 2021, 36, 2343–2357. [Google Scholar] [CrossRef] [PubMed]
- González-Rodríguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gao, L.; et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature. 2021, 599, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M.; Dawson, V.L. Molecular pathways of neurodegeneration in Parkinson’s disease. Science 2003, 302, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Dorszewska, J.; Kowalska, M.; Prendecki, M.; Piekut, T.; Kozłowska, J.; Kozubski, W. Oxidative stress factors in Parkinson’s disease. Neural Regen. Res. 2021, 16, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, M.; Miyazaki, I. Glutathione and Related Molecules in Parkinsonism. Int. J. Mol. Sci. 2021, 22, 8689. [Google Scholar] [CrossRef]
- Watanabe, H.; Dijkstra, J.M.; Nagatsu, T. Parkinson’s Disease: Cells Succumbing to Lifelong Dopamine-Related Oxidative Stress and Other Bioenergetic Challenges. Int. J. Mol. Sci. 2024, 25, 2009. [Google Scholar] [CrossRef]
- Thomas, K.J.; McCoy, M.K.; Blackinton, J.; Beilina, A.; Van der Brug, M.; Sandebring, A.; Miller, D.; Maric, D.; Cedazo-Minguez, A.; Cookson, M.R. DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum. Mol. Genet. 2011, 20, 40–50. [Google Scholar] [CrossRef]
- Williamson, M.G.; Madureira, M.; McGuinness, W.; Heon-Roberts, R.; Mock, E.D.; Naidoo, K.; Cramb, K.M.L.; Caiazza, M.C.; Malpartida, A.B.; Lavelle, M.; et al. Mitochondrial dysfunction and mitophagy defects in LRRK2-R1441C Parkinson’s disease models. Hum. Mol. Genet. 2023, 32, 2808–2821. [Google Scholar] [CrossRef]
- Hur, E.-M.; Lee, B.D. LRRK2 at the Crossroad of Aging and Parkinson’s Disease. Genes 2021, 12, 505. [Google Scholar] [CrossRef]
- Nguyen, D.; Bharat, V.; Conradson, D.M.; Nandakishore, P.; Wang, X. Miro1 Impairment in a Parkinson’s At-Risk Cohort. Front. Mol. Neurosci. 2021, 14, 734273. [Google Scholar] [CrossRef]
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010, 468, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Lawless, C.; Greaves, L.; Reeve, A.K.; Turnbull, D.M.; Vincent, A.E. The rise and rise of mitochondrial DNA mutations. Open Biol. 2020, 10, 200061. [Google Scholar] [CrossRef] [PubMed]
- Pacelli, C.; Giguère, N.; Bourque, M.-J.; Lévesque, M.; Slack, R.S.; Trudeau, L.-É. Elevated mitochondrial bioenergetics and axonal arborization size are key contributors to the vulnerability of dopamine neurons. Curr. Biol. 2015, 25, 2349–2360. [Google Scholar] [CrossRef] [PubMed]
- Michel, P.P.; Hirsch, E.C.; Hunot, S. Understanding dopaminergic cell death pathways in Parkinson disease. Neuron 2016, 90, 675–691. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, W.; Furuta, T.; Nakamura, K.C.; Hioki, H.; Fujiyama, F.; Arai, R.; Kaneko, T. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J. Neurosci. 2009, 29, 444–453. [Google Scholar] [CrossRef]
- Bolam, J.P.; Pissadaki, E.K. Living on the edge with too many mouths to feed: Why dopamine neurons die. Mov. Disord. 2012, 27, 1478–1483. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The Role of Mitochondria in Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2017, 710, 132933. [Google Scholar] [CrossRef]
- Cunha-Oliveira, T.; Montezinho, L.; Mendes, C.; Firuzi, O.; Saso, L.; Oliveira, P.J.; Silva, F.S.G. Oxidative Stress in Amyotrophic Lateral Sclerosis: Pathophysiology and Opportunities for Pharmacological Intervention. Oxidative Med. Cell. Longev. 2020, 2020, 1–29. [Google Scholar] [CrossRef]
- Smirnova, J.; Gavrilova, J.; Noormägi, A.; Valmsen, K.; Pupart, H.; Luo, J.; Tõugu, V.; Palumaa, P. Evaluation of Zn2+- and Cu2+-Binding Affinities of Native Cu,Zn-SOD1 and Its G93A Mutant by LC-ICP MS. Molecules 2022, 27, 3160. [Google Scholar] [CrossRef]
- Martin, L.J.; Koh, S.J.; Price, A.; Park, D.; Kim, B.W. Nuclear Localization of Human SOD1 in Motor Neurons in Mouse Model and Patient Amyotrophic Lateral Sclerosis: Possible Links to Cholinergic Phenotype, NADPH Oxidase, Oxidative Stress, and DNA Damage. Int. J. Mol. Sci. 2024, 25, 9106. [Google Scholar] [CrossRef]
- Healy, E.F.; Roth-Rodriguez, A.; Toledo, S. A model for gain of function in superoxide dismutase. Biochem. Biophys. Rep. 2020, 21, 100728. [Google Scholar] [CrossRef] [PubMed]
- Sanghai, N.; Tranmer, G.K. Hydrogen Peroxide and Amyotrophic Lateral Sclerosis: From Biochemistry to Pathophysiology. Antioxidants 2022, 11, 52. [Google Scholar] [CrossRef] [PubMed]
- Dangoumau, A.; Marouillat, S.; Coelho, R.; Wurmser, F.; Brulard, C.; Haouari, S.; Laumonnier, F.; Corcia, P.; Andres, C.R.; Blasco, H.; et al. Dysregulations of Expression of Genes of the Ubiquitin/SUMO Pathways in an In Vitro Model of Amyotrophic Lateral Sclerosis Combining Oxidative Stress and SOD1 Gene Mutation. Int. J. Mol. Sci. 2021, 22, 1796. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.B. Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, W.; Mitra, J.; Hegde, P.M.; Vandoorne, T.; Eckelmann, B.J.; Mitra, S.; Tomkinson, A.E.; Van Den Bosch, L.; Hegde, M.L. Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Lateral Sclerosis. Nat. Commun. 2018, 9, 3683. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS genetics, mechanisms, and therapeutics: Where are we now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef]
- Diaz-Garcia, C.M.; Mongeon, R.; Lahmann, C.; Koveal, D.; Zucker, H.; Yellen, G. Neuronal stimulation triggers neuronal glycolysis and not lactate uptake. Cell Metab. 2017, 26, 361–374. [Google Scholar] [CrossRef]
- Neuhaus, J.F.; Baris, O.R.; Hess, S.; Moser, N.; Schroder, H.; Chinta, S.J.; Andersen, J.K.; Kloppenburg, P.; Wiesner, R.J. Catecholamine metabolism drives generation of mitochondrial DNA deletions in dopaminergic neurons. Brain 2014, 137, 354–365. [Google Scholar] [CrossRef]
- Hardiman, O.; van den Berg, L.H. Edaravone: A New Treatment for ALS on the Horizon? Lancet Neurol. 2017, 16, 490–491. [Google Scholar] [CrossRef]
- Gao, M.; Zhu, L.; Chang, J.; Cao, T.; Song, L.; Wen, C.; Chen, Y.-X.; Zhuo, Y.; Chen, F. Safety and Efficacy of Edaravone in Patients with Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Clin. Drug Investig. 2022, 43, 1–11. [Google Scholar] [CrossRef]
- Sala, G.; Arosio, A.; Conti, E.; Beretta, S.; Lunetta, C.; Riva, N.; Ferrarese, C.; Tremolizzo, L. Riluzole Selective Antioxidant Effects in Cell Models Expressing Amyotrophic Lateral Sclerosis Endophenotypes. Clin. Psychopharmacol. Neurosci. 2019, 17, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Casati, M.; Boccardi, V.; Ferri, E.; Bertagnoli, L.; Bastiani, P.; Ciccone, S.; Mansi, M.; Scamosci, M.; Rossi, P.D.; Mecocci, P.; et al. Vitamin E and Alzheimer’s disease: The mediating role of cellular aging. Aging Clin. Exp. Res. 2020, 32, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Lloret, A.; Esteve, D.; Monllor, P.; Cervera-Ferri, A.; Lloret, A. The Effectiveness of Vitamin E Treatment in Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 879. [Google Scholar] [CrossRef] [PubMed]
- Cowan, C.M.; Sealey, M.A.; Mudher, A. Suppression of tau-induced phenotypes by vitamin E demonstrates the dissociation of oxidative stress and phosphorylation in mechanisms of tau toxicity. J. Neurochem. 2021, 157, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Pourhanifeh, M.H.; Hosseinzadeh, A.; Koosha, F.; Reiter, R.J.; Mehrzadi, S. Therapeutic Effects of Melatonin in the Regulation of Ferroptosis: A Review of Current Evidence. Curr. Drug Targets 2024, 25, 543–557. [Google Scholar] [CrossRef]
- Faisal, Z.; Mazhar, A.; Batool, S.A.; Akram, N.; Hassan, M.; Khan, M.U.; Afzaal, M.; Hassan, U.U.; Shah, Y.A.; Desta, D.T. Exploring the multimodal health-promoting properties of resveratrol: A comprehensive review. Food Sci. Nutr. 2024, 12, 2240–2258. [Google Scholar] [CrossRef]
- Kaur, D.; Behl, T.; Sehgal, A.; Singh, S.; Sharma, N.; Chigurupati, S.; Alhowail, A.; Abdeen, A.; Ibrahim, S.F.; Vargas-De-La-Cruz, C.; et al. Decrypting the potential role of α-lipoic acid in Alzheimer’s disease. Life Sci. 2021, 284, 119899. [Google Scholar] [CrossRef]
- Speisky, H.; Shahidi, F.; Costa de Camargo, A.; Fuentes, J. Revisiting the Oxidation of Flavonoids: Loss, Conservation or Enhancement of Their Antioxidant Properties. Antioxidants 2022, 11, 133. [Google Scholar] [CrossRef]
- Kabir, M.T.; Rahman, M.H.; Shah, M.; Jamiruddin, M.R.; Basak, D.; Al-Harrasi, A.; Bhatia, S.; Ashraf, G.M.; Najda, A.; El-Kott, A.F.; et al. Therapeutic promise of carotenoids as antioxidants and anti-inflammatory agents in neurodegenerative disorders. Biomed. Pharmacother. 2022, 146, 112610. [Google Scholar] [CrossRef]
- Jiang, Q.; Yin, J.; Chen, J.; Ma, X.; Wu, M.; Liu, G.; Yao, K.; Tan, B.; Yin, Y. Mitochondria-Targeted Antioxidants: A Step towards Disease Treatment. Oxidative Med. Cell. Longev. 2020, 2020, 8837893. [Google Scholar]
- Zinovkin, R.A.; Zamyatnin, A.A. Mitochondria-Targeted Drugs. Curr. Mol. Pharmacol. 2019, 12, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Oliver, D.M.A.; Reddy, P.H. Small molecules as therapeutic drugs for Alzheimer’s disease. Mol. Cell. Neurosci. 2019, 96, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Young, M.L.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ inhibits memory loss, neuropathology, and extends lifespan in aged 3xTg-AD mice. Mol. Cell. Neurosci. 2019, 101, 103409. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Smith, R.A.J. Targeting Antioxidants to Mitochondria by Conjugation to Lipophilic Cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar] [CrossRef]
- Hu, H.; Li, M. Mitochondria-targeted antioxidant mitotempo protects mitochondrial function against amyloid beta toxicity in primary cultured mouse neurons. Biochem. Biophys. Res. Commun. 2016, 478, 174–180. [Google Scholar] [CrossRef]
- Du, F.; Yu, Q.; Kanaan, N.M.; Yan, S.S. Mitochondrial oxidative stress contributes to the pathological aggregation and accumulation of tau oligomers in Alzheimer’s disease. Hum. Mol. Genet. 2022, 31, 2498–2507. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef]
- Abdul-Rahman, T.; Awuah, W.A.; Mikhailova, T.; Kalmanovich, J.; Mehta, A.; Ng, J.C.; Coghlan, M.A.; Zivcevska, M.; Tedeschi, A.J.; de Oliveira, E.C.; et al. Antioxidant, anti-inflammatory and epigenetic potential of curcumin in Alzheimer’s disease. Biofactors 2024, 50, 693–708. [Google Scholar] [CrossRef]
- Tang, S.; Zhang, Y.; Botchway, B.O.A.; Wang, X.; Huang, M.; Liu, X. Epigallocatechin-3-Gallate Inhibits Oxidative Stress Through the Keap1/Nrf2 Signaling Pathway to Improve Alzheimer Disease. Mol. Neurobiol. 2024; ahead of print. [Google Scholar] [CrossRef]
- Shi, Z.; Chen, H.; Zhou, X.; Yang, W.; Lin, Y. Pharmacological effects of natural medicine ginsenosides against Alzheimer’s disease. Front. Pharmacol. 2022, 13, 952332. [Google Scholar] [CrossRef]
- Sharif, R.; Aghsami, M.; Gharghabi, M.; Sanati, M.; Khorshidahmad, T.; Vakilzadeh, G.; Mehdizadeh, H.; Gholizadeh, S.; Taghizadeh, G.; Sharifzadeh, M. Melatonin reverses H-89 induced spatial memory deficit: Involvement of oxidative stress and mitochondrial function. Behav. Brain Res. 2017, 316, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Hantikainen, E.; Trolle Lagerros, Y.; Ye, W.; Serafini, M.; Adami, H.O.; Bellocco, R.; Bonn, S. Dietary Antioxidants and the Risk of Parkinson Disease: The Swedish National March Cohort. Neurology 2021, 96, e895–e903. [Google Scholar] [CrossRef] [PubMed]
- Group, P.S. Effects of tocopherol and deprenyl on the progression of disability in early Parkinson’s disease. N. Engl. J. Med. 1993, 328, 176–183. [Google Scholar]
- Shults, C.W.; Oakes, D.; Kieburtz, K.; Beal, M.F.; Haas, R.; Plumb, S.; Juncos, J.L.; Nutt, J.; Shoulson, I.; Carter, J.; et al. Effects of coenzyme Q10 in early Parkinson disease: Evidence of slowing of the functional decline. Arch. Neurol. 2002, 59, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Oakes, D.; Shoulson, I.; Henchcliffe, C.; Galpern, W.R.; Haas, R.; Juncos, J.L.; Nutt, J.G.; Voss, T.S.; Ravina, B.; et al. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: No evidence of benefit. JAMA Neurol. 2014, 71, 543–552. [Google Scholar]
- Su, L.Y.; Li, H.; Lv, L.; Feng, Y.M.; Li, G.D.; Luo, R.; Zhou, H.J.; Lei, X.G.; Ma, L.; Li, J.L.; et al. Melatonin attenuates MPTP-induced neurotoxicity via preventing CDK5-mediated autophagy and SNCA/α-synuclein aggregation. Autophagy 2015, 11, 1745–1759. [Google Scholar] [CrossRef]
- Ozsoy, O.; Yildirim, F.B.; Ogut, E.; Kaya, Y.; Tanriover, G.; Parlak, H.; Agar, A.; Aslan, M. Melatonin is protective against 6-hydroxydopamine-induced oxidative stress in a hemiparkinsonian rat model. Free Radic. Res. 2015, 49, 1004–1014. [Google Scholar] [CrossRef]
- Saravanan, K.S.; Sindhu, K.M.; Mohanakumar, K.P. Melatonin protects against rotenone-induced oxidative stress in a hemiparkinsonian rat model. J. Pineal. Res. 2007, 42, 247–253. [Google Scholar] [CrossRef]
- Ghosh, A.; Chandran, K.; Kalivendi, S.V.; Joseph, J.; Antholine, W.E.; Hillard, C.J.; Kanthasamy, A.; Kanthasamy, A.; Kalyanaraman, B. Neuroprotection by a mitochondria-targeted drug in a Parkinson’s disease model. Free Radic. Biol. Med. 2010, 49, 1674–1684. [Google Scholar] [CrossRef]
- Snow, B.J.; Rolfe, F.L.; Lockhart, M.M.; Frampton, C.M.; O’Sullivan, J.D.; Fung, V.; Smith, R.A.; Murphy, M.P.; Taylor, K.M. Protect Study Group. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord. 2010, 25, 670–674. [Google Scholar] [CrossRef]
- Fields, M.; Marcuzzi, A.; Gonelli, A.; Celeghini, C.; Maximova, N.; Rimondi, E. Mitochondria-Targeted Antioxidants, an Innovative Class of Antioxidant Compounds for Neurodegenerative Diseases: Perspectives and Limitations. Int. J. Mol. Sci. 2023, 24, 3739. [Google Scholar] [CrossRef] [PubMed]
- Shinn, L.J.; Lagalwar, S. Treating Neurodegenerative Disease with Antioxidants: Efficacy of the Bioactive Phenol Resveratrol and Mitochondrial-Targeted MitoQ and SkQ. Antioxidants 2021, 10, 573. [Google Scholar] [CrossRef] [PubMed]
- Pavshintsev, V.V.; Podshivalova, L.S.; Frolova, O.Y.; Belopolskaya, M.V.; Averina, O.A.; Kushnir, E.A.; Marmiy, N.V.; Lovat, M.L. Effects of Mitochondrial Antioxidant SkQ1 on Biochemical and Behavioral Parameters in a Parkinsonism Model in Mice. Biochemistry 2017, 82, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Zhang, Y.; Hu, Q.; Wu, J.; Ren, H.; Liu, C.-F.; Wang, G. P7C3 inhibits GSK3β activation to protect dopaminergic neurons against neurotoxin-induced cell death in vitro and in vivo. Cell Death. Dis. 2017, 8, e2858. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; O’Reilly, É.J.; Weisskopf, M.G.; Logroscino, G.; McCullough, M.L.; Schatzkin, A.; Kolonel, L.N.; Ascherio, A. Vitamin E intake and risk of amyotrophic lateral sclerosis: A pooled analysis of data from 5 prospective cohort studies. Am. J. Epidemiol. 2011, 173, 595–602. [Google Scholar] [CrossRef]
- Desnuelle, C.; Dib, M.; Garrel, C.; Favier, A. A double-blind, placebo-controlled randomized clinical trial of alpha-tocopherol (vitamin E) in the treatment of amyotrophic lateral sclerosis. ALS riluzole-tocopherol Study Group. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2001, 2, 9–18. [Google Scholar] [CrossRef]
- Lanznaster, D.; Bejan-Angoulvant, T.; Gandía, J.; Blasco, H.; Corcia, P. Is There a Role for Vitamin D in Amyotrophic Lateral Sclerosis? A Systematic Review and Meta-Analysis. Front. Neurol. 2020, 11, 697. [Google Scholar] [CrossRef]
- Matthews, R.T.; Yang, L.; Browne, S.; Baik, M.; Beal, M.F. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc. Natl. Acad. Sci. USA 1998, 95, 8892–8897. [Google Scholar] [CrossRef]
- Kawasaki, T.; Singh, R.B.; Germaine, C.; Franz, H. Effects of Coenzyme Q10 Administration in Amyotrophic Lateral Sclerosis (ALS). Report of a Case and Review. Open Nutraceuticals J. 2012, 5, 187–192. [Google Scholar] [CrossRef]
- Rauchová, H. Coenzyme Q10 effects in neurological diseases. Physiol. Res. 2021, 70, S683–S714. [Google Scholar] [CrossRef] [PubMed]
- Miquel, E.; Cassina, A.; Martínez-Palma, L.; Souza, J.M.; Bolatto, C.; Rodríguez-Bottero, S.; Logan, A.; Smith, R.A.; Murphy, M.P.; Barbeito, L.; et al. Neuroprotective effects of the mitochondria-targeted antioxidant MitoQ in a model of inherited amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2014, 70, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cook, A.; Kim, J.; Baranov, S.V.; Jiang, J.; Smith, K.; Cormier, K.; Bennett, E.; Browser, R.P.; Day, A.L.; et al. Melatonin inhibits the caspase-1/cytochrome c/caspase-3 cell death pathway, inhibits MT1 receptor loss and delays disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2013, 55, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Dardiotis, E.; Panayiotou, E.; Feldman, M.L.; Hadjisavvas, A.; Malas, S.; Vonta, I.; Hadjigeorgiou, G.; Kyriakou, K.; Kyriakides, T. Intraperitoneal melatonin is not neuroprotective in the G93ASOD1 transgenic mouse model of familial ALS and may exacerbate neurodegeneration. Neurosci. Lett. 2013, 548, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Crosstalk between Oxidative Stress and SIRT1: Impact on the Aging Process. Int. J. Mol. Sci. 2013, 14, 3834–3859. [Google Scholar] [CrossRef]
- Song, L.; Chen, L.; Zhang, X.; Li, J.; Le, W. Resveratrol ameliorates motor neuron degeneration and improves survival in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Biomed. Res Int. 2014, 2014, 483501. [Google Scholar] [CrossRef]
- Andreassen, O.A.; Dedeoglu, A.; Klivenyi, P.; Beal, M.F.; Bush, A.I. N-acetyl-L-cysteine improves survival and preserves motor performance in an animal model of familial amyotrophic lateral sclerosis. Neuroreport 2000, 11, 2491–2493. [Google Scholar] [CrossRef]
- Louwerse, E.S.; Weverling, G.J.; Bossuyt, P.M.; Meyjes, F.E.; de Jong, J.M. Randomized, double-blind, controlled trial of acetylcysteine in amyotrophic lateral sclerosis. Arch. Neurol. 1995, 52, 559–564. [Google Scholar] [CrossRef]
- Kurano, T.; Kanazawa, T.; Iioka, S.; Kondo, H.; Kosuge, Y.; Suzuki, T. Intranasal Administration of N-acetyl-L-cysteine Combined with Cell-Penetrating Peptide-Modified Polymer Nanomicelles as a Potential Therapeutic Approach for Amyotrophic Lateral Sclerosis. Pharmaceutics 2022, 14, 2590. [Google Scholar] [CrossRef]
- Arslanbaeva, L.; Bisaglia, M. Activation of the Nrf2 Pathway as a Therapeutic Strategy for ALS Treatment. Molecules 2022, 27, 1471. [Google Scholar] [CrossRef]
- Choi, H.J.; Cha, S.J.; Lee, J.W.; Kim, H.J.; Kim, K. Recent advances on the role of gsk3β in the pathogenesis of amyotrophic lateral sclerosis. Brain Sci. 2020, 10, 675. [Google Scholar] [CrossRef] [PubMed]
- Van Daele, S.H.; Masrori, P.; Van Damme, P.; Van Den Bosch, L. The sense of antisense therapies in ALS. Trends Mol. Med. 2024, 30, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A.; Jurcau, M.C. Therapeutic Strategies in Huntington’s Disease: From Genetic Defect to Gene Therapy. Biomedicines 2022, 10, 1895. [Google Scholar] [CrossRef] [PubMed]
- Available online: www.fda.gov/drugs/news-events-human-drugs/fda-approves-treatment-amyotrophic-lateral-sclerosis-associated-mutation-sod1-gene (accessed on 25 October 2024).
- Bartolome, F.; Carro, E.; Alquezar, C. Oxidative Stress in Tauopathies: From Cause to Therapy. Antioxidants 2022, 11, 1421. [Google Scholar] [CrossRef] [PubMed]
- Michalska, P.; León, R. When It Comes to an End: Oxidative Stress Crosstalk with Protein Aggregation and Neuroinflammation Induce Neurodegeneration. Antioxidants 2020, 9, 740. [Google Scholar] [CrossRef]
- Muller, M.; Banning, A.; Brigelius-Flohe, R.; Kipp, A. Nrf2 target genes are induced under marginal selenium-deficiency. Genes Nutr. 2010, 5, 297–307. [Google Scholar] [CrossRef]
- Logan, S.; Arzua, T.; Canfield, S.G.; Seminary, E.R.; Sison, S.L.; Ebert, A.D.; Bai, X. Studying Human Neurological Disorders Using Induced Pluripotent Stem Cells: From 2D Monolayer to 3D Organoid and Blood Brain Barrier Models. Compr. Physiol. 2019, 9, 565–611. [Google Scholar]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018, 285, 3576–3590. [Google Scholar] [CrossRef]
- Armeli, F.; Mengoni, B.; Laskin, D.L.; Businaro, R. Interplay among Oxidative Stress, Autophagy, and the Endocannabinoid System in Neurodegenerative Diseases: Role of the Nrf2- p62/SQSTM1 Pathway and Nutraceutical Activation. Curr. Issues Mol. Biol. 2024, 46, 6868–6884. [Google Scholar] [CrossRef]
- Profumo, E.; Maggi, E.; Arese, M.; Di Cristofano, C.; Salvati, B.; Saso, L.; Businaro, R.; Buttari, B. Neuropeptide Y Promotes Human M2 Macrophage Polarization and Enhances p62/SQSTM1-Dependent Autophagy and NRF2 Activation. Int. J. Mol. Sci. 2022, 23, 13009. [Google Scholar] [CrossRef]
- Zhang, W.; Feng, C.; Jiang, H. Novel target for treating Alzheimer’s Diseases: Crosstalk between the Nrf2 pathway and autophagy. Ageing Res. Rev. 2021, 65, 101207. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, C.; Lastres-Becker, I.; Demirdöğen, B.C.; Costa, V.M.; Daiber, A.; Foresti, R.; Motterlini, R.; Kalyoncu, S.; Arioz, B.I.; Genc, S.; et al. Biomarkers of NRF2 signaling: Current status and future challenges. Redox Biol. 2024, 72, 103134. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Murphy, M.P.; Bayir, H.; Belousov, V.; Chang, C.J.; Davies, K.J.A.; Davies, M.J.; Dick, T.P.; Finkel, T.; Forman, H.J.; Janssen-Heininger, Y.; et al. Guidelines for Measuring Reactive Oxygen Species and Oxidative Damage in Cells and in Vivo. Nat. Metab. 2022, 4, 651–662. [Google Scholar] [CrossRef]
- Stefanatos, R.; Sanz, A. The Role of Mitochondrial ROS in the Aging Brain. FEBS Lett. 2018, 592, 743–758. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting Oxidative Stress in Disease: Promise and Limitations of Antioxidant Therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Gammon, K. Neurodegenerative Disease: Brain Windfall. Nature 2014, 515, 299–300. [Google Scholar] [CrossRef]
- Korovesis, D.; Rubio-Tomás, T.; Tavernarakis, N. Oxidative Stress in Age-Related Neurodegenerative Diseases: An Overview of Recent Tools and Findings. Antioxidants 2023, 12, 131. [Google Scholar] [CrossRef]
- Morén, C.; deSouza, R.M.; Giraldo, D.M.; Uff, C. Antioxidant Therapeutic Strategies in Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 9328. [Google Scholar] [CrossRef]
- Rao, Y.L.; Ganaraja, B.; Suresh, P.K.; Joy, T.; Ullal, S.D.; Manjrekar, P.A.; Murlimanju, B.V.; Sharma, B.G.; Massand, A.; Agrawal, A. Outcome of resveratrol and resveratrol with donepezil combination on the β-amyloid plaques and neurofibrillary tangles in Alzheimer’s disease. 3 Biotech. 2024, 14, 190. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Delgado, A.; Ortiz, G.G.; Delgado-Lara, D.L.; González-Usigli, H.A.; González-Ortiz, L.J.; Cid-Hernández, M.; Cruz-Serrano, J.A.; Pacheco-Moisés, F.P. Effect of Melatonin Administration on Mitochondrial Activity and Oxidative Stress Markers in Patients with Parkinson’s Disease. Oxidative Med. Cell. Longev. 2021, 2021, 5577541. [Google Scholar] [CrossRef] [PubMed]
- Paknahad, Z.; Sheklabadi, E.; Moravejolahkami, A.R.; Chitsaz, A.; Hassanzadeh, A. The effects of Mediterranean diet on severity of disease and serum Total Antioxidant Capacity (TAC) in patients with Parkinson’s disease: A single center, randomized controlled trial. Nutr. Neurosci. 2022, 25, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Dhana, K.; Furtado, J.D.; Agarwal, P.; Aggarwal, N.T.; Tangney, C.; Laranjo, N.; Carey, V.; Barnes, L.L.; Sacks, F.M. Higher circulating α-carotene was associated with better cognitive function: An evaluation among the MIND trial participants. J. Nutr. Sci. 2021, 10, e64. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A. The Role of Natural Antioxidants in the Prevention of Dementia—Where Do We Stand and Future Perspectives. Nutrients 2021, 13, 282. [Google Scholar] [CrossRef] [PubMed]
- Lamptey, R.N.L.; Chaulagain, B.; Trivedi, R.; Gothwal, A.; Layek, B.; Singh, J. A Review of the Common Neurodegenerative Disorders: Current Therapeutic Approaches and the Potential Role of Nanotherapeutics. Int. J. Mol. Sci. 2022, 23, 1851. [Google Scholar] [CrossRef]
- Martinelli, C.; Pucci, C.; Battaglini, M.; Marino, A.; Ciofani, G. Antioxidants and Nanotechnology: Promises and Limits of Potentially Disruptive Approaches in the Treatment of Central Nervous System Diseases. Adv. Healthc. Mater. 2020, 9, e1901589. [Google Scholar] [CrossRef]
- Zielińska, A.; Costa, B.; Ferreira, M.V.; Miguéis, D.; Louros, J.; Durazzo, A.; Lucarini, M.; Eder, P.; V Chaud, M.; Morsink, M. Nanotoxicology and nanosafety: Safety-by-design and testing at a glance. Int. J. Environ. Res. Public Health 2020, 17, 4657. [Google Scholar] [CrossRef]
- Lee, D.; Choi, Y.H.; Seo, J.; Kim, J.K.; Lee, S.B. Discovery of new epigenomics-based biomarkers and the early diagnosis of neurodegenerative diseases. Ageing Res. Rev. 2020, 61, 101069. [Google Scholar] [CrossRef]
- Sun, J.; Roy, S. Gene-based therapies for neurodegenerative diseases. Nat. Neurosci. 2021, 24, 297–311. [Google Scholar] [CrossRef]

{kind=link}
| Categorization of GPx Isoenzymes Based on Selenium Dependence | |
|---|---|
| Selenium-dependent | Selenium-independent |
| GPx1 | |
| GPx2 | GPx5 |
| GPx3 | GPx7 |
| GPx4 | GPx8 |
| GPx6 | |
| Protein | Function | Reference |
|---|---|---|
| Glutamate dehydrogenase 1 | TCA cycle | [98] |
| Malate dehydrogenase | TCA cycle | [99] |
| Subunit Va of cytochrome c oxidase | ETC | [100] |
| Ubiquinone (NADH dehydrogenase) | ETC | [101] |
| Core protein 1 of ubiquinol-cytochrome c reductase complex | ETC | [102] |
| ATP synthase | OXPHOS | [103] |
| Drug | Mechanisms of Action | Outcomes | References |
|---|---|---|---|
| Vitamin E | Maintains membrane integrity in mitochondria | Antioxidant properties in AD | [161] |
| Alpha-lipoic acid | Scavenges the toxic byproducts of lipid peroxidation | Antioxidant properties in AD | [161] |
| Curcumin | Suppresses TNF-α activity | Antioxidant and amyloid disaggregating properties in AD | [162] |
| Epigallocatechin-3-gallate (Camellia sinensis) | Inhibits oxidative stress via the Keap1/Nrf2 signaling pathway | Antioxidant effects in AD | [163] |
| Ginsenosides | Suppress Aβ-associated generation of ROS by enhancing the activity of endogenous antioxidants and reducing the expression of NOX2 | Inhibit Aβ and neurofibrillary tangle formation | [164] |
| Melatonin | Direct scavenger of many ROS species | Protective role against H-89-induced memory impairment in mouse brain | [165] |
| MitoQ | Scavenges peroxyl, peroxynitrite, and superoxide ROS | Lower levels of lipid peroxidation | [157] |
| Drug | Mechanisms of Action | Outcomes | References |
|---|---|---|---|
| Vitamins E and C | Maintain the integrity of mitochondrial membranes | Antioxidant and neuroprotective activities in PD | [161] |
| P7C3 (aminopropyl carbazole) | Acts by protecting mitochondria | Stabilized mitochondrial membrane potential in PD (dopaminergic cell lines), reduced ROS production, inhibited GSK3β activation and p53 activity, Bax upregulation, cytochrome c release exposed to MPTP, and prevented neuronal loss in the substantia nigra (mouse brain) | [178] |
| Terpene lactones and flavonoids from Ginkgo biloba | Stabilize mitochondrial functions and interact with the mitochondrial electron transport chain | Antioxidant effects in PD | [161] |
| Triterpene saponin and phenol from Glycyrrhiza | Reduces oxidative stress and damage to brain cells | Antioxidant and neuroprotective effects in PD | [161] |
| MitoQ | Scavenges peroxyl, peroxynitrite, and superoxide ROS | Antioxidant effects in PD | [179] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jurcau, M.-C.; Jurcau, A.; Diaconu, R.-G. Oxidative Stress in the Pathogenesis of Neurodegenerative Diseases. Stresses 2024, 4, 827-849. https://doi.org/10.3390/stresses4040055
Jurcau M-C, Jurcau A, Diaconu R-G. Oxidative Stress in the Pathogenesis of Neurodegenerative Diseases. Stresses. 2024; 4(4):827-849. https://doi.org/10.3390/stresses4040055
Chicago/Turabian StyleJurcau, Maria-Carolina, Anamaria Jurcau, and Razvan-Gabriel Diaconu. 2024. "Oxidative Stress in the Pathogenesis of Neurodegenerative Diseases" Stresses 4, no. 4: 827-849. https://doi.org/10.3390/stresses4040055
APA StyleJurcau, M.-C., Jurcau, A., & Diaconu, R.-G. (2024). Oxidative Stress in the Pathogenesis of Neurodegenerative Diseases. Stresses, 4(4), 827-849. https://doi.org/10.3390/stresses4040055