
Chemistry of Hydrogen Peroxide Formation and Elimination in Mammalian Cells, and Its Role in Various Pathologies

 ,
,  and
and
Abstract
:1. Introduction
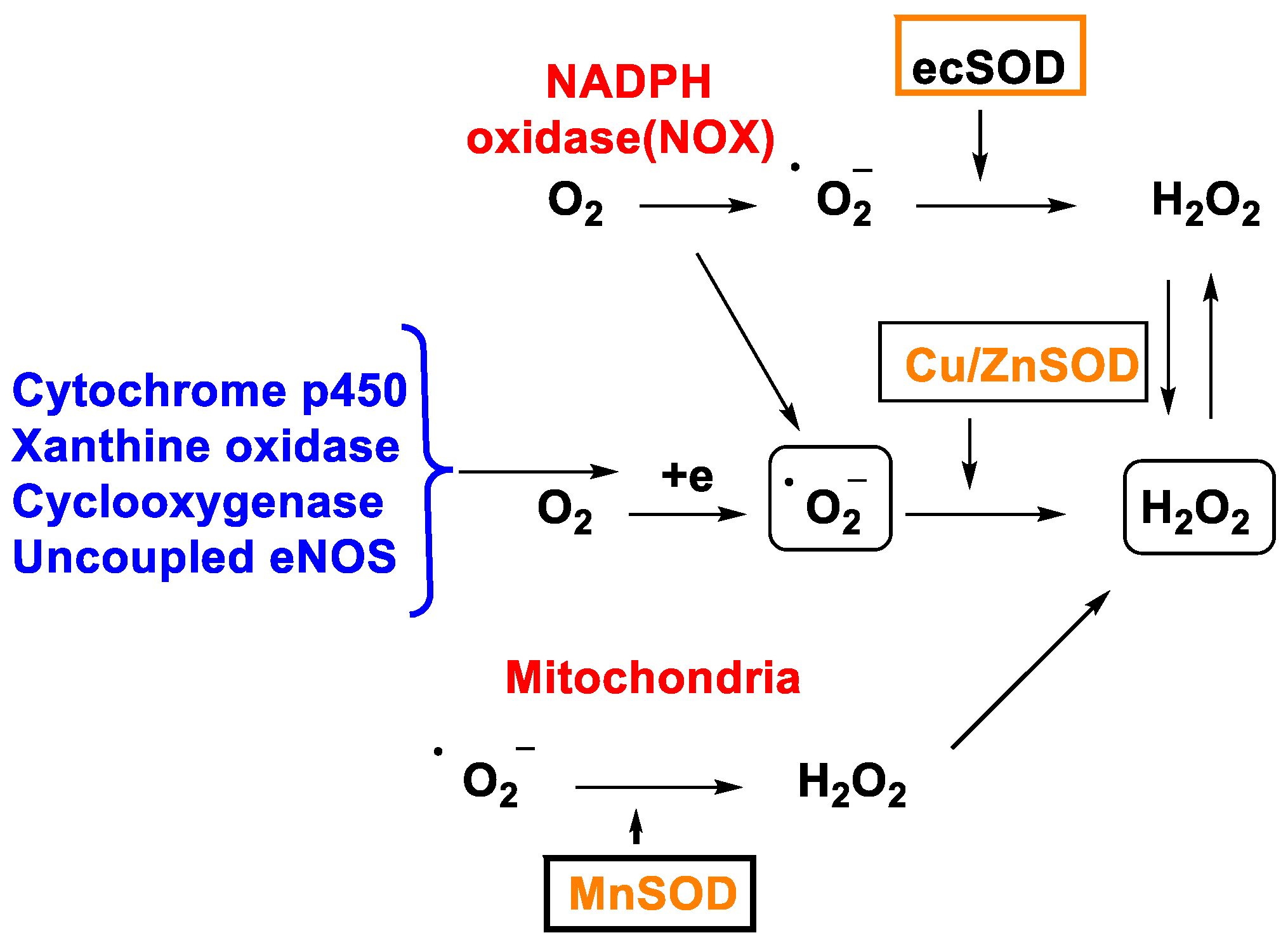
2. Hydrogen Peroxide Formation
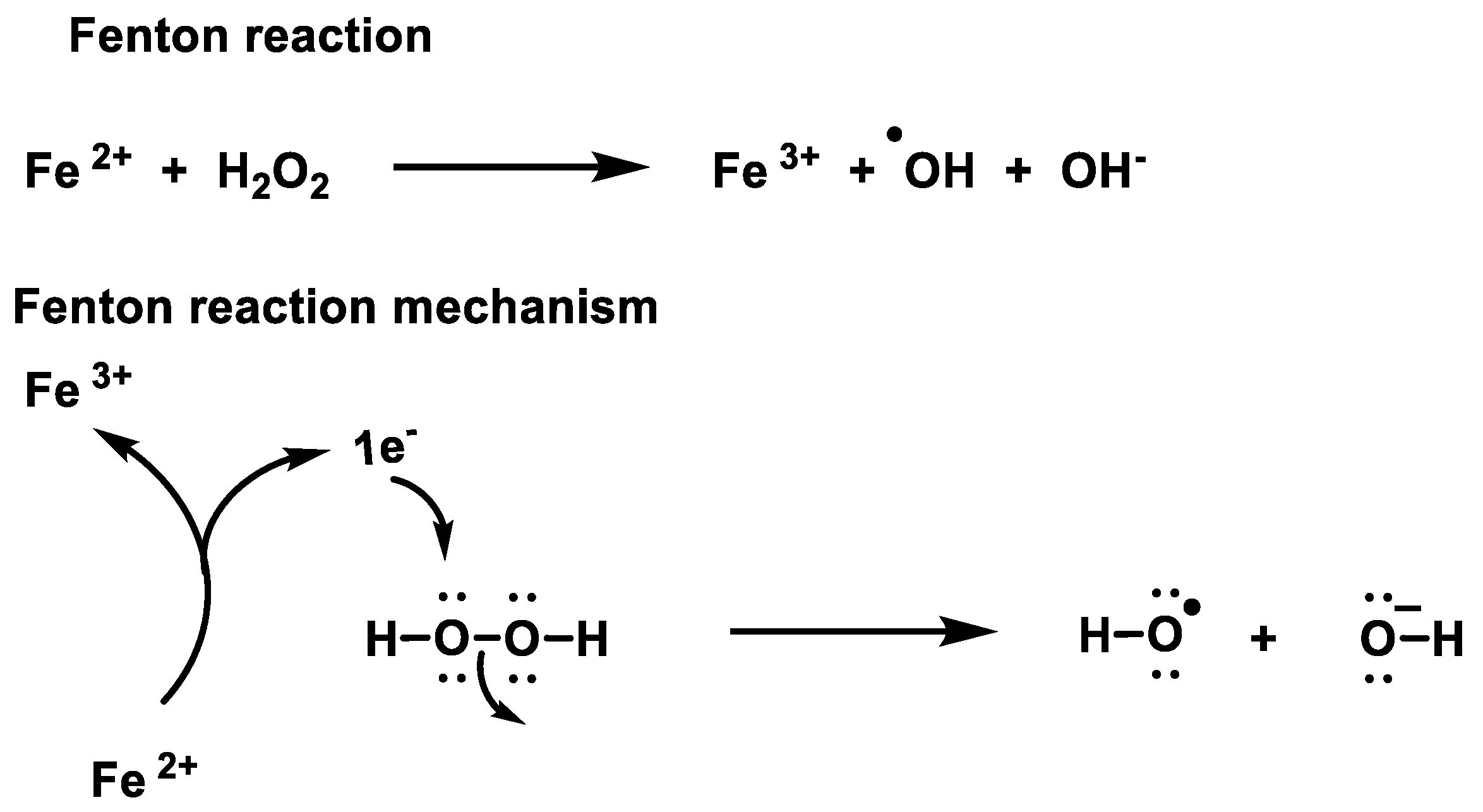
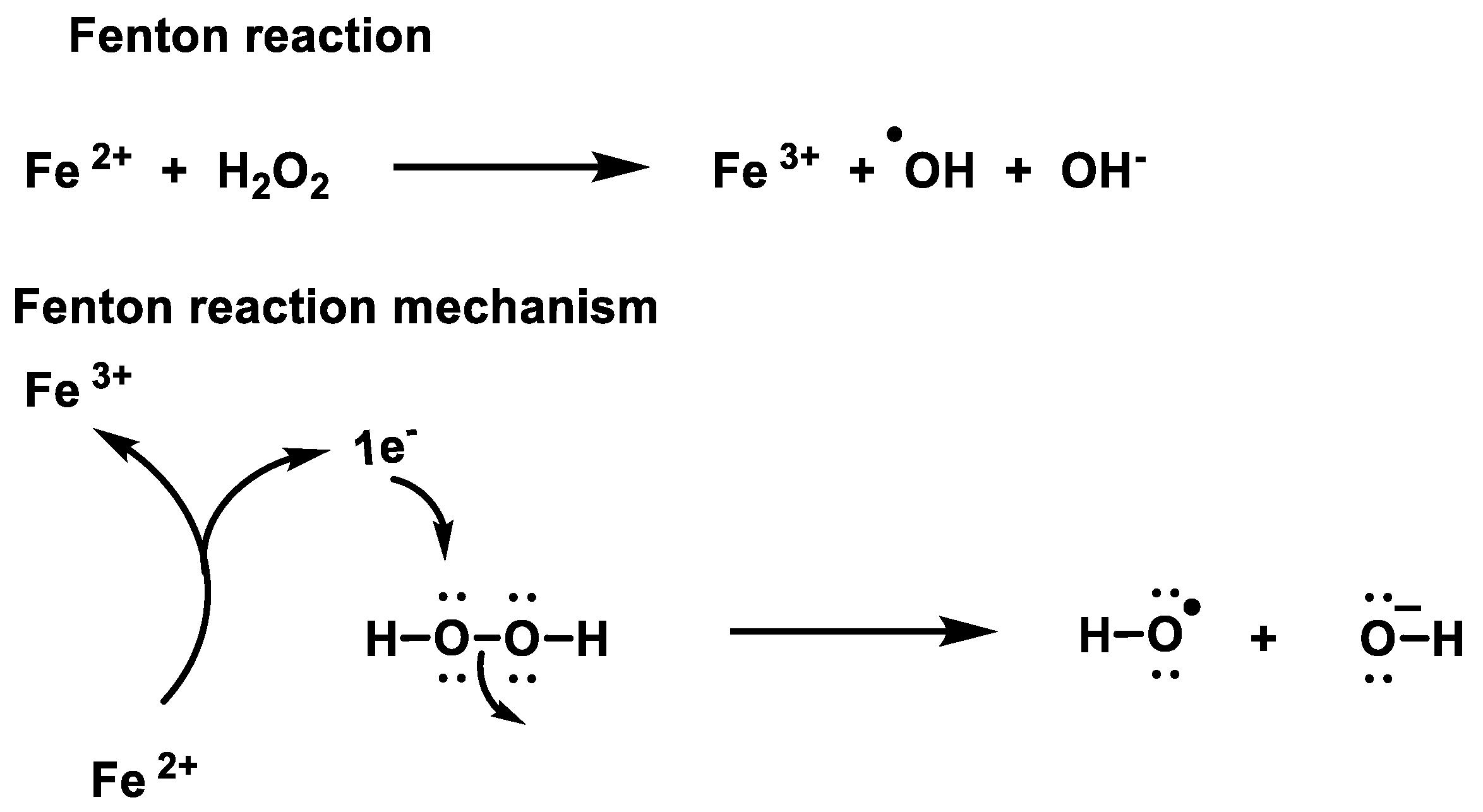
2.1. Non-Enzymatic Generation of H2O2
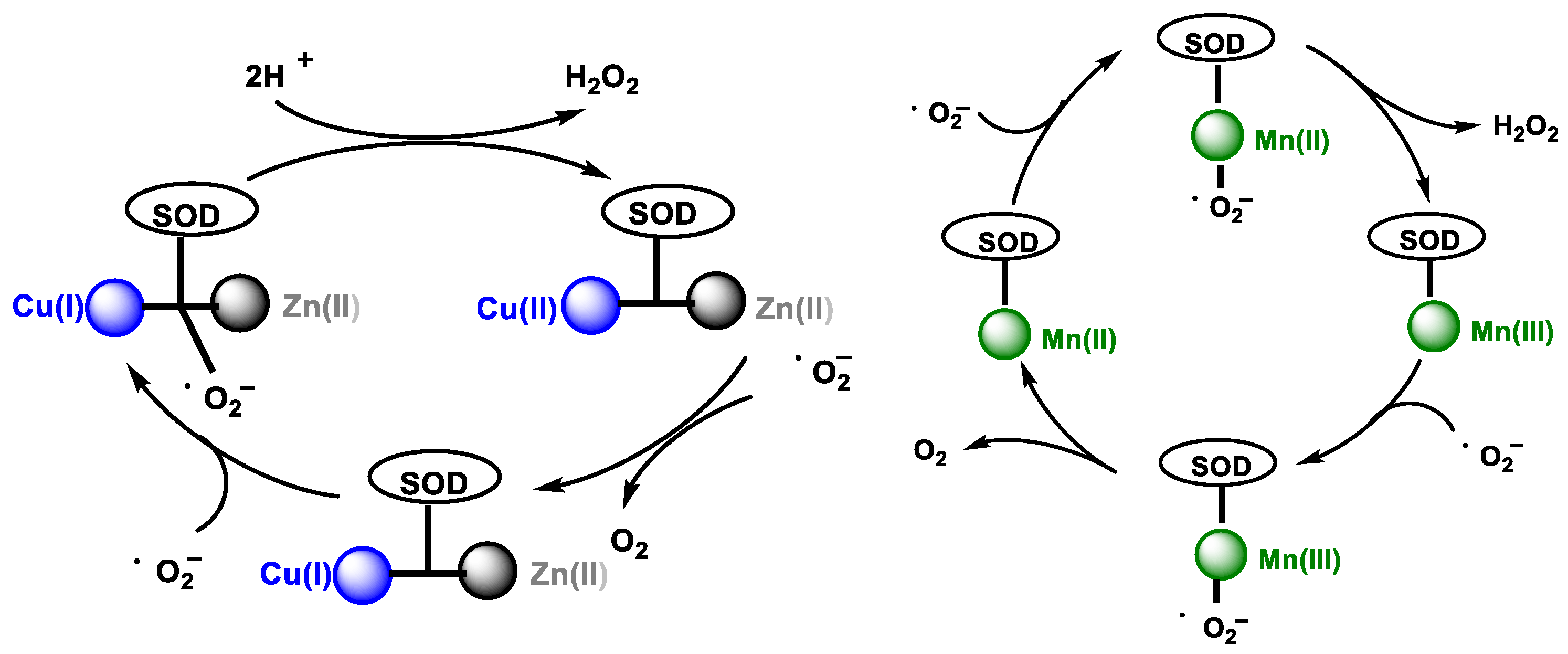
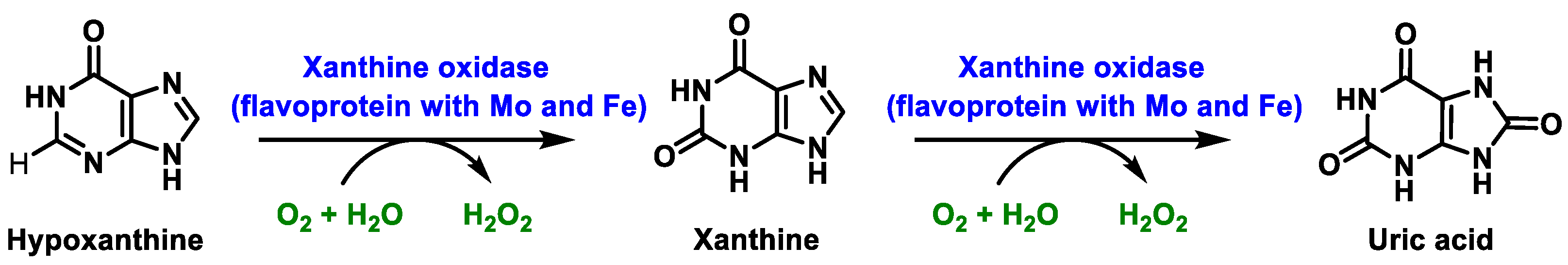
2.2. Enzymatic Generation of H2O2
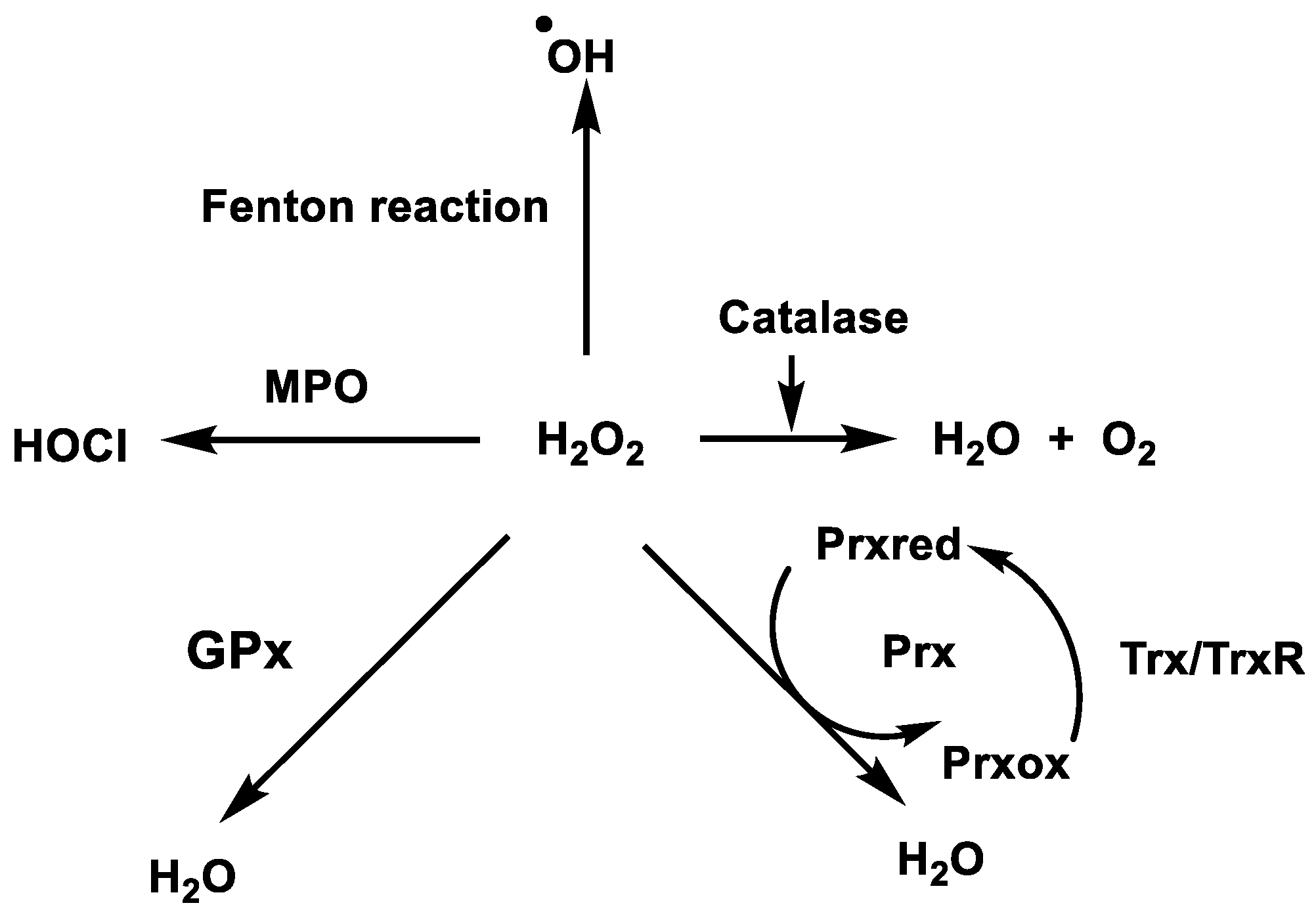
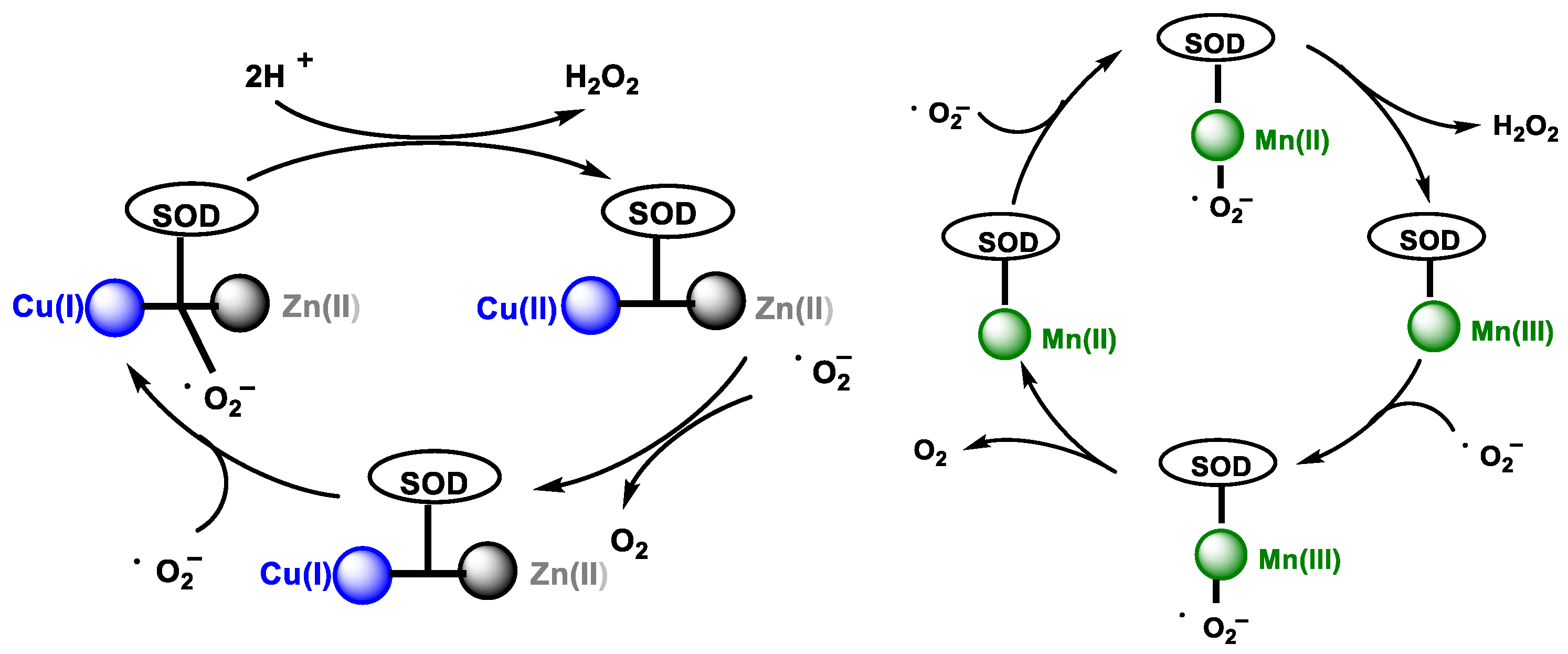
3. Removal of Hydrogen Peroxide
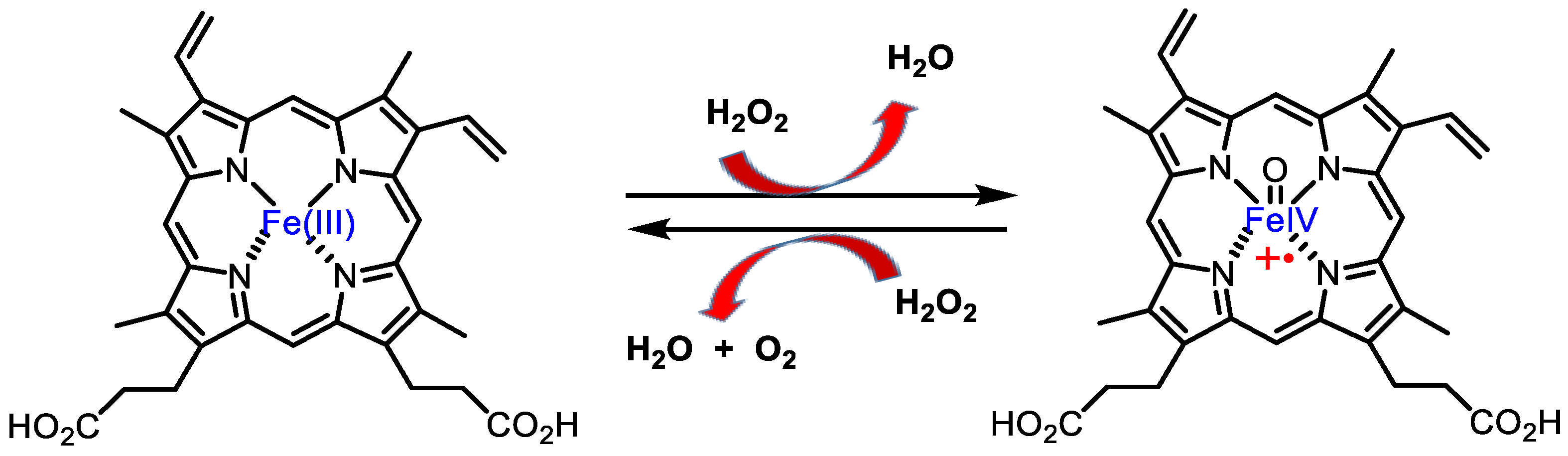
3.1. Catalase
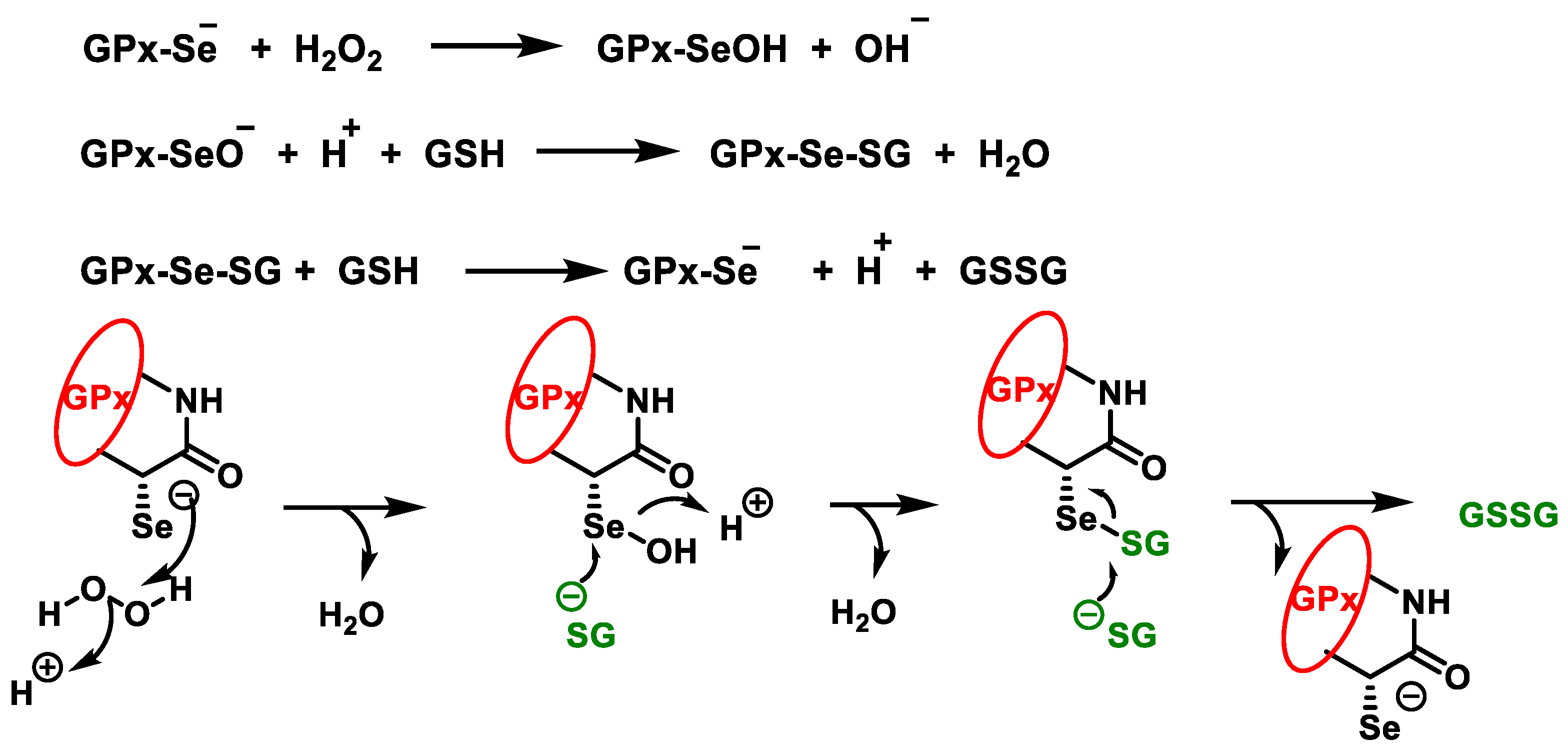
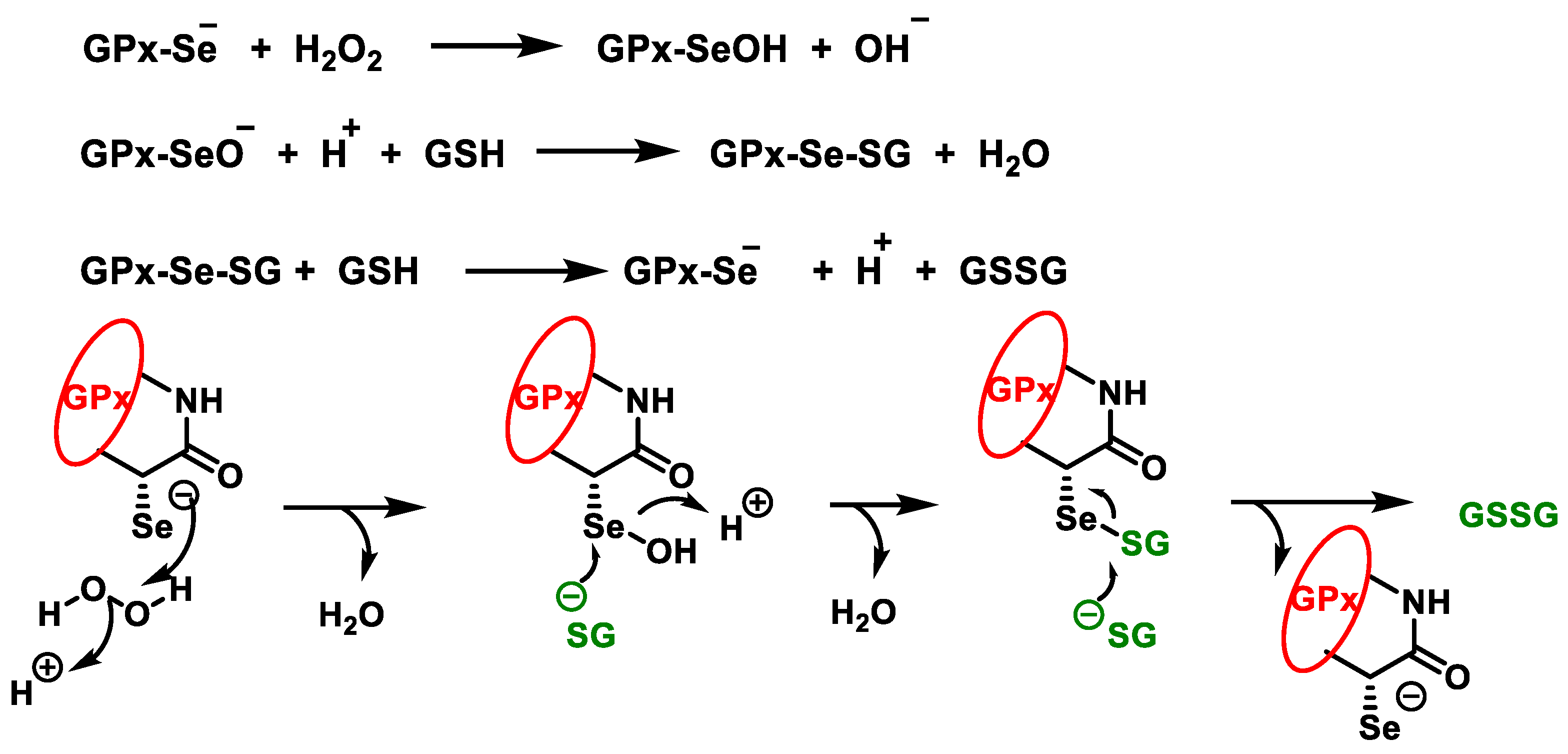
3.2. Glutathione Peroxidases GPx
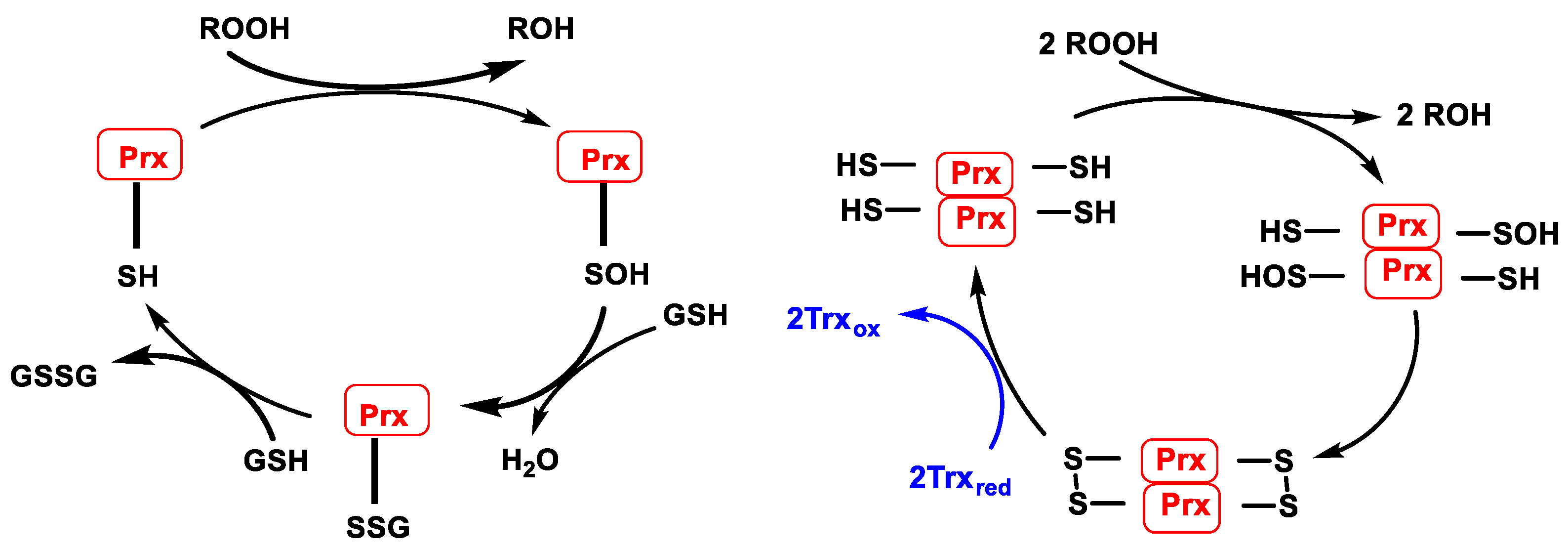
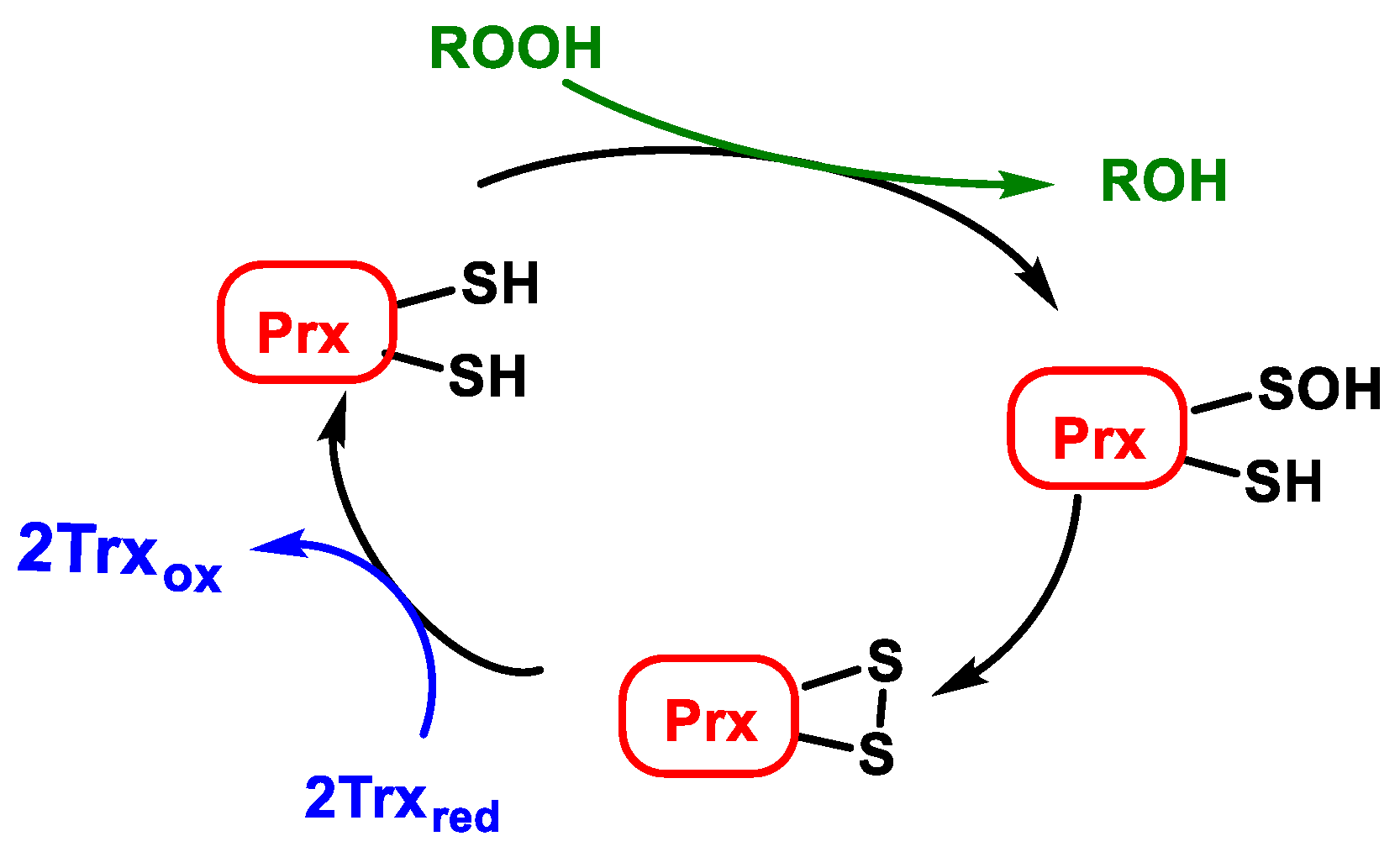
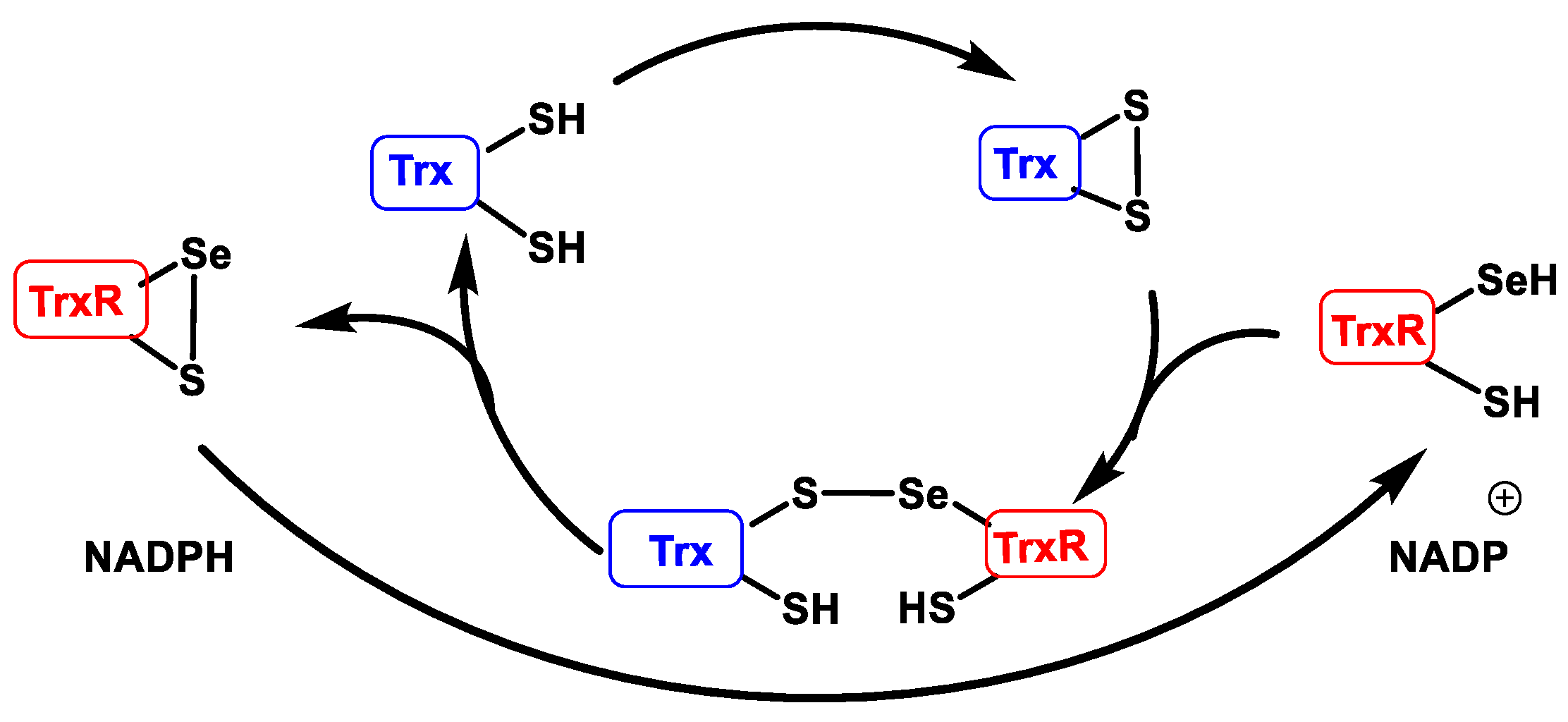
3.3. Peroxiredoxins (Prx) and Thioredoxins (Trx)
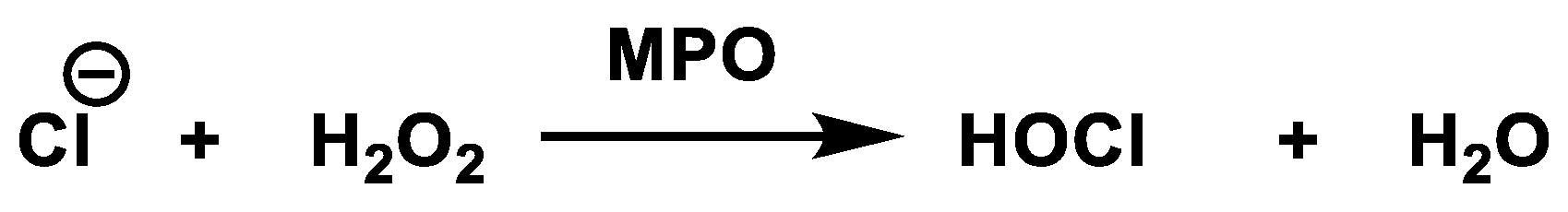
3.4. Hypochlorous Acid Formation (HOCl)
4. H2O2 and Inflammation
5. H2O2 and Cancer
6. H2O2 and Related Diseases
7. Measurement of Hydrogen Peroxide in Human Body
7.1. Presence in Blood
7.2. Presence in Urine
7.3. Presence in the Exhaled Air
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Briffa, J.; Sinagra, E.; Blundell, R. Heavy metal pollution in the environment and their toxicological effects on humans. Heliyon 2020, 6, e04691. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.L.; Milligan, T.W.; Joyner, R.E.; Jefferson, M.M. Antibacterial activity of hydrogen peroxide and the lactoperoxidase-hydrogen peroxide-thiocyanate system against oral streptococci. Infect. Immun. 1994, 62, 529–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H. Role of Metabolic H2O2 Generation: Redox Signaling And Oxidative Stress. J. Biol. Chem. 2014, 289, 8735–8741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickinson, B.C.; Chang, C.J. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol. 2011, 7, 504–511. [Google Scholar] [CrossRef] [Green Version]
- Castagna, R.; Eiserich, J.P.; Budamagunta, M.S.; Stipa, P.; Cross, C.E.; Proietti, E.; Voss, J.C.; Greci, L. Hydroxyl radical from the reaction between hypochlorite and hydrogen peroxide. Atmos. Environ. 2008, 42, 6551–6554. [Google Scholar] [CrossRef]
- Hyslop, P.A.; Chaney, M.O. Mechanism of GAPDH Redox Signaling by H2O2 Activation of a Two−Cysteine Switch. Int. J. Mol. Sci. 2022, 23, 4604. [Google Scholar] [CrossRef]
- Mahaseth, T.; Kuzminov, A. Potentiation of hydrogen peroxide toxicity: From catalase inhibition to stable DNA-iron complexes. Mutat. Res. Rev. Mutat. Res. 2017, 773, 274–281. [Google Scholar] [CrossRef]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide–production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 2015, 13, 39. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M. Free Radicals in Biology and Medicine, 4th ed.; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Messner, K.R.; Imlay, J.A. Mechanism of Superoxide and Hydrogen Peroxide Formation by Fumarate Reductase, Succinate Dehydrogenase, and Aspartate Oxidase. J. Biol. Chem. 2002, 277, 42563–42571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez, M.; Lissarrague, M.S.; Bechthold, P.; González, E.; Jasen, P.; Juan, A. Ethanol adsorption on Ni doped Mo2C (001): A theoretical study. Top. Catal. 2022, 104, 839–847. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, A.M.; Brown, J.D.; Taylor, S.R.; Rand, J.D.; Morgan, B.A.; Veal, E.A. Inactivation of a Peroxiredoxin by Hydrogen Peroxide Is Critical for Thioredoxin-Mediated Repair of Oxidized Proteins and Cell Survival. Mol. Cell 2012, 45, 398–408. [Google Scholar] [CrossRef] [Green Version]
- Dada, L.A.; Chandel, N.S.; Ridge, K.M.; Pedemonte, C.; Bertorello, A.M.; Sznajder, J.I. Hypoxia-induced endocytosis of Na,K-ATPase in alveolar epithelial cells is mediated by mitochondrial reactive oxygen species and PKC-ζ. J. Clin. Investig. 2003, 111, 1057–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 Functionally Interacts with the Metabolic Regulator and Transcriptional Coactivator PGC-1α. J. Biol. Chem. 2005, 280, 16456–16460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell. Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef]
- Fridovich, I. The Biology of Oxygen Radicals. Science 1978, 201, 875–880. [Google Scholar] [CrossRef]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Valle, N.R.-D.; Huang, P. Redox Regulation of Cell Survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [Green Version]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guengerich, F.P.; Munro, A.W. Unusual Cytochrome P450 Enzymes and Reactions. J. Biol. Chem. 2013, 288, 17065–17073. [Google Scholar] [CrossRef] [Green Version]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Sowers, J.R.; Zhang, Y. Autophagy and Cardiometabolic Diseases: From Molecular Mechanisms to Translational Medicine; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Nriagu, J.O. Encyclopedia of Environmental Health; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Battelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine Oxidoreductase-Derived Reactive Species: Physiological and Pathological Effects. Oxid. Med. Cell. Longev. 2016, 2016, 3527579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hille, R. Xanthine oxidoreductase. In xPharm: The Comprehensive Pharmacology Reference; Elsevier: Amsterdam, The Netherlands, 2007; pp. 1–10. [Google Scholar]
- Becker, B.F. Towards the physiological function of uric acid. Free Radic. Biol. Med. 1993, 14, 615–631. [Google Scholar] [CrossRef]
- Cooper, G. The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2000. [Google Scholar]
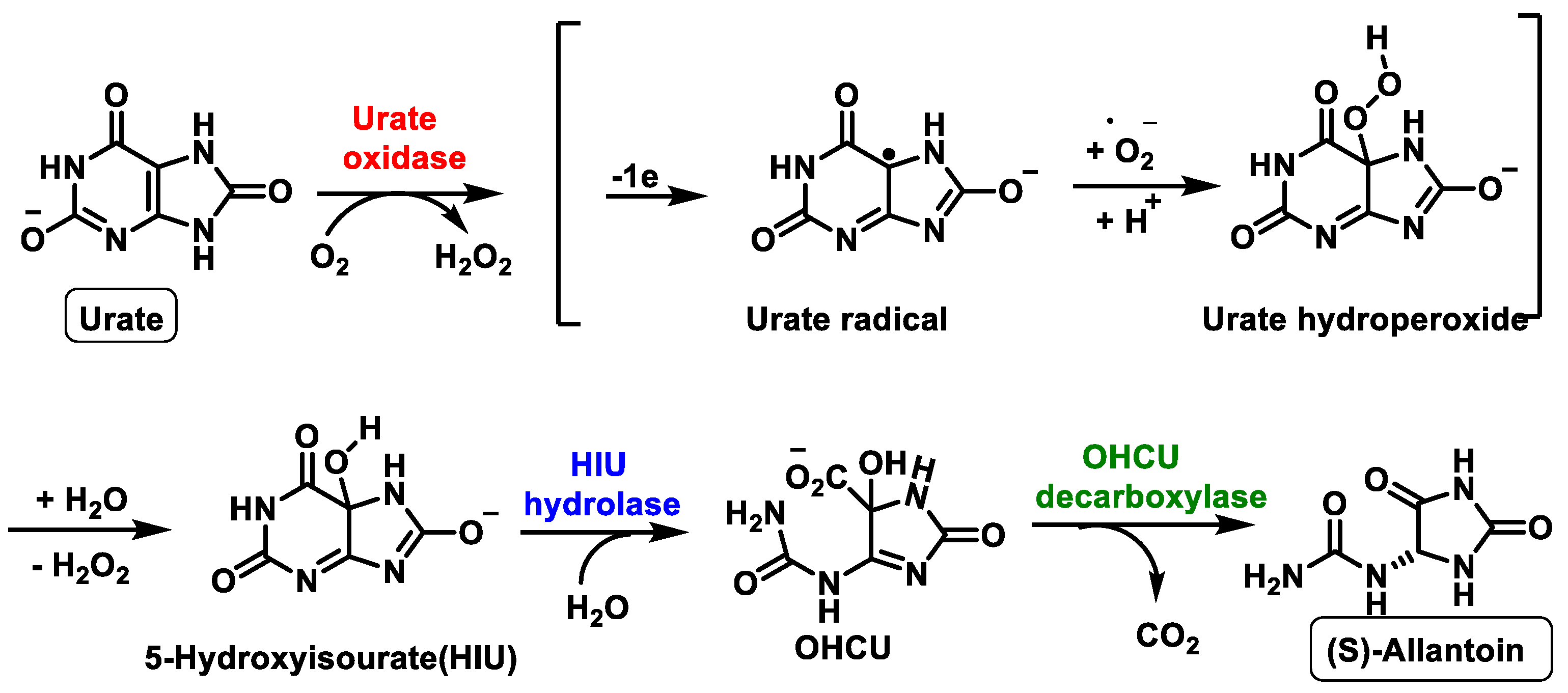
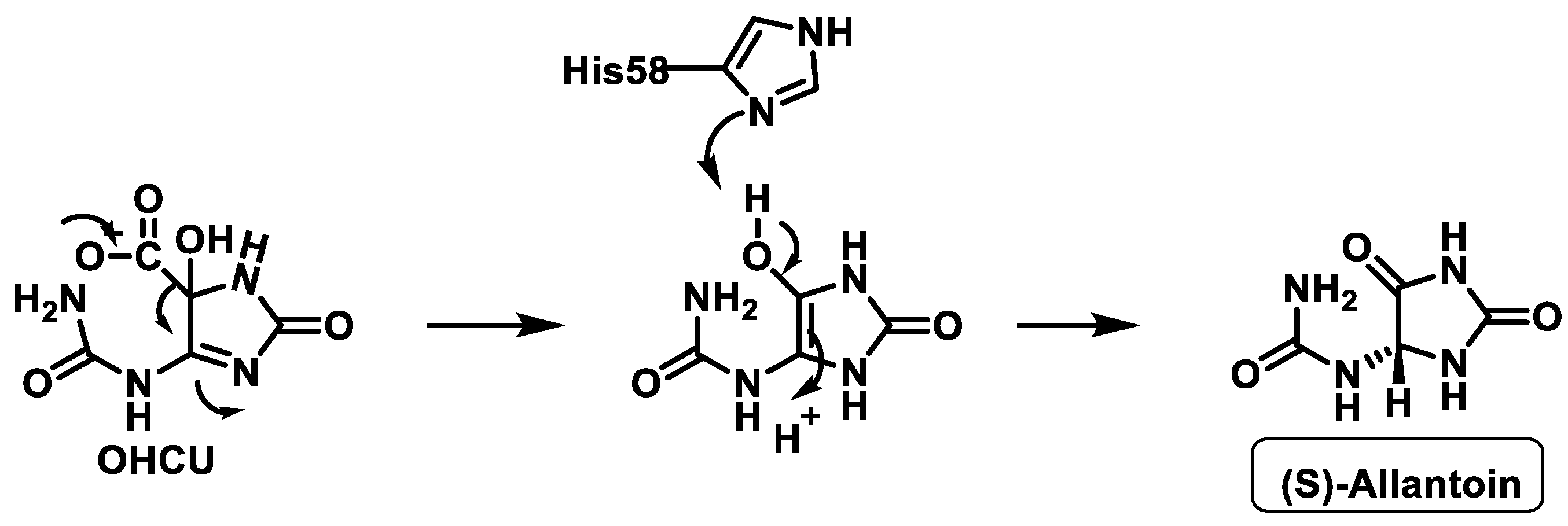
- Wu, X.W.; Lee, C.C.; Muzny, D.M.; Caskey, C.T. Urate oxidase: Primary structure and evolutionary implications. Proc. Natl. Acad. Sci. USA 1989, 86, 9412–9416. [Google Scholar] [CrossRef] [Green Version]
- Kratzer, J.T.; Lanaspa, M.A.; Murphy, M.N.; Cicerchi, C.; Graves, C.L.; Tipton, P.A.; Ortlund, E.A.; Johnson, R.J.; Gaucher, E.A. Evolutionary history and metabolic insights of ancient mammalian uricases. Proc. Natl. Acad. Sci. USA 2014, 111, 3763–3768. [Google Scholar] [CrossRef] [Green Version]
- Liu-Bryan, R.; Terkeltaub, R. Tophus Biology and Pathogenesis of Monosodium Urate Crystal–Induced Inflammation. In Gout & Other Crystal Arthropathies; Terkeltaub, R., Ed.; W.B. Saunders: Philadelphia, PA, USA, 2012; pp. 59–71. [Google Scholar]
- Kim, K.; Park, J.; Rhee, S. Structural and Functional Basis for (S)-Allantoin Formation in the Ureide Pathway. J. Biol. Chem. 2007, 282, 23457–23464. [Google Scholar] [CrossRef] [Green Version]
- Zito, E. ERO1: A protein disulfide oxidase and H2O2 producer. Free Radic. Biol. Med. 2015, 83, 299–304. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82, 969–974. [Google Scholar] [CrossRef]
- Di Giorgio, F.P.; Carrasco, M.A.; Siao, M.C.; Maniatis, T.; Eggan, K. Non–cell autonomous effect of glia on motor neurons in an embryonic stem cell–based ALS model. Nat. Neurosci. 2007, 10, 608–614. [Google Scholar] [CrossRef] [Green Version]
- Otera, H.; Fujiki, Y. Pex5p imports folded tetrameric catalase by interaction with Pex13p. Traffic 2012, 13, 1364–1377. [Google Scholar] [CrossRef] [PubMed]
- Okumoto, K.; El Shermely, M.; Natsui, M.; Kosako, H.; Natsuyama, R.; Marutani, T.; Fujiki, Y. The peroxisome counteracts oxidative stresses by suppressing catalase import via Pex14 phosphorylation. eLife 2020, 9, e55896. [Google Scholar] [CrossRef]
- Benner, P.; Hooper-Kyriakidis, P.; Stannard, D. Clinical Wisdom and Interventions in Acute and Critical Care; Springer: New York, NY, USA, 2011. [Google Scholar]
- Margis, R.; Dunand, C.; Teixeira, F.K.; Margis-Pinheiro, M. Glutathione peroxidase family—An evolutionary overview. FEBS J. 2008, 275, 3959–3970. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological Roles of Mitochondrial Reactive Oxygen Species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ďuračková, Z. Free Radicals and Antioxidants for Non-Experts. In Systems Biology of Free Radicals and Antioxidants; Laher, I., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 3–38. [Google Scholar]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta BBA Gen. Subj. 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Rhee, S.G.; Chae, H.Z.; Kim, K. Peroxiredoxins: A historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic. Biol. Med. 2005, 38, 1543–1552. [Google Scholar] [CrossRef]
- Hall, A.; Nelson, K.; Poole, L.B.; Karplus, P.A. Structure-based Insights into the Catalytic Power and Conformational Dexterity of Peroxiredoxins. Antioxid. Redox Signal. 2010, 15, 795–815. [Google Scholar] [CrossRef] [Green Version]
- Nordberg, J.; Arnér, E.S.J. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Review based on “Thioredoxin reductase—Interactions with the redox active compounds 1-chloro-2,4-dinitrobenzene and lipoic acid”. Free Radic. Biol. Med. 2001, 31, 1287–1312. [Google Scholar] [CrossRef]
- Berndt, C.; Lillig, C.H.; Holmgren, A. Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: Implications for diseases in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1227–H1236. [Google Scholar] [CrossRef]
- Tvrdá, E.; Benko, F. Chapter 1—Free radicals: What they are and what they do. In Pathology; Preedy, V.R., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 3–13. [Google Scholar]
- Casciaro, M.; Di Salvo, E.; Pace, E.; Ventura-Spagnolo, E.; Navarra, M.; Gangemi, S. Chlorinative stress in age-related diseases: A literature review. Immun. Ageing 2017, 14, 21. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.J. Myeloperoxidase-derived oxidation: Mechanisms of biological damage and its prevention. J. Clin. Biochem. Nutr. 2010, 48, 8–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittmann, C.; Chockley, P.; Singh, S.K.; Pase, L.; Lieschke, G.J.; Grabher, C. Hydrogen Peroxide in Inflammation: Messenger, Guide, and Assassin. Adv. Hematol. 2012, 2012, 541471. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Wang, K.; Deng, L.; Chen, Y.; Nice, E.C.; Huang, C. Redox Regulation of Inflammation: Old Elements, a New Story. Med. Res. Rev. 2015, 35, 306–340. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Hüll, M.; Berger, M.; Heneka, M. Disease-Modifying Therapies in Alzheimer’s Disease. Drugs 2006, 66, 2075–2093. [Google Scholar] [CrossRef]
- Gunawardena, D.; Raju, R.; Münch, G. Hydrogen peroxide mediates pro-inflammatory cell-to-cell signaling: A new therapeutic target for inflammation? Neural Regen. Res. 2019, 14, 1430. [Google Scholar]
- De Oliveira-Marques, V.; Cyrne, L.; Marinho, H.S.; Antunes, F. A Quantitative Study of NF-κB Activation by H2O2: Relevance in Inflammation and Synergy with TNF-α. J. Immunol. 2007, 178, 3893–3902. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Michael, J.; May, A.; Kopp, E.B. NF-κB AND REL PROTEINS: Evolutionarily Conserved Mediators of Immune Responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef]
- Nauseef, W.M. Assembly of the phagocyte NADPH oxidase. Histochem. Cell Biol. 2004, 122, 277–291. [Google Scholar] [CrossRef]
- Burdon, R.H.; Gill, V.; Rice-Evans, C. Oxidative Stress and Tumour Cell Proliferation. Free Radic. Res. Commun. 1990, 11, 65–76. [Google Scholar] [CrossRef]
- Reddy, K.B.; Glaros, S. Inhibition of the MAP kinase activity suppresses estrogen-induced breast tumor growth both in vitro and in vivo. Int. J. Oncol. 2007, 30, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.M.; Bae, Y.S.; Lee, S.Y. Molecular ordering of ROS production, mitochondrial changes, and caspase activation during sodium salicylate-induced apoptosis. Free Radic. Biol. Med. 2003, 34, 434–442. [Google Scholar] [CrossRef]
- Oberley, T.D. Antioxidant enzyme levels in cancer. Histol. Histopathol. 1997, 12, 525–535. [Google Scholar]
- Doskey, C.M.; Buranasudja, V.; Wagner, B.A.; Wilkes, J.G.; Du, J.; Cullen, J.J.; Buettner, G.R. Tumor cells have decreased ability to metabolize H2O2: Implications for pharmacological ascorbate in cancer therapy. Redox Biol. 2016, 10, 274–284. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Lasky, J.L.; Chang, C.-J.; Mosessian, S.; Lewis, X.; Xiao, Y.; Yeh, J.E.; Chen, J.Y.; Iruela-Arispe, M.L.; Varella-Garcia, M. Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature 2008, 453, 529–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greaves, M.F. Biological models for leukaemia and lymphoma. IARC Sci. Publ. 2004, 156, 351–372. [Google Scholar]
- Marincevic-Zuniga, Y.; Dahlberg, J.; Nilsson, S.; Raine, A.; Nystedt, S.; Lindqvist, C.M.; Berglund, E.C.; Abrahamsson, J.; Cavelier, L.; Forestier, E. Transcriptome sequencing in pediatric acute lymphoblastic leukemia identifies fusion genes associated with distinct DNA methylation profiles. J. Hematol. Oncol. 2017, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Perry, J.M.; Tao, F.; Roy, A.; Lin, T.; He, X.C.; Chen, S.; Lu, X.; Nemechek, J.; Ruan, L.; Yu, X. Overcoming Wnt–β-catenin dependent anticancer therapy resistance in leukaemia stem cells. Nat. Cell Biol. 2020, 22, 689–700. [Google Scholar] [CrossRef]
- Carden, D.L.; Granger, D.N. Pathophysiology of ischaemia–reperfusion injury. J. Pathol. 2000, 190, 255–266. [Google Scholar] [CrossRef]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [Green Version]
- Grisham, M.B.; Granger, D.N.; Lefer, D.J. Modulation of leukocyte–endothelial interactions by reactive metabolites of oxygen and nitrogen: Relevance to ischemic heart disease. Free Radic. Biol. Med. 1998, 25, 404–433. [Google Scholar] [CrossRef]
- Magro, F.; Gionchetti, P.; Eliakim, R.; Ardizzone, S.; Armuzzi, A.; Barreiro-de Acosta, M.; Burisch, J.; Gecse, K.B.; Hart, A.L.; Hindryckx, P.; et al. Third European Evidence-based Consensus on Diagnosis and Management of Ulcerative Colitis. Part 1: Definitions, Diagnosis, Extra-intestinal Manifestations, Pregnancy, Cancer Surveillance, Surgery, and Ileo-anal Pouch Disorders. J. Crohn’s Colitis 2017, 11, 649–670. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, K.; Feuerstein, J.D. New developments in ulcerative colitis: Latest evidence on management, treatment, and maintenance. Drugs Context 2019, 8, 212572. [Google Scholar] [CrossRef] [PubMed]
- Kirsner, J.B. Historical Aspects of Inflammatory Bowel Disease. J. Clin. Gastroenterol. 1988, 10, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Kirsner, J. Historical origins of current IBD concepts. World J. Gastroenterol. 2001, 7, 175–184. [Google Scholar] [CrossRef]
- Santhanam, S.; Venkatraman, A.; Ramakrishna, B.S. Impairment of mitochondrial acetoacetyl CoA thiolase activity in the colonic mucosa of patients with ulcerative colitis. Gut 2007, 56, 1543–1549. [Google Scholar] [CrossRef] [Green Version]
- van Asbeck, B.S.; Braams, R.; Aarsman, J.M.; Sprong, R.C.; Groenewegen, G.A. Hydrogen Peroxide in Blood of Patients with Sepsis Syndrome: A Realistic Phenomenon. Crit. Care Med. 1995, 23, A169. [Google Scholar] [CrossRef]
- Forman, H.J.; Bernardo, A.; Davies, K.J.A. What is the concentration of hydrogen peroxide in blood and plasma? Arch. Biochem. Biophys. 2016, 603, 48–53. [Google Scholar] [CrossRef]
- Pravda, J. Hydrogen peroxide and disease: Towards a unified system of pathogenesis and therapeutics. Mol. Med. 2020, 26, 41. [Google Scholar] [CrossRef]
- Tretter, L.; Adam-Vizi, V. Inhibition of Krebs Cycle Enzymes by Hydrogen Peroxide: A Key Role of α-Ketoglutarate Dehydrogenase in Limiting NADH Production under Oxidative Stress. J. Neurosci. 2000, 20, 8972–8979. [Google Scholar] [CrossRef]
- Tretter, L.; Adam-Vizi, V. Alpha-ketoglutarate dehydrogenase: A target and generator of oxidative stress. Philos. Trans. R. Soc. B Biol. Sci. 2005, 360, 2335–2345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nulton-Persson, A.C.; Szweda, L.I. Modulation of Mitochondrial Function by Hydrogen Peroxide. J. Biol. Chem. 2001, 276, 23357–23361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, T.; Martinou, J.-C. The mitochondrial pyruvate carrier in health and disease: To carry or not to carry? Biochim. Biophys. Acta BBA Mol. Cell Res. 2016, 1863, 2436–2442. [Google Scholar] [CrossRef] [PubMed]
- Bonnefont, J.-P.; Chretien, D.; Rustin, P.; Robinson, B.; Vassault, A.; Aupetit, J.; Charpentier, C.; Rabier, D.; Saudubray, J.-M.; Munnich, A. Alpha-ketoglutarate dehydrogenase deficiency presenting as congenital lactic acidosis. J. Pediatr. 1992, 121, 255–258. [Google Scholar] [CrossRef]
- Grankvist, K.; Marklund, S.L.; Täljedal, I.B. CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochem. J. 1981, 199, 393–398. [Google Scholar] [CrossRef]
- Lenzen, S.; Drinkgern, J.; Tiedge, M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic. Biol. Med. 1996, 20, 463–466. [Google Scholar] [CrossRef]
- Lenzen, S. Oxidative stress: The vulnerable β-cell. Biochem. Soc. Trans. 2008, 36, 343–347. [Google Scholar] [CrossRef]
- Ellis, E.A.; Guberski, D.L.; Somogyi-Mann, M.; Grant, M.B. Increased H2O2, vascular endothelial growth factor and receptors in the retina of the BBZ/WOR diabetic rat. Free Radic. Biol. Med. 2000, 28, 91–101. [Google Scholar] [CrossRef]
- Wang, W.-X.; Jiang, W.-L.; Mao, G.-J.; Tan, M.; Fei, J.; Li, Y.; Li, C.-Y. Monitoring the Fluctuation of Hydrogen Peroxide in Diabetes and Its Complications with a Novel Near-Infrared Fluorescent Probe. Anal. Chem. 2021, 93, 3301–3307. [Google Scholar] [CrossRef]
- Tanaka, Y.; Tran, P.O.T.; Harmon, J.; Robertson, R.P. A role for glutathione peroxidase in protecting pancreatic β cells against oxidative stress in a model of glucose toxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 12363–12368. [Google Scholar] [CrossRef] [Green Version]
- Kubisch, H.M.; Wang, J.; Luche, R.; Carlson, E.; Bray, T.M.; Epstein, C.J.; Phillips, J.P. Transgenic copper/zinc superoxide dismutase modulates susceptibility to type I diabetes. Proc. Natl. Acad. Sci. USA 1994, 91, 9956–9959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, B.; Moritz, J.T.; Epstein, P.N. Overexpression of catalase provides partial protection to transgenic mouse beta cells. Free Radic. Biol. Med. 1999, 27, 830–837. [Google Scholar] [CrossRef]
- Hotta, M.; Tashiro, F.; Ikegami, H.; Niwa, H.; Ogihara, T.; Yodoi, J.; Miyazaki, J.-i. Pancreatic β Cell–specific Expression of Thioredoxin, an Antioxidative and Antiapoptotic Protein, Prevents Autoimmune and Streptozotocin-induced Diabetes. J. Exp. Med. 1998, 188, 1445–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, F.; Wang, Q. The protective effect of peroxiredoxin II on oxidative stress induced apoptosis in pancreatic β-cells. Cell Biosci. 2012, 2, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bast, A.; Wolf, G.; Oberbäumer, I.; Walther, R. Oxidative and nitrosative stress induces peroxiredoxins in pancreatic beta cells. Diabetologia 2002, 45, 867–876. [Google Scholar] [CrossRef] [Green Version]
- Bachmeier, E.; Wietz, F.M.; Linares, J.A.; Goitea, M.E.M.; Jarchum, M.S.; Jarchum, G.; Brunotto, M.N.; Mazzeo, M.A. Determinación de algunos marcadores de estrés oxidativo, funcionales e inmunológicos en saliva de pacientes sometidos a trasplante de médula ósea (TMO). Rev. Fac. Cienc. Méd. 2021, 78, 384. [Google Scholar] [CrossRef]
- Chazelas, P.; Steichen, C.; Favreau, F.; Trouillas, P.; Hannaert, P.; Thuillier, R.; Giraud, S.; Hauet, T.; Guillard, J. Oxidative Stress Evaluation in Ischemia Reperfusion Models: Characteristics, Limits and Perspectives. Int. J. Mol. Sci. 2021, 22, 2366. [Google Scholar] [CrossRef]
- Lipcsey, M.; Bergquist, M.; Sirén, R.; Larsson, A.; Huss, F.; Pravda, J.; Furebring, M.; Sjölin, J.; Janols, H. Urine Hydrogen Peroxide Levels and Their Relation to Outcome in Patients with Sepsis, Septic Shock, and Major Burn Injury. Biomedicines 2022, 10, 848. [Google Scholar] [CrossRef]
- Pravda, J. Radical induction theory of ulcerative colitis. World J. Gastroenterol. WJG 2005, 11, 2371. [Google Scholar] [CrossRef]
- Bautista-Leon, M.R.; Alanís-García, E.; Cansino, N.D.S.C.; Delgado, L. Papel del estrés oxidativo en la infección por SAR-coV-2, y uso de antioxidantes como mecanismo de prevención. Educ. Salud Bol. Cient. Inst. Cienc. Salud Univ. Autón. Estado Hidalgo 2021, 9, 232–237. [Google Scholar] [CrossRef]
- Goud, P.T.; Bai, D.; Abu-Soud, H.M. A multiple-hit hypothesis involving reactive oxygen species and myeloperoxidase explains clinical deterioration and fatality in COVID-19. Int. J. Biol. Sci. 2021, 17, 62. [Google Scholar] [CrossRef] [PubMed]
- Paula, J.I.O.; da Silva Pinto, J.; Rossini, A.; Nogueira, N.P.; Paes, M.C. New perspectives for hydrogen peroxide in the amastigogenesis of Trypanosoma cruzi in vitro. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2020, 1866, 165951. [Google Scholar] [CrossRef] [PubMed]
- Chandramathi, S.; Suresh, K.; Shuba, S.; Mahmood, A.; Kuppusamy, U. High levels of oxidative stress in rats infected with Blastocystis hominis. Parasitology 2010, 137, 605–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauriasari, R.; Zulfa, A.I.; Sekar, A.P.; Azmi, N.U.; Tan, X.W.; Matsuura, E. Role of urinary H2O2, 8-iso-PGF2α, and serum oxLDL/β2GP1 complex in the diabetic kidney disease. PLoS ONE 2022, 17, e0263113. [Google Scholar] [CrossRef] [PubMed]
- Stancill, J.S.; Broniowska, K.A.; Oleson, B.J.; Naatz, A.; Corbett, J.A. Pancreatic β-cells detoxify H2O2 through the peroxiredoxin/thioredoxin antioxidant system. J. Biol. Chem. 2019, 294, 4843–4853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliwell, B.; Clement, M.V.; Long, L.H. Hydrogen peroxide in the human body. FEBS Lett. 2000, 486, 10–13. [Google Scholar] [CrossRef] [Green Version]
- Gaikwad, R.; Thangaraj, P.R.; Sen, A.K. Direct and rapid measurement of hydrogen peroxide in human blood using a microfluidic device. Sci. Rep. 2021, 11, 2960. [Google Scholar] [CrossRef]
- Banerjee, D.; Jacob, J.; Kunjamma, G.; Madhusoodanan, U.; Ghosh, S. Measurement of urinary hydrogen peroxide by FOX-1 method in conjunction with catalase in diabetes mellitus—A sensitive and specific approach. Clin. Chim. Acta Int. J. Clin. Chem. 2004, 350, 233–236. [Google Scholar] [CrossRef]
- Konstantinidi, E.M.; Lappas, A.S.; Tzortzi, A.S.; Behrakis, P.K. Exhaled breath condensate: Technical and diagnostic aspects. Sci. World J. 2015, 2015, 435160. [Google Scholar] [CrossRef] [Green Version]
- Carraro, S.; Giordano, G.; Piacentini, G.; Kantar, A.; Moser, S.; Cesca, L.; Berardi, M.; Di Gangi, I.M.; Baraldi, E. Asymmetric dimethylarginine in exhaled breath condensate and serum of children with asthma. Chest 2013, 144, 405–410. [Google Scholar] [CrossRef]
- Ganas, K.; Loukides, S.; Papatheodorou, G.; Panagou, P.; Kalogeropoulos, N. Total nitrite/nitrate in expired breath condensate of patients with asthma. Respir. Med. 2001, 95, 649–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, K.; Fujimoto, K.; Kitaguchi, Y.; Horiuchi, T.; Kubo, K.; Honda, T. Hydrogen peroxide content and pH of expired breath condensate from patients with asthma and COPD. COPD J. Chronic Obstr. Pulm. Dis. 2014, 11, 81–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]






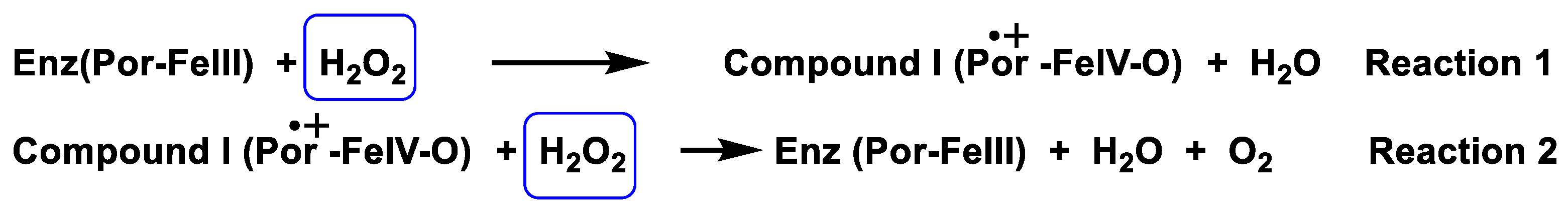


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathology | Levels of H2O2 | Location | Mechanism |
|---|---|---|---|
| Cancer | Plasma increases 2–3 times higher than normal | Mitochondrial membrane of neoplastic cells | Stimulation of cell proliferation by increased metabolic activity [95] |
| Ischaemia-reperfusion injury (I/R) | Plasma levels higher than NO concentration | Extra- and intracellular spaces | Oxidative stress secondary to tissue damage [96] |
| Sepsis and septic shock | Plasma increases 18 times higher than normal | Endothelial cells | Increased oxidative phosphorylation by metabolic hyperdemand [97] |
| Ulcerative colitis | Significantly increased urinary excretion levels | Colon epithelium | Mechanism under study (not known at present, also implicated in other autoimmune diseases), although a causative role is suggested [98] |
| COVID-19 and respiratory distress syndrome RDS | Very high plasma levels especially in combination with urinary sepsis | Endothelial cells mainly from the lung | Expansion of ACE 2 protein leading to increased cellular oxidative status [99,100] |
| Intestinal parasitic infection | Urinary excretion 4 times higher | Intestinal endothelial cells | Oxidative stress secondary to phagocytosis [101,102] |
| Diabetes mellitus type II | 3-fold increase in superoxide dismutase with associated decrease in erythrocyte catalase leading to increases in peroxide excretion. | Pancreatic beta cells | Increased oxidative Phosphorylation [103,104] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrés, C.M.C.; Pérez de la Lastra, J.M.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. Chemistry of Hydrogen Peroxide Formation and Elimination in Mammalian Cells, and Its Role in Various Pathologies. Stresses 2022, 2, 256-274. https://doi.org/10.3390/stresses2030019
Andrés CMC, Pérez de la Lastra JM, Juan CA, Plou FJ, Pérez-Lebeña E. Chemistry of Hydrogen Peroxide Formation and Elimination in Mammalian Cells, and Its Role in Various Pathologies. Stresses. 2022; 2(3):256-274. https://doi.org/10.3390/stresses2030019
Chicago/Turabian StyleAndrés, Celia María Curieses, José Manuel Pérez de la Lastra, Celia Andrés Juan, Francisco J. Plou, and Eduardo Pérez-Lebeña. 2022. "Chemistry of Hydrogen Peroxide Formation and Elimination in Mammalian Cells, and Its Role in Various Pathologies" Stresses 2, no. 3: 256-274. https://doi.org/10.3390/stresses2030019
APA StyleAndrés, C. M. C., Pérez de la Lastra, J. M., Juan, C. A., Plou, F. J., & Pérez-Lebeña, E. (2022). Chemistry of Hydrogen Peroxide Formation and Elimination in Mammalian Cells, and Its Role in Various Pathologies. Stresses, 2(3), 256-274. https://doi.org/10.3390/stresses2030019