
Decomposition of Individual SNP Patterns from Mixed DNA Samples

 , , , and
, , , and
Abstract
:1. Introduction
2. Methods
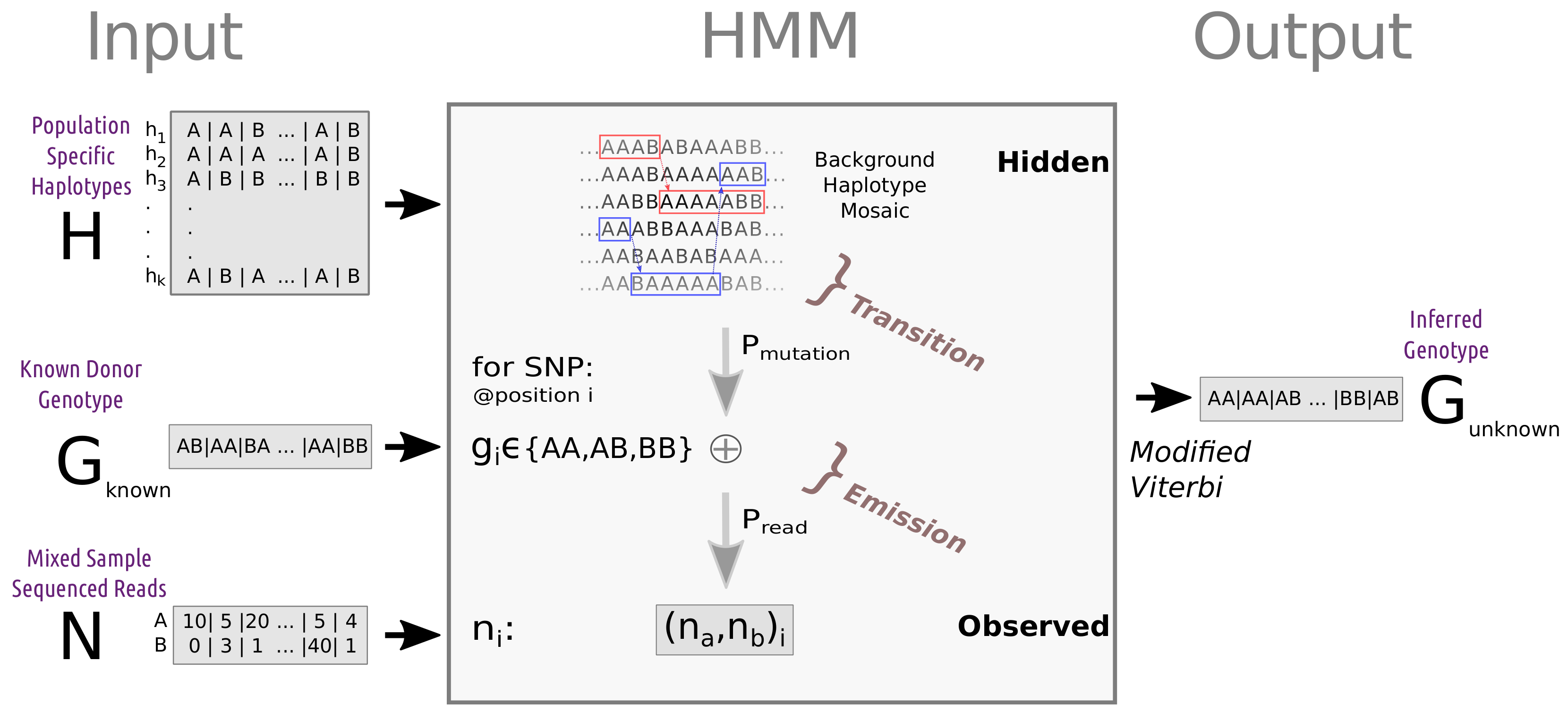
2.1. Problem Setup
- The number of individuals in the mixed sample is known to be two.
- The ethnic origin of the unknown person could be determined by preliminary steps.
- The victim has been genotyped with very high accuracy.
- All indels have been removed.
- The inference is designed for bi-allelic SNPs. Having the value of reference allele (tagged as ‘A’) or alternate allele (tagged as ‘B’), also called “REF” or “ALT”, respectively, in this paper.
2.2. Data Sets
2.3. Data Processing
2.4. Algorithms
2.4.1. Per SNP Bayesian Model (BYS)
2.4.2. Next SNP Based HMM (NS-HMM)
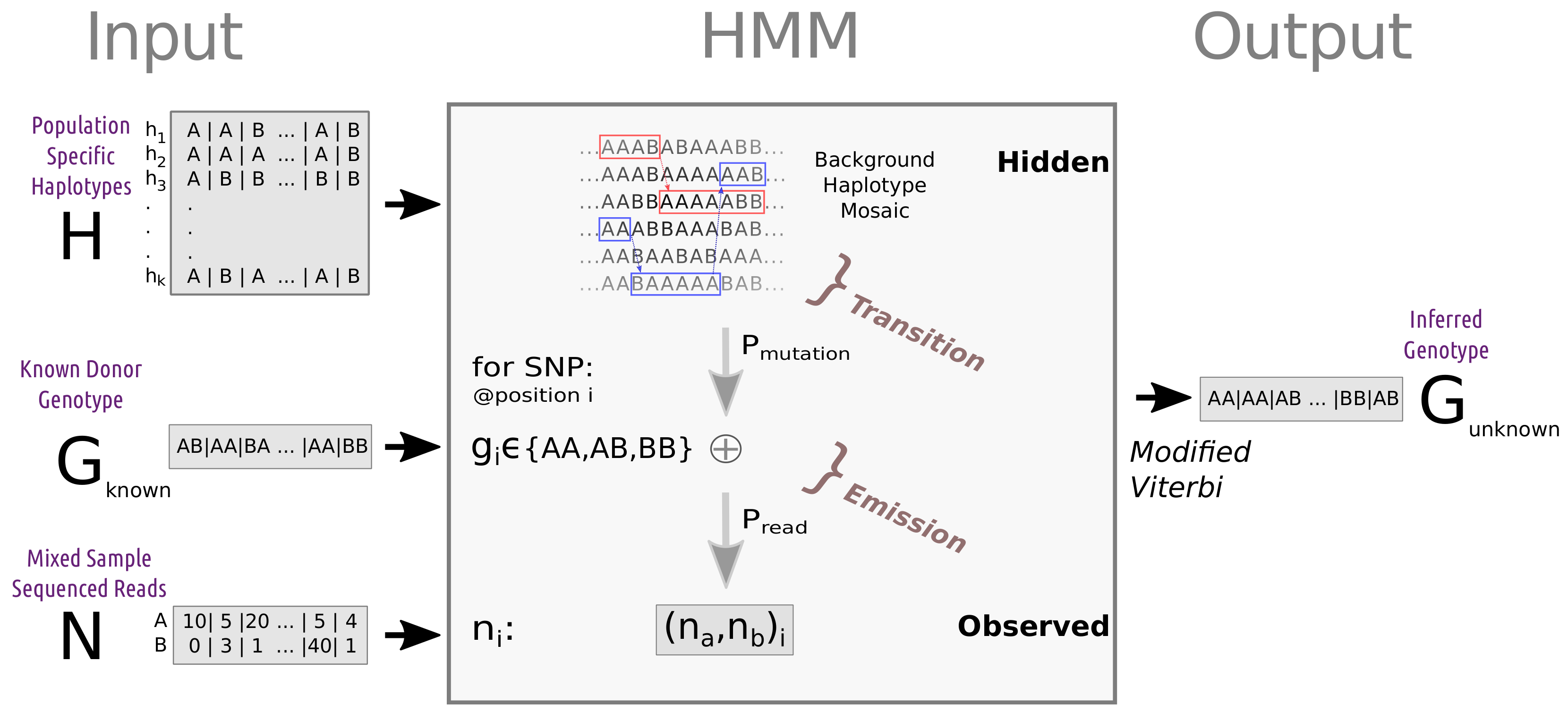
2.4.3. Simple and Advanced Haplotype-Based HMM Algorithm (SH-HMM and AH-HA)
- states —a haplotype pair from the reference panel.
- transition probability - representing transitions between haplotypes due to ancestral recombinations.
- observed data—REF-ALT read count table.
- emission probability—representing differences between the reference haplotype and the target genome, as well as sequencing errors. These parameters can be fixed or estimated (see Section 2.5).
2.5. Parameter Estimation
- —represents the effective population size. This parameter is used in the transition probability, as described in Appendix B. We used CHROMOPAINTER [29], an implementation of the Li and Stephens HMM for representing a target haplotype as a sequence of haplotypes from a reference panel. We used CHROMOPAINTER’s built-in E-M functionality to optimize this parameter in the relevant reference panel. For the benchmark case (chromosome 22, for the 240 haplotypes AJ reference panel) .
- —is the probability of an allele to differ between the nearest haplotype in the reference panel and the target [29]. It represents any process that would lead to a difference between the genotype of the target and the genotype of the most similar reference haplotype at this site. Similar to , this parameter was also optimized by using CHROMOPAINTER. For the benchmark case (chromosome 22, for the 240 haplotypes AJ reference panel) = 2.37 × 10−3.
- —represents the per base pair error rate, caused by amplification, alignment, and sequencing errors. In modern NGS technologies (ILLUMINA and CG) there is at least a 0.1% discordance rate [36]. This parameter, along with , determines the emission probability in HMM.
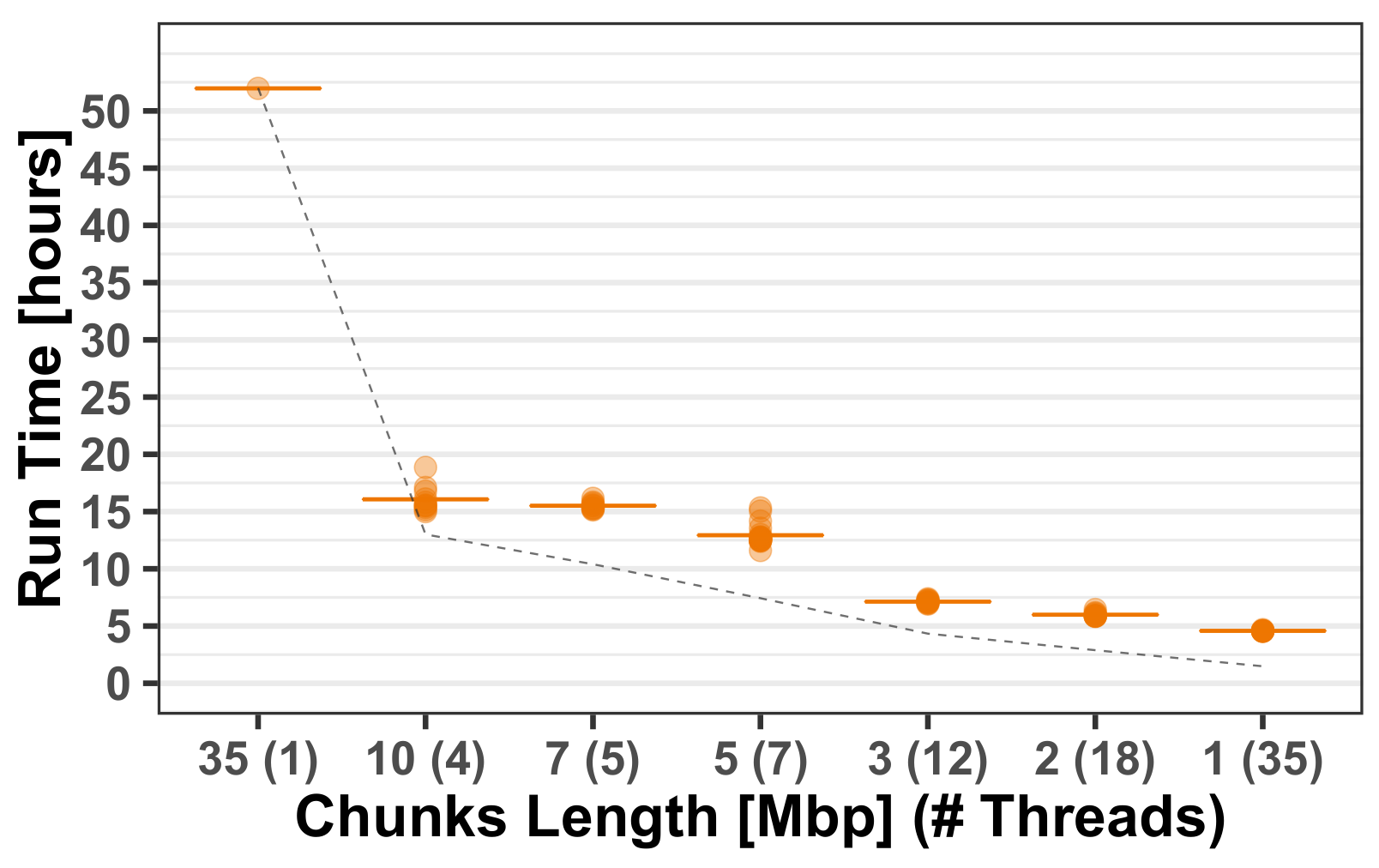
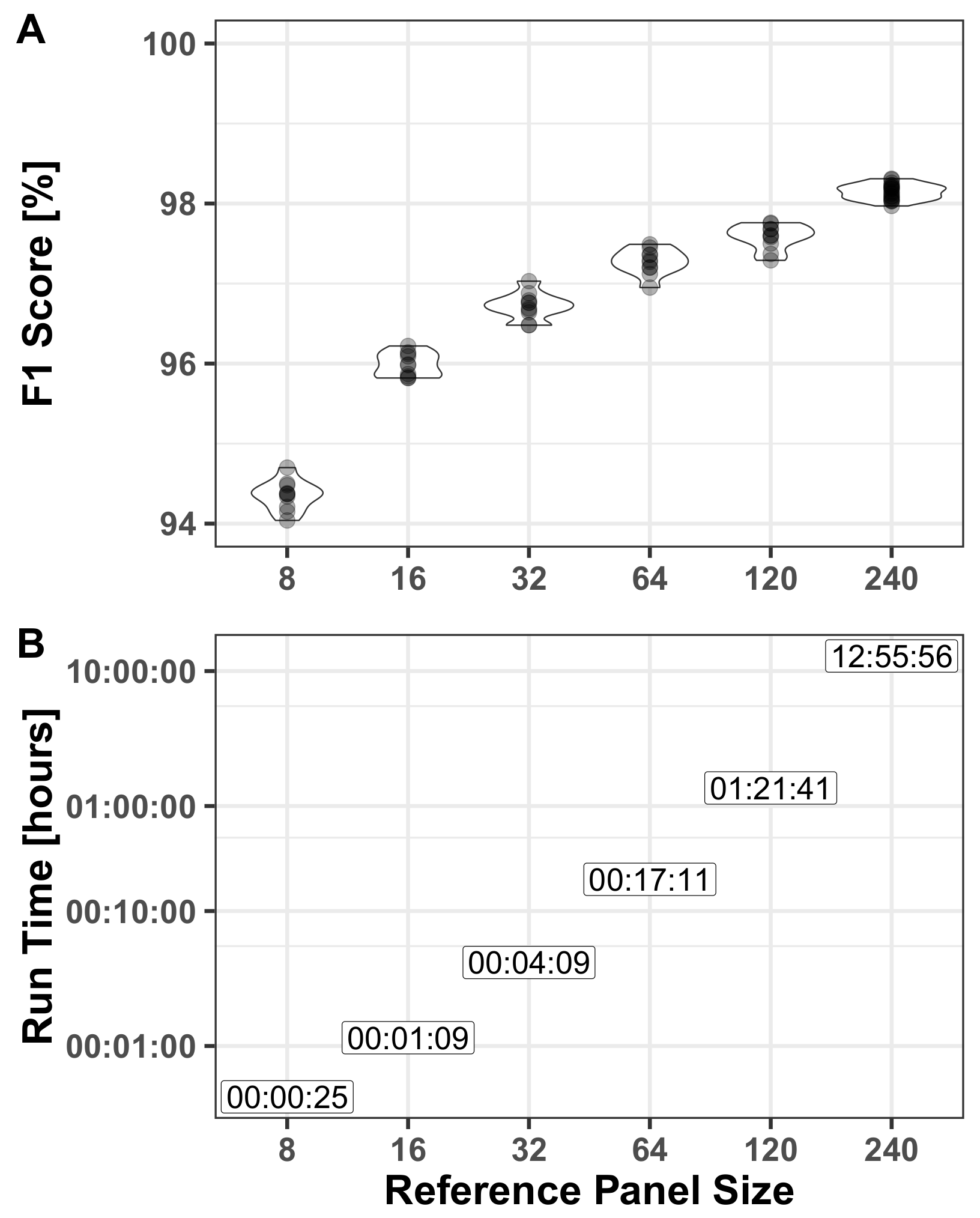
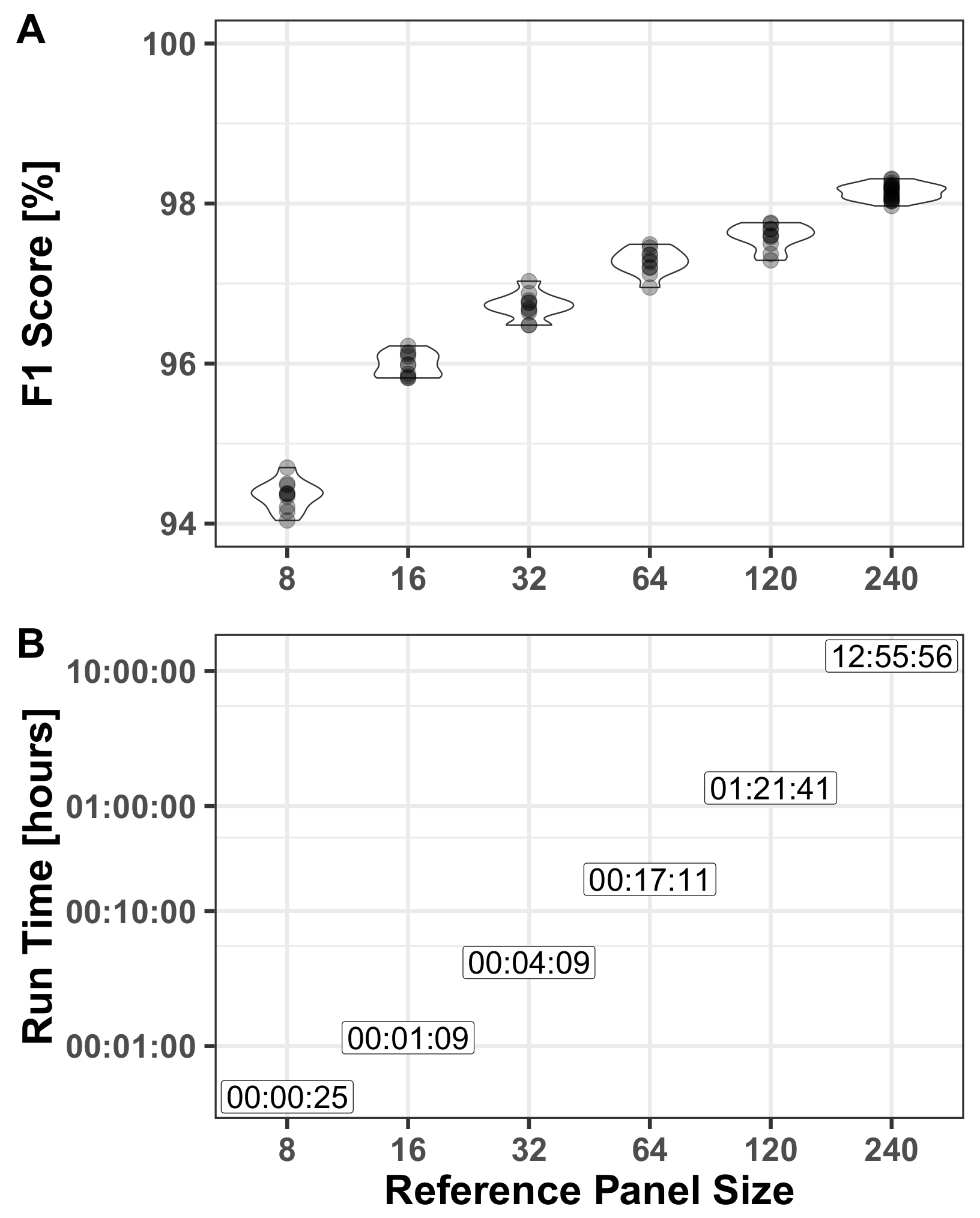
- Reference panel—derived as described above (Section 2.3). For assessing the effect on run-time and performance we used different panel sizes with different realizations of the haploids used in the panel.
2.6. F1 Score Calculation
3. Results
3.1. Performance Evaluation
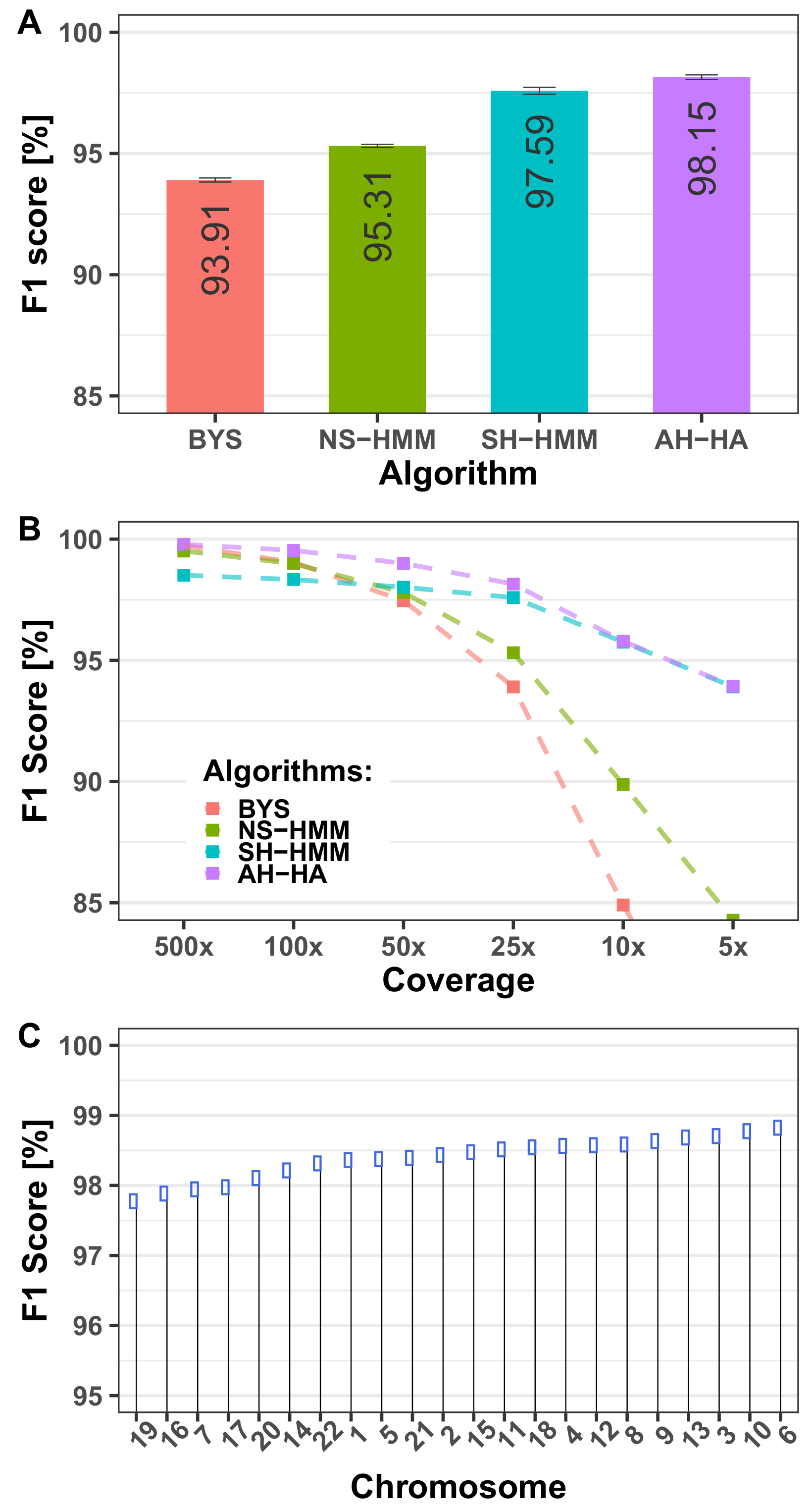
3.1.1. Algorithm Performance
- a.
- The AA–AA case accounts for 66.4% of all SNPs. This number is likely to remain high for all mixed samples. This case (together with the BB–BB case) is the simplest case of all algorithms, and hence the absolute performance of all algorithms is generally high.
- b.
- The unknown genotype is AA in 74.4% of all SNPs. A trivial algorithm that always outputs AA gives a good concordance score (74.4%), but an F1 score of 0 %. That is the main motivation behind using an F1 score instead of simple concordance.
- c.
- Two other simple inferences we looked at for baseline purposes are: 1. Infer the genotype most probable allele, based on the allele frequency of the reference panel for each SNP. This predictor yields an F1 score of 52.87% for the benchmark case. 2. Infer the genotype to be that of the known individual. This predictor yields an F1 score of 72.31% for the benchmark case.
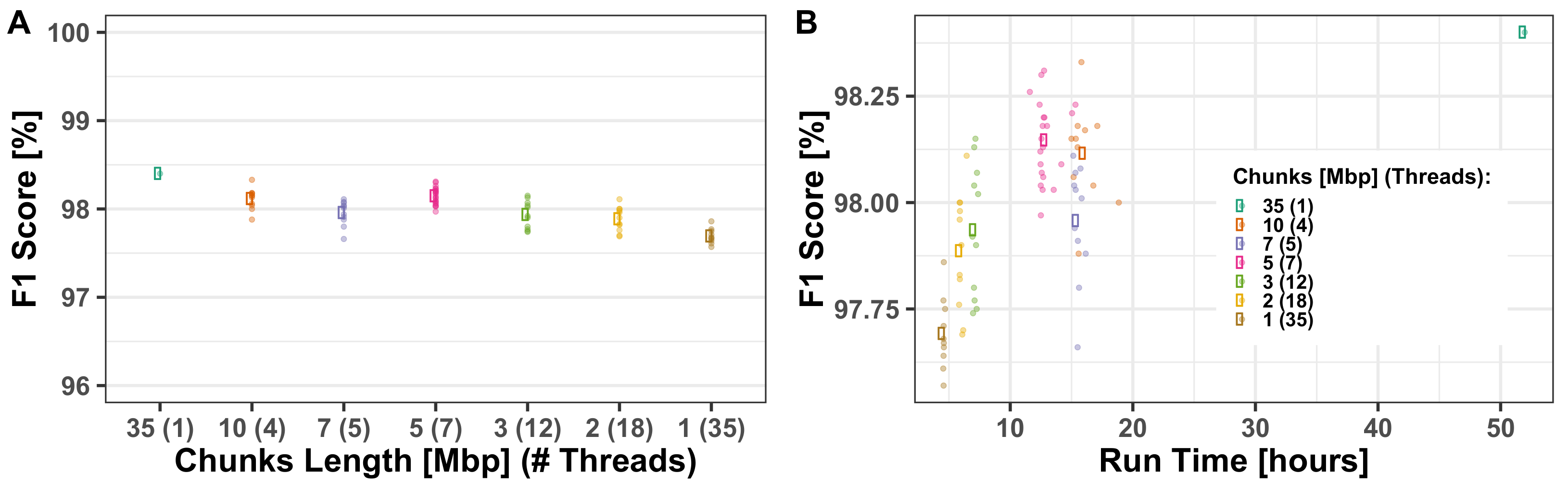
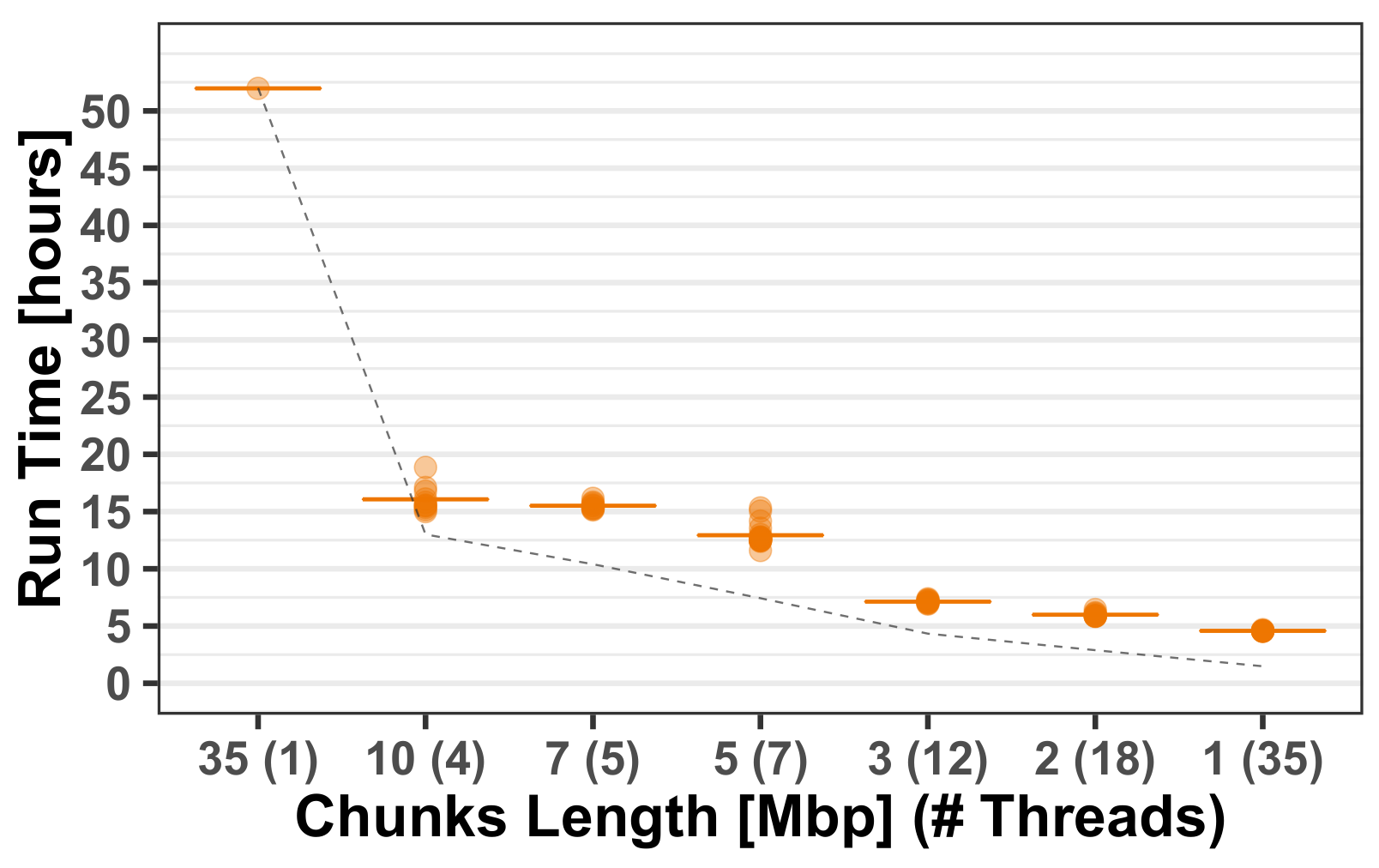
3.1.2. Computational Run-Times
3.2. Model Configuration
3.2.1. HMM Parameters
3.2.2. Reference Panel
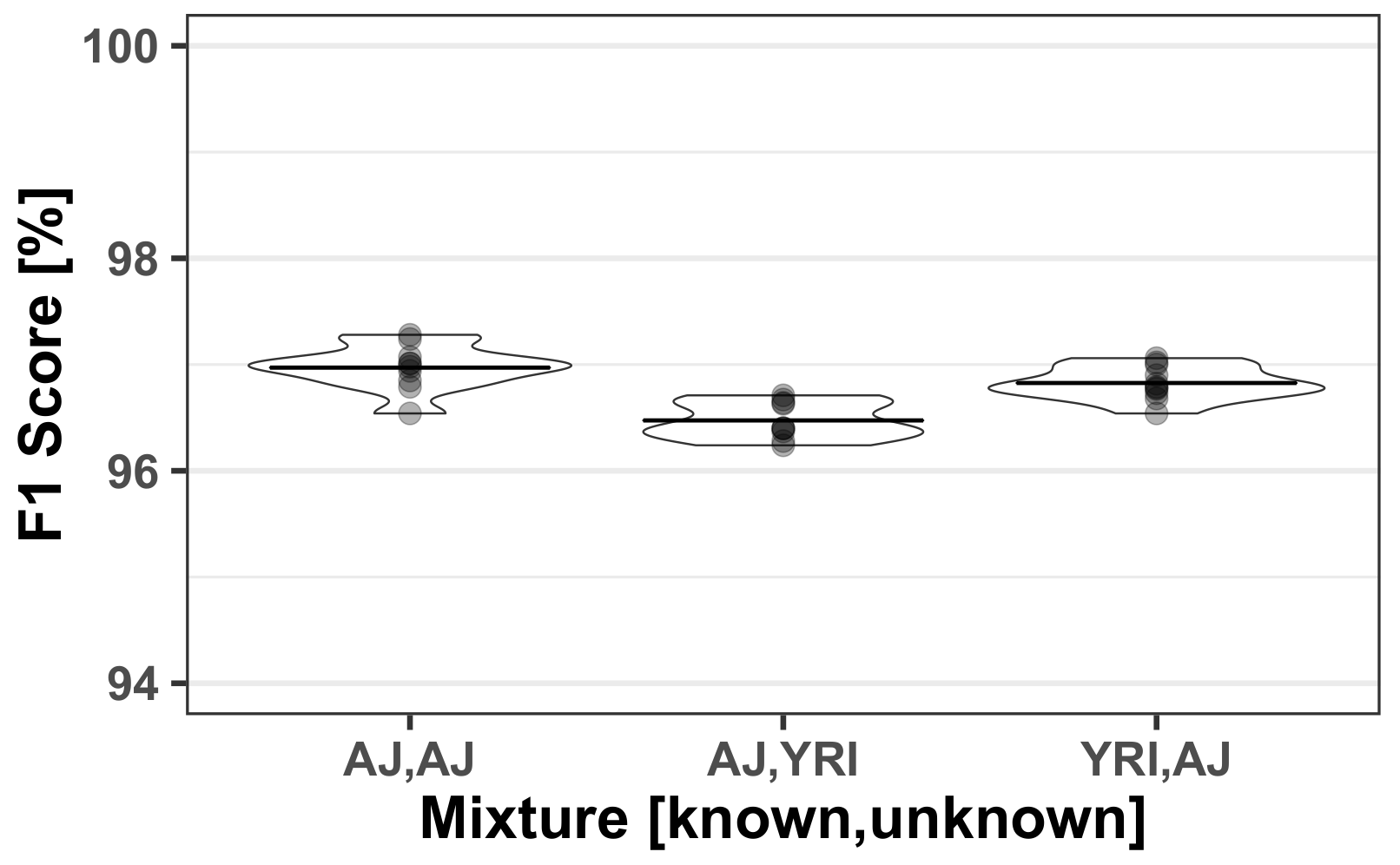
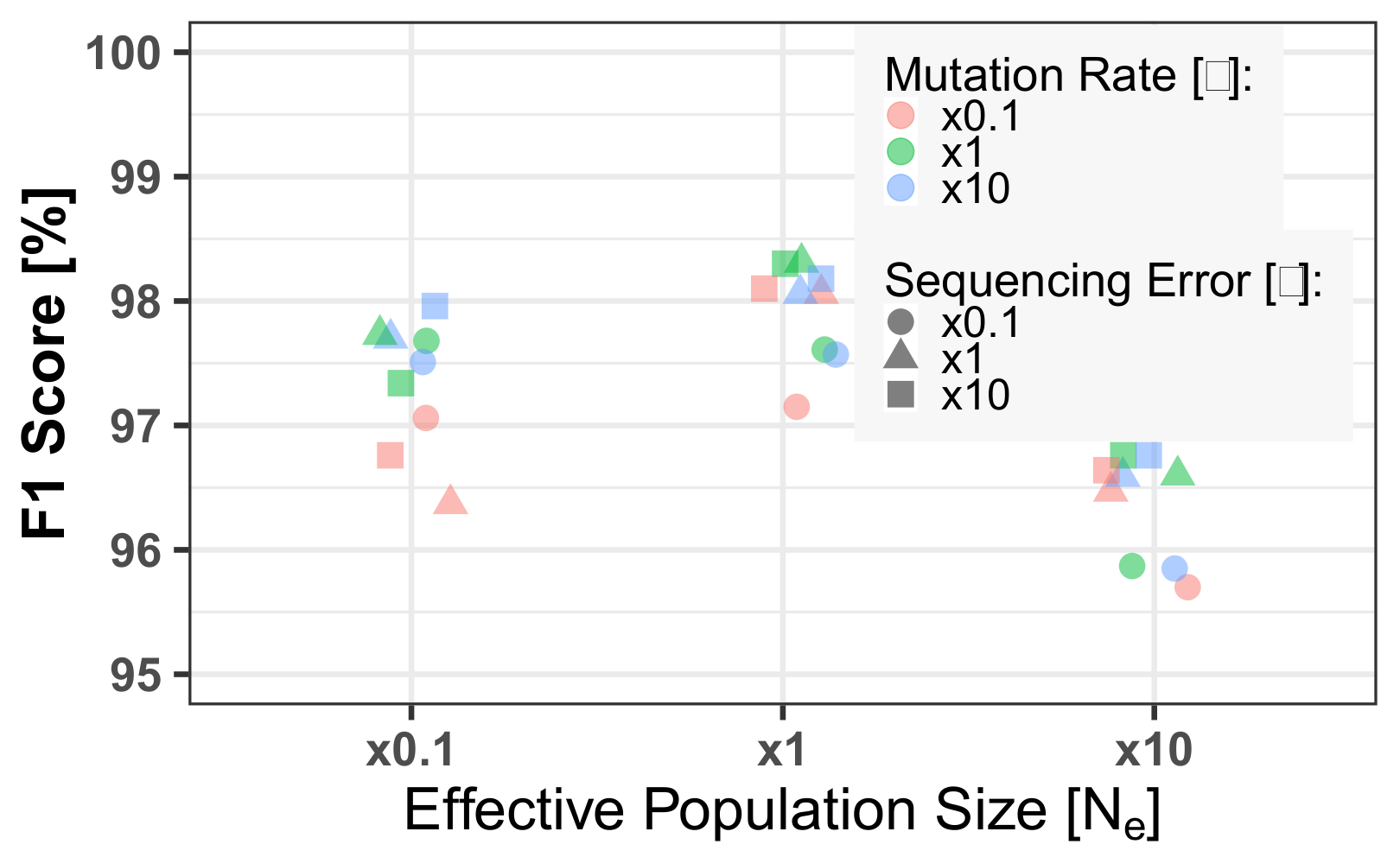
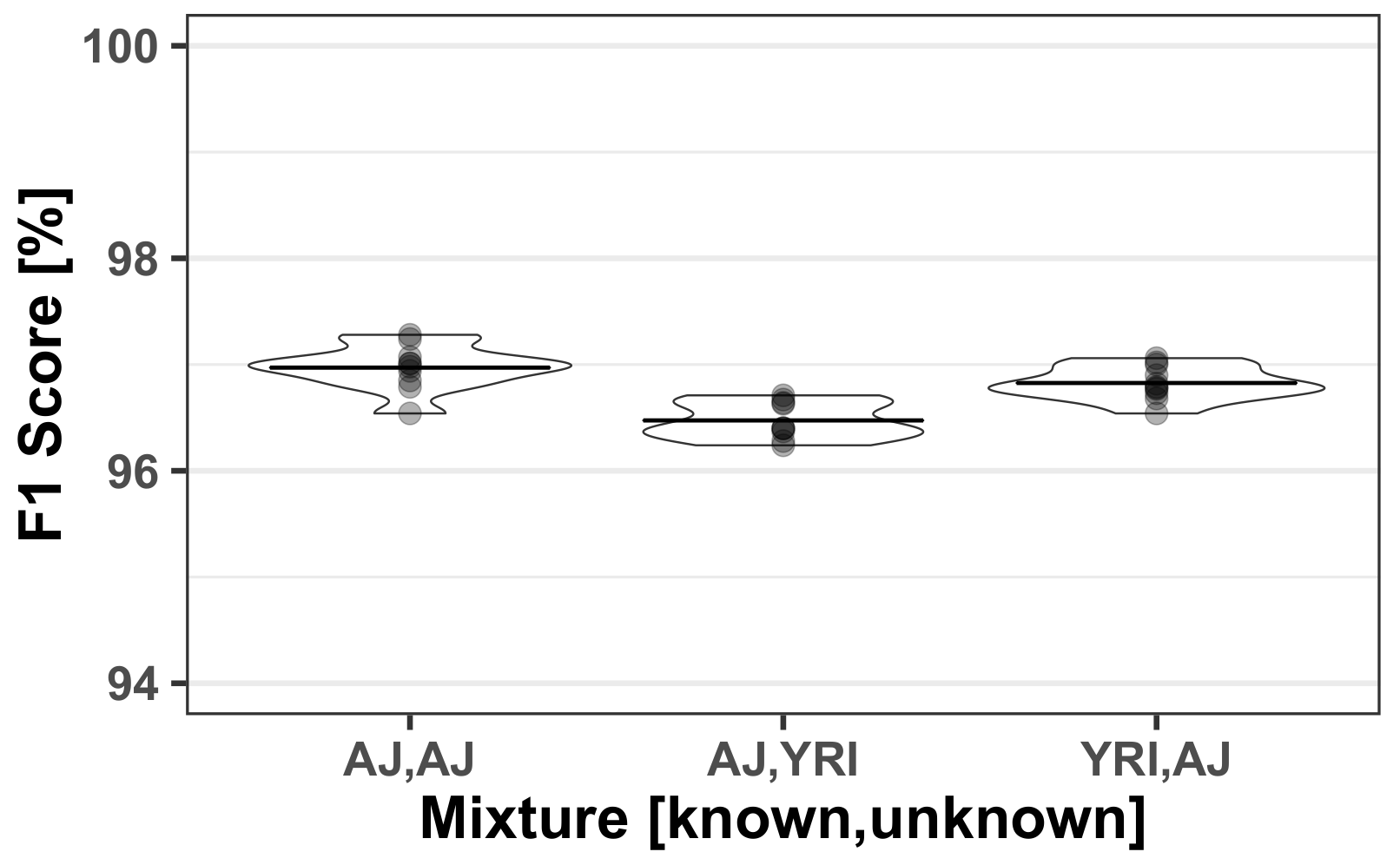
3.3. Mixed Population
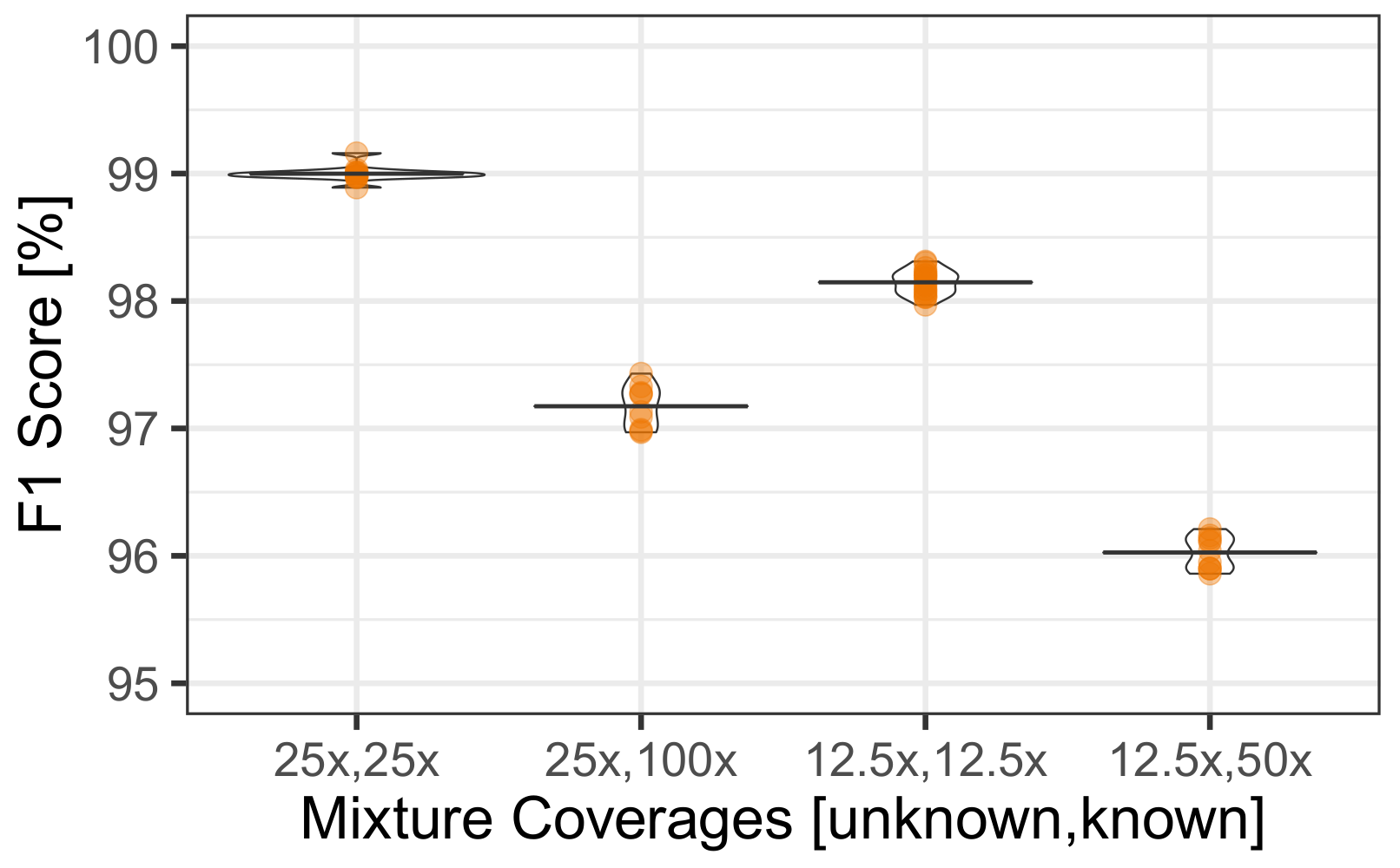
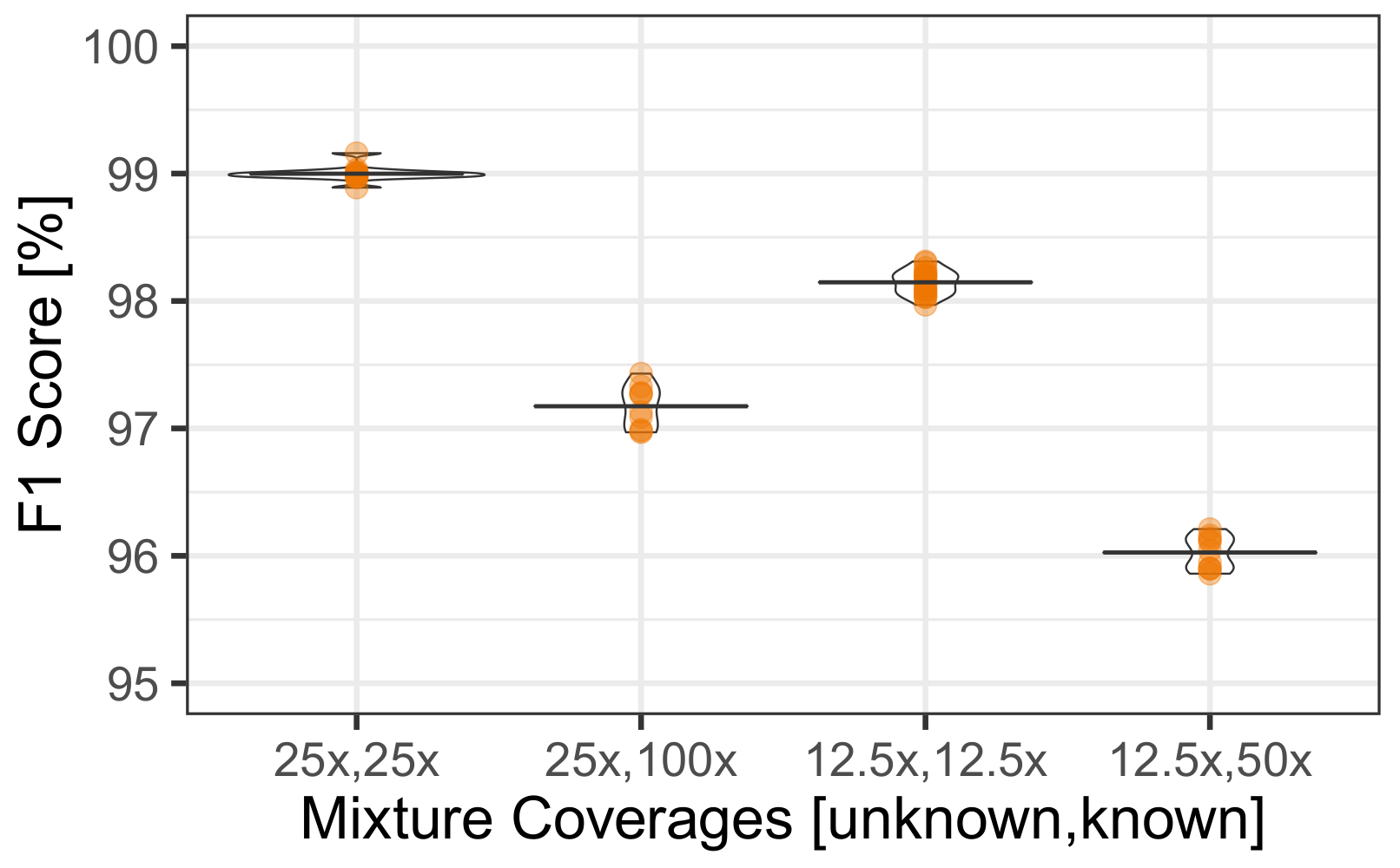
3.4. Uneven Mixtures
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SNP | Single-Nucleotide Polymorphism |
| HMM | Hidden Markov Model |
| WGS | Whole Genome Sequencing |
| STR | Short Tandem Repeat |
| HTS | High-Throughput Sequencing |
| MAF | Minor Allele Frequency |
| DNA | Deoxyribonucleic acid |
Appendix A. Additional Figures
Appendix A.1. Multi-Threading Performance
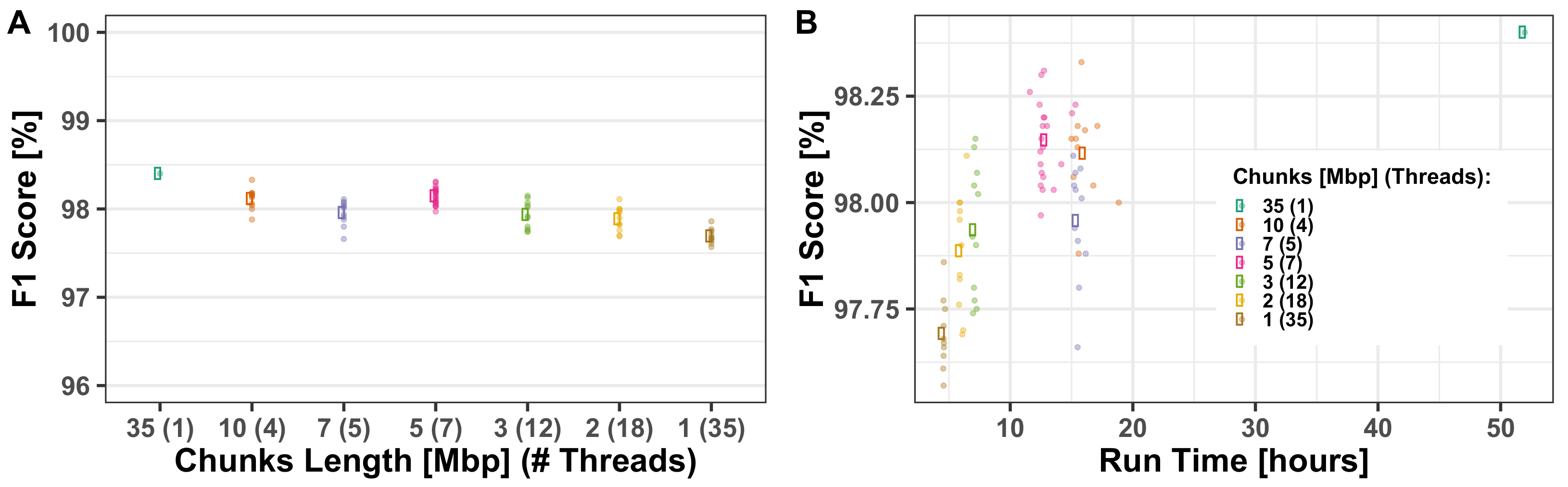
Appendix B. Extended Mathematical Description
Appendix B.1. Simple and Advanced Haplotype Based HMM Algorithm (SH-HMM and AH-HA)
Appendix B.1.1. Transition (Recombination) Model
Appendix B.1.2. Emission (Mutation) Model

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Observed | AAAA | AAAB | AABB | ABBB | BBBB | |
|---|---|---|---|---|---|---|
| Hidden | ||||||
| Known | Unknown | |||||
| AA | AA | 0 | 0 | |||
| AA | AB | 0 | 0 | |||
| AA | BB | 0 | 0 | |||
| AB | AA | 0 | 0 | |||
| AB | AB | 0 | 0 | |||
| AB | BB | 0 | 0 | |||
| BB | AA | 0 | 0 | |||
| BB | AB | 0 | 0 | |||
| BB | BB | 0 | 0 |
| Unknown | AA | AB | BB | |
|---|---|---|---|---|
| Known | ||||
| AA | 1 | |||
| AB | 0.5 | |||
| BB | 0 | |||
Appendix B.1.3. Viterbi Algorithm
References
- Gill, P. An assessment of the utility of single-nucleotide polymorphisms (SNPs) for forensic purposes. Int. J. Leg. Med. 2001, 114, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Sobrino, B.; Bríon, M.; Carracedo, A. SNPs in forensic genetics: A review on SNP typing methodologies. Forensic Sci. Int. 2005, 154, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.M.; Coble, M.D.; Vallone, P.M. STRs vs. SNPs: Thoughts on the future of forensic DNA testing. Forensic Sci. Med. Pathol. 2007, 3, 200–205. [Google Scholar] [CrossRef]
- Butler, J.M.; Budowle, B.; Gill, P.; Kidd, K.; Phillips, C.; Schneider, P.M.; Vallone, P.; Morling, N. Report on ISFG SNP panel discussion. Forensic Sci. Int. Genet. Suppl. Ser. 2008, 1, 471–472. [Google Scholar] [CrossRef]
- Algee-Hewitt, B.F.; Edge, M.D.; Kim, J.; Li, J.Z.; Rosenberg, N.A. Individual identifiability predicts population identifiability in forensic microsatellite markers. Curr. Biol. 2016, 26, 935–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.Y.; Harbison, S. A review of bioinformatic methods for forensic DNA analyses. Forensic Sci. Int. Genet. 2018, 33, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Budowle, B.; Van Daal, A. Forensically relevant SNP classes. Biotechniques 2008, 44, 603–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, R.; Santos, C.; Phillips, C.; Fondevila, M.; Van Oorschot, R.; Carracedo, A.; Lareu, M.; McNevin, D. A SNaPshot of next generation sequencing for forensic SNP analysis. Forensic Sci. Int. Genet. 2015, 14, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Churchill, J.D.; Schmedes, S.E.; King, J.L.; Budowle, B. Evaluation of the Illumina® beta version ForenSeq™ DNA signature prep kit for use in genetic profiling. Forensic Sci. Int. Genet. 2016, 20, 20–29. [Google Scholar] [CrossRef]
- Erlich, Y.; Shor, T.; Pe’er, I.; Carmi, S. Identity inference of genomic data using long-range familial searches. Science 2018, 362, 690–694. [Google Scholar] [CrossRef] [Green Version]
- Katsanis, S.H. Pedigrees and perpetrators: Uses of DNA and genealogy in forensic investigations. Annu. Rev. Genom. Hum. Genet. 2020, 21, 535–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennett, D. Using genetic genealogy databases in missing persons cases and to develop suspect leads in violent crimes. Forensic Sci. Int. 2019, 301, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Gettings, K.B.; Kiesler, K.M.; Vallone, P.M. Performance of a next generation sequencing SNP assay on degraded DNA. Forensic Sci. Int. Genet. 2015, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lin, D.; Deng, C.; Li, Z.; Pu, Y.; Yu, Y.; Li, K.; Li, D.; Chen, P.; Chen, F. The advances in DNA mixture interpretation. Forensic Sci. Int. 2019. [Google Scholar] [CrossRef] [PubMed]
- Buckleton, J.S.; Bright, J.A.; Gittelson, S.; Moretti, T.R.; Onorato, A.J.; Bieber, F.R.; Budowle, B.; Taylor, D.A. The Probabilistic Genotyping Software STR mix: Utility and Evidence for its Validity. J. Forensic Sci. 2019, 64, 393–405. [Google Scholar] [CrossRef]
- Haned, H.; Slooten, K.; Gill, P. Exploratory data analysis for the interpretation of low template DNA mixtures. Forensic Sci. Int. Genet. 2012, 6, 762–774. [Google Scholar] [CrossRef]
- Bleka, Ø.; Storvik, G.; Gill, P. EuroForMix: An open source software based on a continuous model to evaluate STR DNA profiles from a mixture of contributors with artefacts. Forensic Sci. Int. Genet. 2016, 21, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Ricke, D.O.; Isaacson, J.; Watkins, J.; Fremont-Smith, P.; Boettcher, T.; Petrovick, M.; Wack, E.; Schwoebel, E. The Plateau Method for Forensic DNA SNP Mixture Deconvolution. bioRxiv 2017, 225805. [Google Scholar]
- Gill, P.; Haned, H.; Eduardoff, M.; Santos, C.; Phillips, C.; Parson, W. The open-source software LRmix can be used to analyse SNP mixtures. Forensic Sci. Int. Genet. Suppl. Ser. 2015, 5, e50–e51. [Google Scholar] [CrossRef] [Green Version]
- Bleka, Ø.; Eduardoff, M.; Santos, C.; Phillips, C.; Parson, W.; Gill, P. Open source software EuroForMix can be used to analyse complex SNP mixtures. Forensic Sci. Int. Genet. 2017, 31, 105–110. [Google Scholar] [CrossRef]
- Voskoboinik, L.; Ayers, S.B.; LeFebvre, A.K.; Darvasi, A. SNP-microarrays can accurately identify the presence of an individual in complex forensic DNA mixtures. Forensic Sci. Int. Genet. 2015, 16, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, J.; Schwoebel, E.; Shcherbina, A.; Ricke, D.; Harper, J.; Petrovick, M.; Bobrow, J.; Boettcher, T.; Helfer, B.; Zook, C.; et al. Robust detection of individual forensic profiles in DNA mixtures. Forensic Sci. Int. Genet. 2015, 14, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.M.; Gambin, T.; Jhangiani, S.N.; Grove, M.L.; Veeraraghavan, N.; Muzny, D.M.; Shaw, C.A.; Gibbs, R.A.; Boerwinkle, E.; Yu, F.; et al. Multiallelic positions in the human genome: Challenges for genetic analyses. Hum. Mutat. 2016, 37, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Kidd, K.; Pakstis, A.; Speed, W.; Lagace, R.; Chang, J.; Wootton, S.; Ihuegbu, N. Microhaplotype loci are a powerful new type of forensic marker. Forensic Sci. Int. Genet. Suppl. Ser. 2013, 4, e123–e124. [Google Scholar] [CrossRef]
- Voskoboinik, L.; Motro, U.; Darvasi, A. Facilitating complex DNA mixture interpretation by sequencing highly polymorphic haplotypes. Forensic Sci. Int. Genet. 2018, 35, 136–140. [Google Scholar] [CrossRef]
- Zhu, S.J.; Almagro-Garcia, J.; McVean, G. Deconvolution of multiple infections in Plasmodium falciparum from high throughput sequencing data. Bioinformatics 2018, 34, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Smart, U.; Cihlar, J.C.; Mandape, S.N.; Muenzler, M.; King, J.L.; Budowle, B.; Woerner, A.E. A continuous statistical phasing framework for the analysis of forensic mitochondrial DNA mixtures. Genes 2021, 12, 128. [Google Scholar] [CrossRef]
- Li, N.; Stephens, M. Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics 2003, 165, 2213–2233. [Google Scholar] [CrossRef]
- Lawson, D.J.; Hellenthal, G.; Myers, S.; Falush, D. Inference of population structure using dense haplotype data. PLoS Genet. 2012, 8, e1002453. [Google Scholar] [CrossRef] [Green Version]
- Browning, S.R.; Browning, B.L. Haplotype phasing: Existing methods and new developments. Nat. Rev. Genet. 2011, 12, 703–714. [Google Scholar] [CrossRef] [Green Version]
- Howie, B.; Marchini, J.; Stephens, M. Genotype imputation with thousands of genomes. G3 Genes Genomes Genet. 2011, 1, 457–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature 2012, 491, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaneau, O.; Zagury, J.F.; Marchini, J. Improved whole-chromosome phasing for disease and population genetic studies. Nat. Methods 2013, 10, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Carmi, S.; Hui, K.Y.; Kochav, E.; Liu, X.; Xue, J.; Grady, F.; Guha, S.; Upadhyay, K.; Ben-Avraham, D.; Mukherjee, S.; et al. Sequencing an Ashkenazi reference panel supports population-targeted personal genomics and illuminates Jewish and European origins. Nat. Commun. 2014, 5, 4835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zook, J.M.; Catoe, D.; McDaniel, J.; Vang, L.; Spies, N.; Sidow, A.; Weng, Z.; Liu, Y.; Mason, C.E.; Alexander, N.; et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. Sci. Data 2016, 3. [Google Scholar] [CrossRef] [Green Version]
- Wall, J.D.; Tang, L.F.; Zerbe, B.; Kvale, M.N.; Kwok, P.Y.; Schaefer, C.; Risch, N. Estimating genotype error rates from high-coverage next-generation sequence data. Genome Res. 2014, 24, 1734–1739. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, R.A.; Belmont, J.W.; Hardenbol, P.; Willis, R.A.; Gibbs, T.D.; Yu, F.L.; Yang, H.M.; Ch’ang, L.Y.; Huang, W.; Liu, B.; et al. The international HapMap project. Nature 2003, 426, 789–796. [Google Scholar] [CrossRef] [Green Version]
- Rabiner, L.R. A tutorial on hidden Markov models and selected applications in speech recognition. Proc. IEEE 1989, 77, 257–286. [Google Scholar] [CrossRef] [Green Version]
- Kidd, K.K.; Speed, W.C.; Pakstis, A.J.; Furtado, M.R.; Fang, R.; Madbouly, A.; Maiers, M.; Middha, M.; Friedlaender, F.R.; Kidd, J.R. Progress toward an efficient panel of SNPs for ancestry inference. Forensic Sci. Int. Genet. 2014, 10, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.L.; Wei, L.; Zhao, L.; Sun, Q.F.; Jiang, L.; Zhang, T.; Liu, H.B.; Chen, J.G.; Ye, J.; Hu, L.; et al. A single-tube 27-plex SNP assay for estimating individual ancestry and admixture from three continents. Int. J. Leg. Med. 2016, 130, 27–37. [Google Scholar] [CrossRef]
- Delaneau, O.; Howie, B.; Cox, A.J.; Zagury, J.F.; Marchini, J. Haplotype estimation using sequencing reads. Am. J. Hum. Genet. 2013, 93, 687–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browning, S.R.; Browning, B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007, 81, 1084–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunter, G. Haplotype matching in large cohorts using the Li and Stephens model. Bioinformatics 2019, 35, 798–806. [Google Scholar] [CrossRef] [PubMed]
| AA | AAAA | AAAB | AABB | 1 | 0.75 | 0.5 |
| AB | AAAB | AABB | ABBB | 0.75 | 0.5 | 0.25 |
| BB | AABB | ABBB | BBBB | 0.5 | 0.75 | 0 |
| Known | Unknown | AA | AB | BB | Total | Percentage | Discordance |
|---|---|---|---|---|---|---|---|
| [#] | [#] | [#] | [#] | [%] | [%] | ||
| AA | AA | 95,057 | 284 | 1 | 95,342 | 66.42 | 0.30 |
| AA | AB | 73 | 12,068 | 66 | 12,207 | 8.5 | 1.14 |
| AA | BB | 8 | 210 | 1744 | 1962 | 1.37 | 11.11 |
| AB | AA | 9120 | 284 | 0 | 9404 | 6.55 | 3.02 |
| AB | AB | 190 | 8803 | 73 | 9066 | 6.32 | 2.90 |
| AB | BB | 0 | 249 | 3524 | 3773 | 2.63 | 6.60 |
| BB | AA | 2034 | 82 | 0 | 2116 | 1.47 | 3.88 |
| BB | AB | 47 | 3774 | 9 | 3830 | 2.67 | 1.46 |
| BB | BB | 3 | 39 | 5798 | 5840 | 4.07 | 0.72 |
| Predicted | AA | AB | BB | |
|---|---|---|---|---|
| Truth | ||||
| AA | TN | 1/2TN + 1/2FP | FP | |
| AB | 1/2TN + 1/2FN | 1/2TN + 1/2TP | 1/2FP + 1/2TP | |
| BB | FN | 1/2FN + 1/2TP | TP | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azhari, G.; Waldman, S.; Ofer, N.; Keller, Y.; Carmi, S.; Yaari, G. Decomposition of Individual SNP Patterns from Mixed DNA Samples. Forensic Sci. 2022, 2, 455-472. https://doi.org/10.3390/forensicsci2030034
Azhari G, Waldman S, Ofer N, Keller Y, Carmi S, Yaari G. Decomposition of Individual SNP Patterns from Mixed DNA Samples. Forensic Sciences. 2022; 2(3):455-472. https://doi.org/10.3390/forensicsci2030034
Chicago/Turabian StyleAzhari, Gabriel, Shamam Waldman, Netanel Ofer, Yosi Keller, Shai Carmi, and Gur Yaari. 2022. "Decomposition of Individual SNP Patterns from Mixed DNA Samples" Forensic Sciences 2, no. 3: 455-472. https://doi.org/10.3390/forensicsci2030034
APA StyleAzhari, G., Waldman, S., Ofer, N., Keller, Y., Carmi, S., & Yaari, G. (2022). Decomposition of Individual SNP Patterns from Mixed DNA Samples. Forensic Sciences, 2(3), 455-472. https://doi.org/10.3390/forensicsci2030034