
Reactive Polymer Composite Microparticles Based on Glycidyl Methacrylate and Magnetite Nanoparticles


Abstract
1. Introduction
2. Experimental Part
2.1. Materials
2.2. Characterization
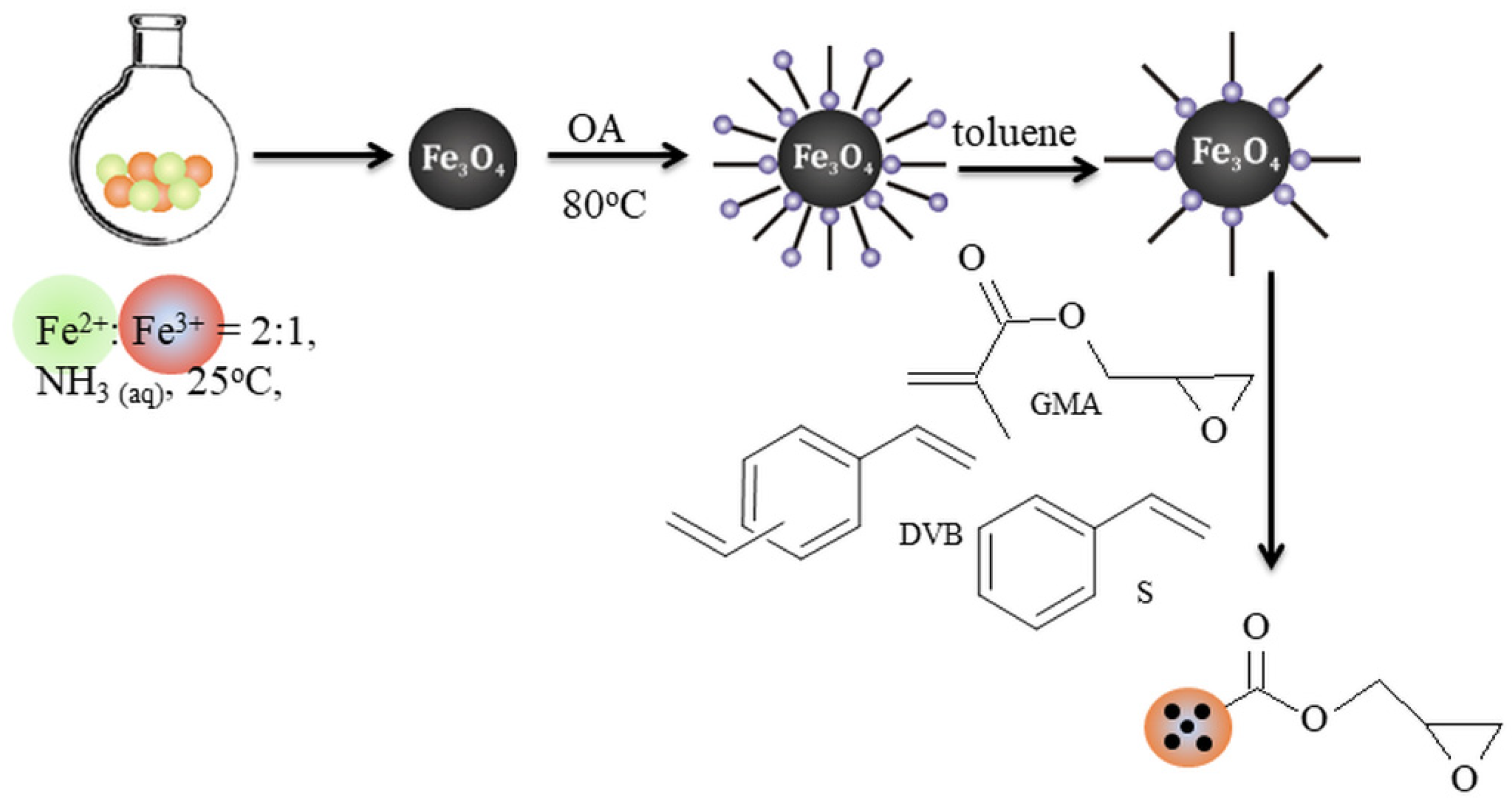
2.3. General Procedure of Preparation of Glycidyl-Functionalized Polymer Composite Microparticles
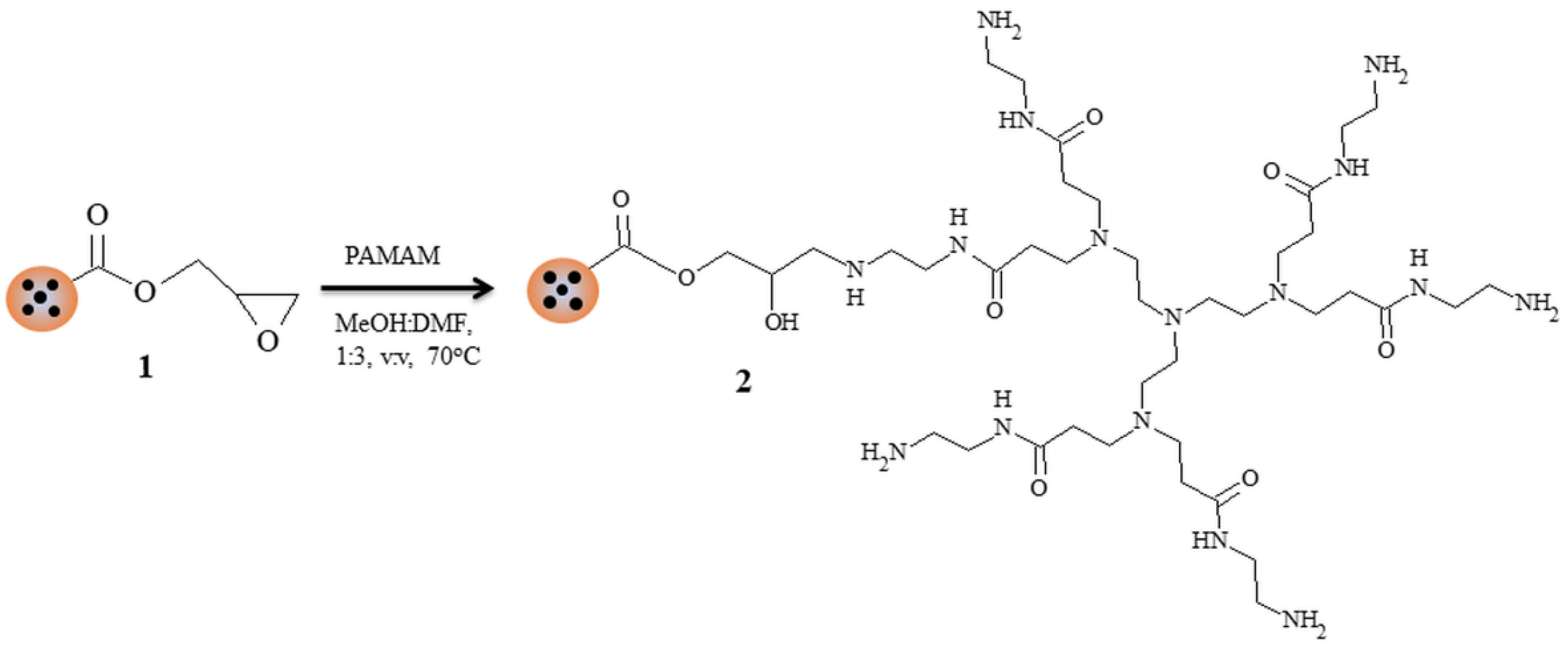
2.4. Post-Modification of Composite Microparticles with PAMAM Type of Polyamidoamine
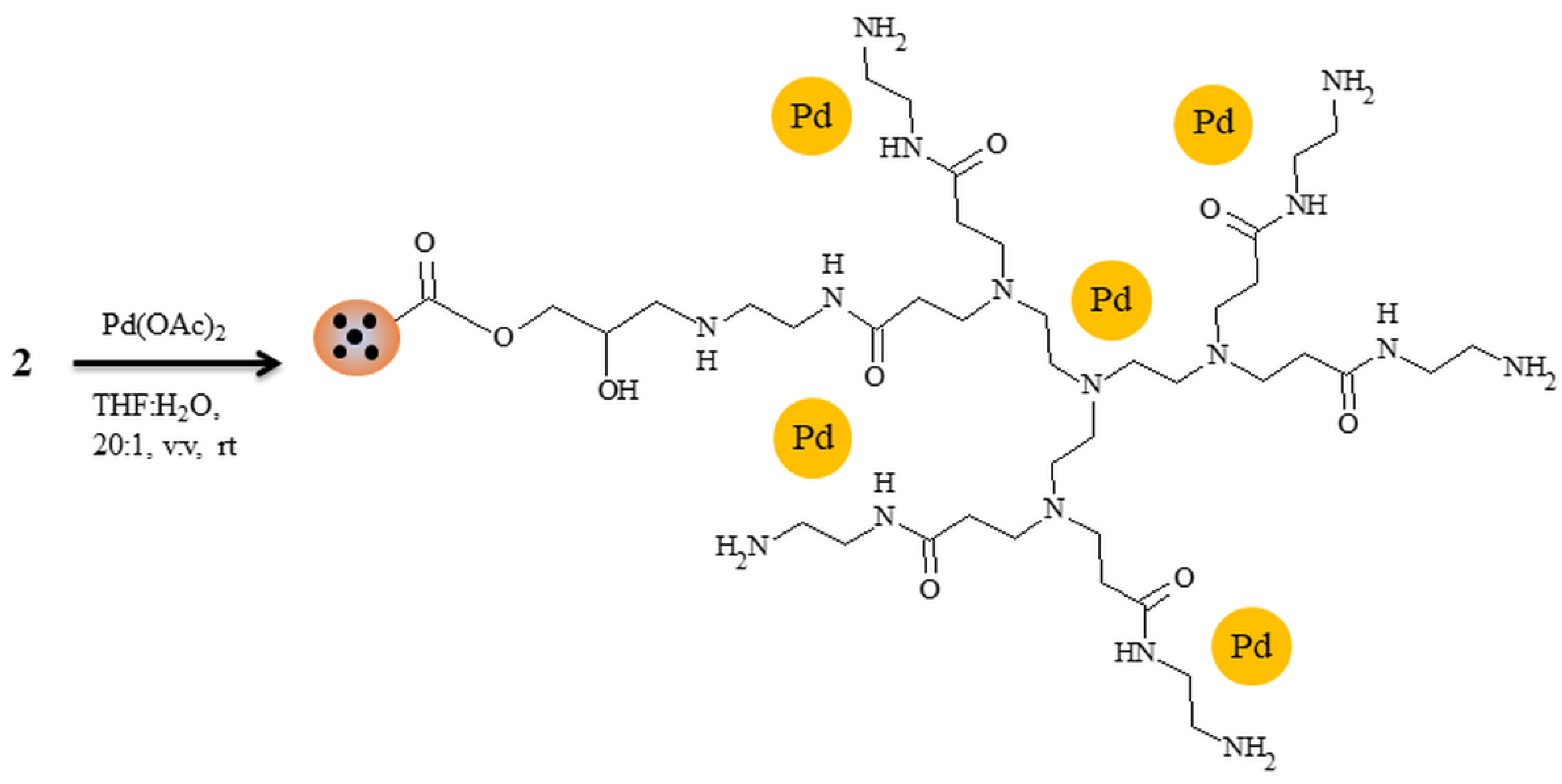
2.5. Palladium(II) Acetate Immobilization
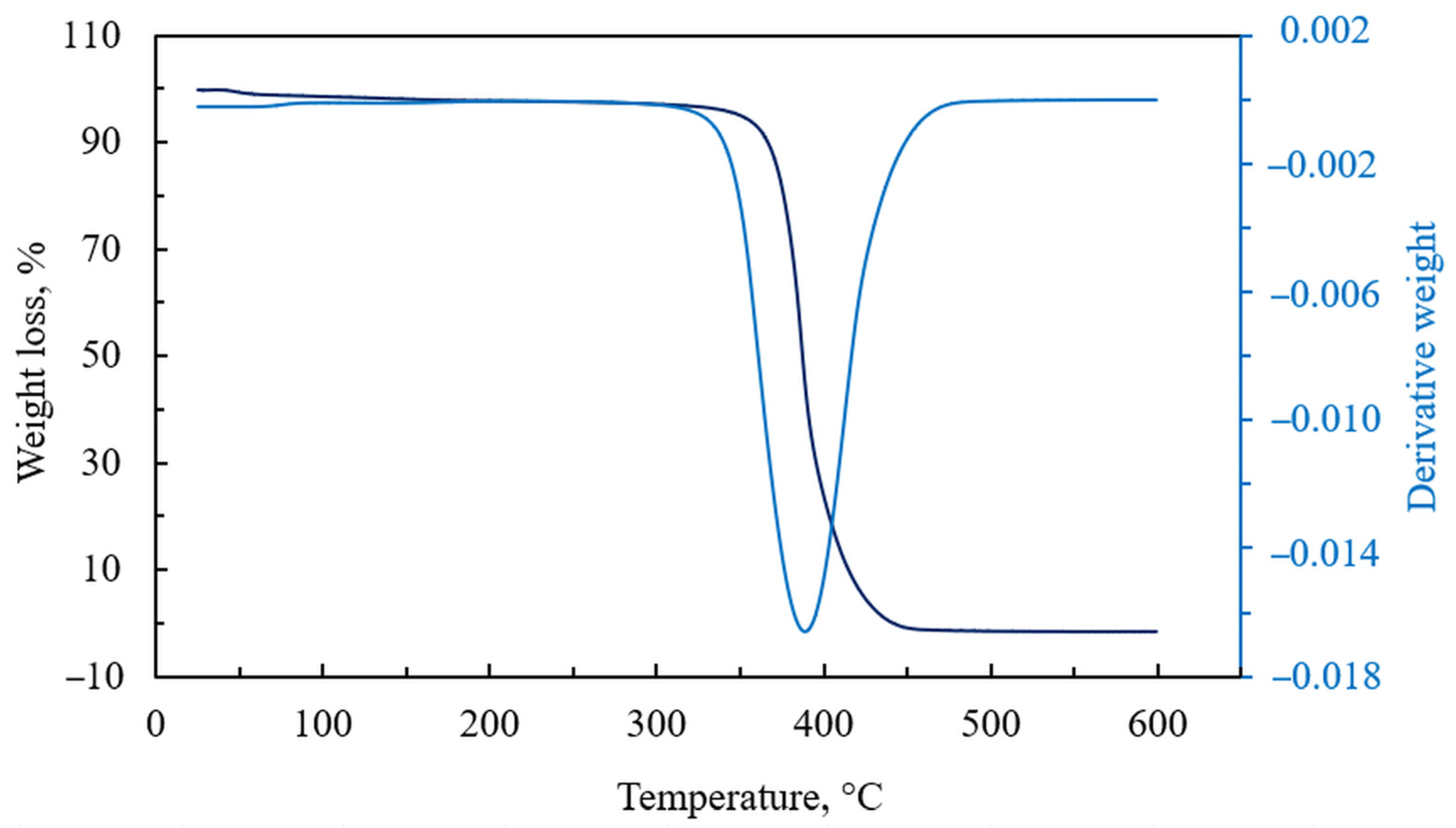
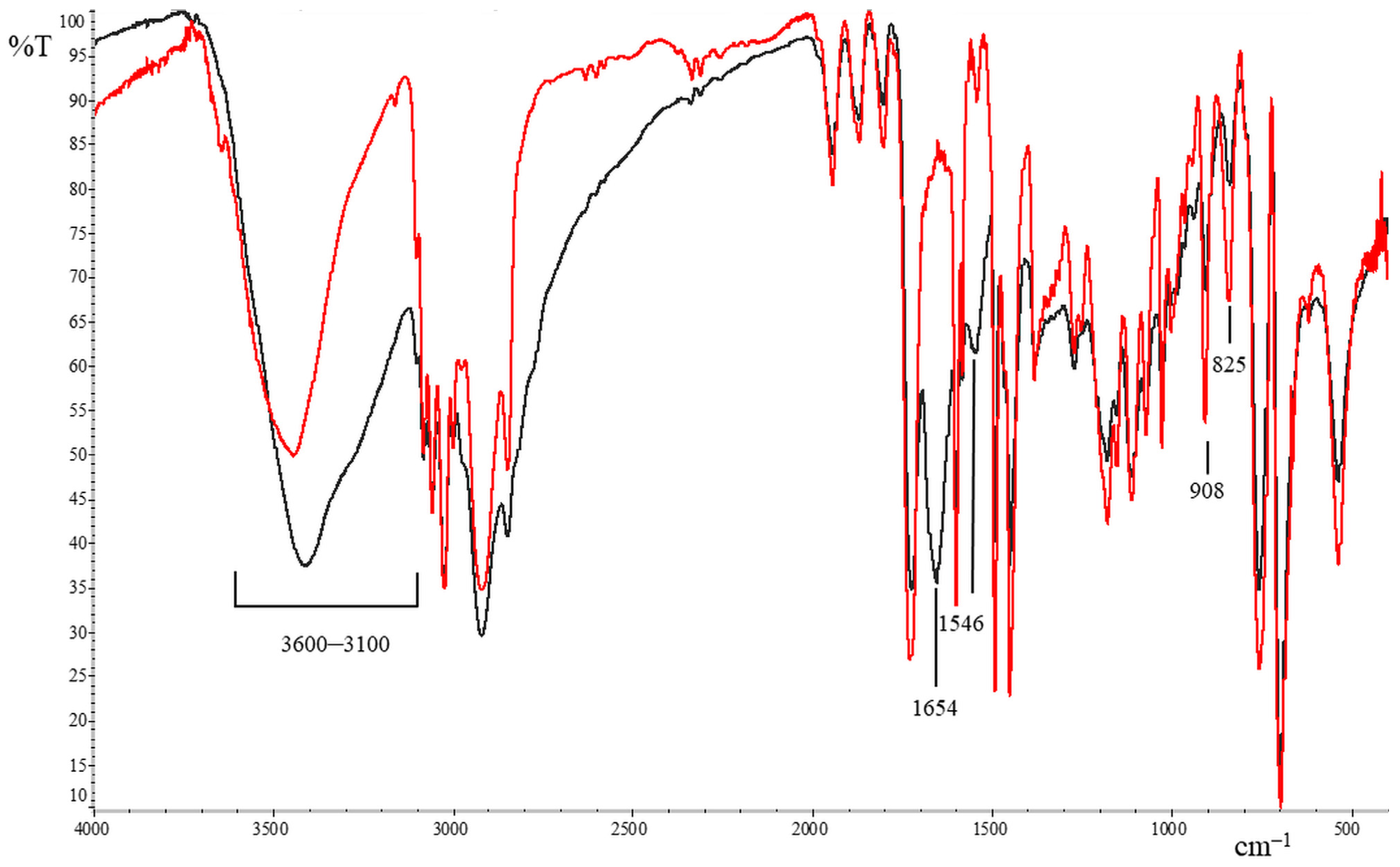
3. Results and Discussion
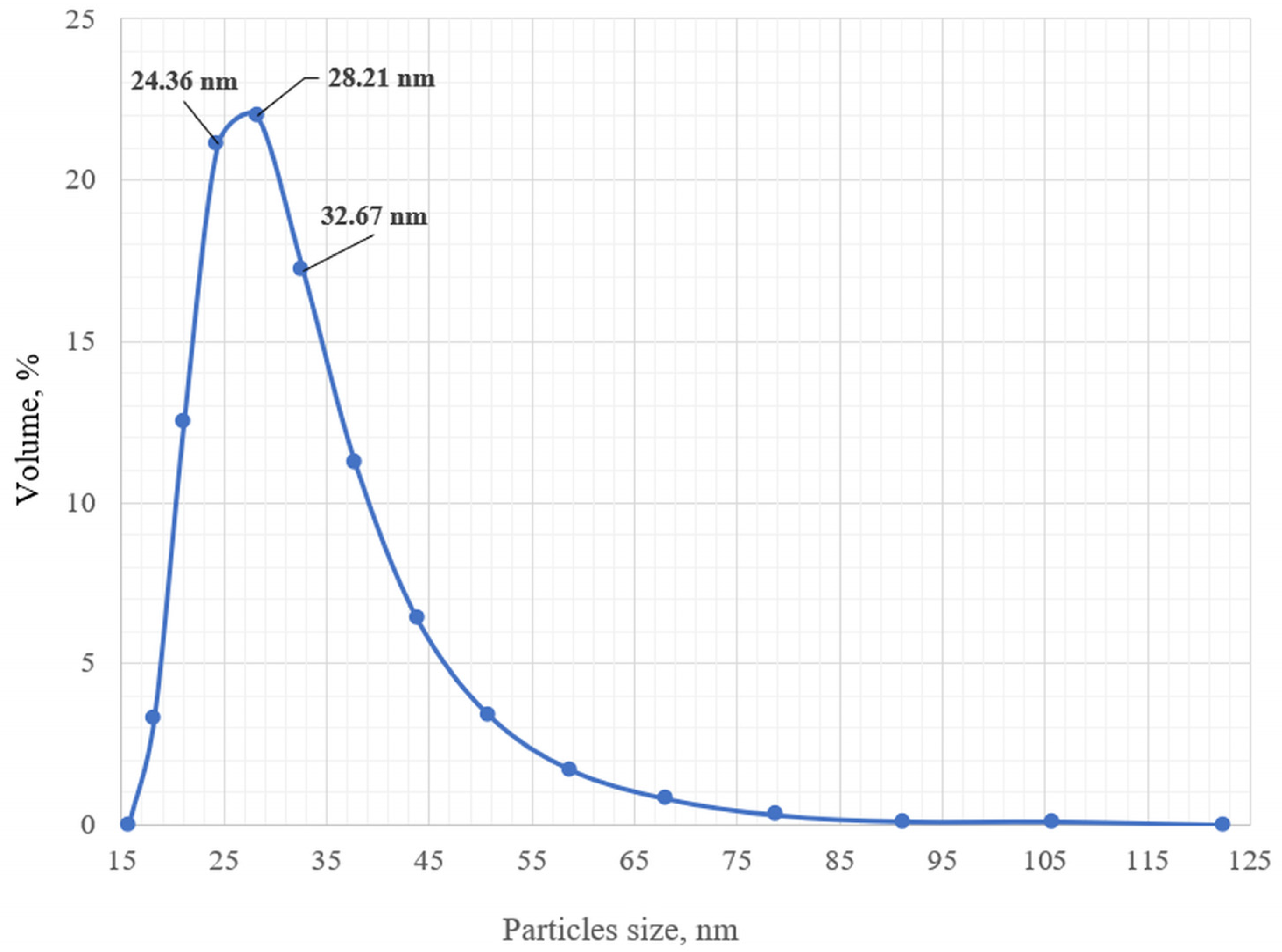
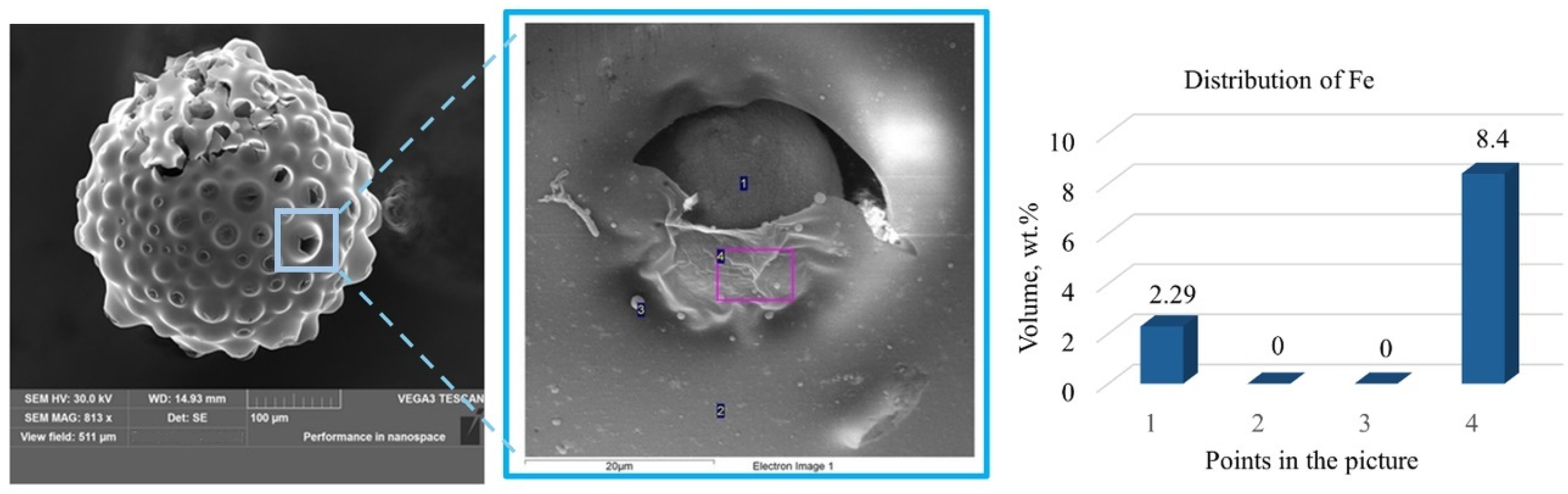
3.1. Preparation of Epoxy Functionalized Composite Microparticles
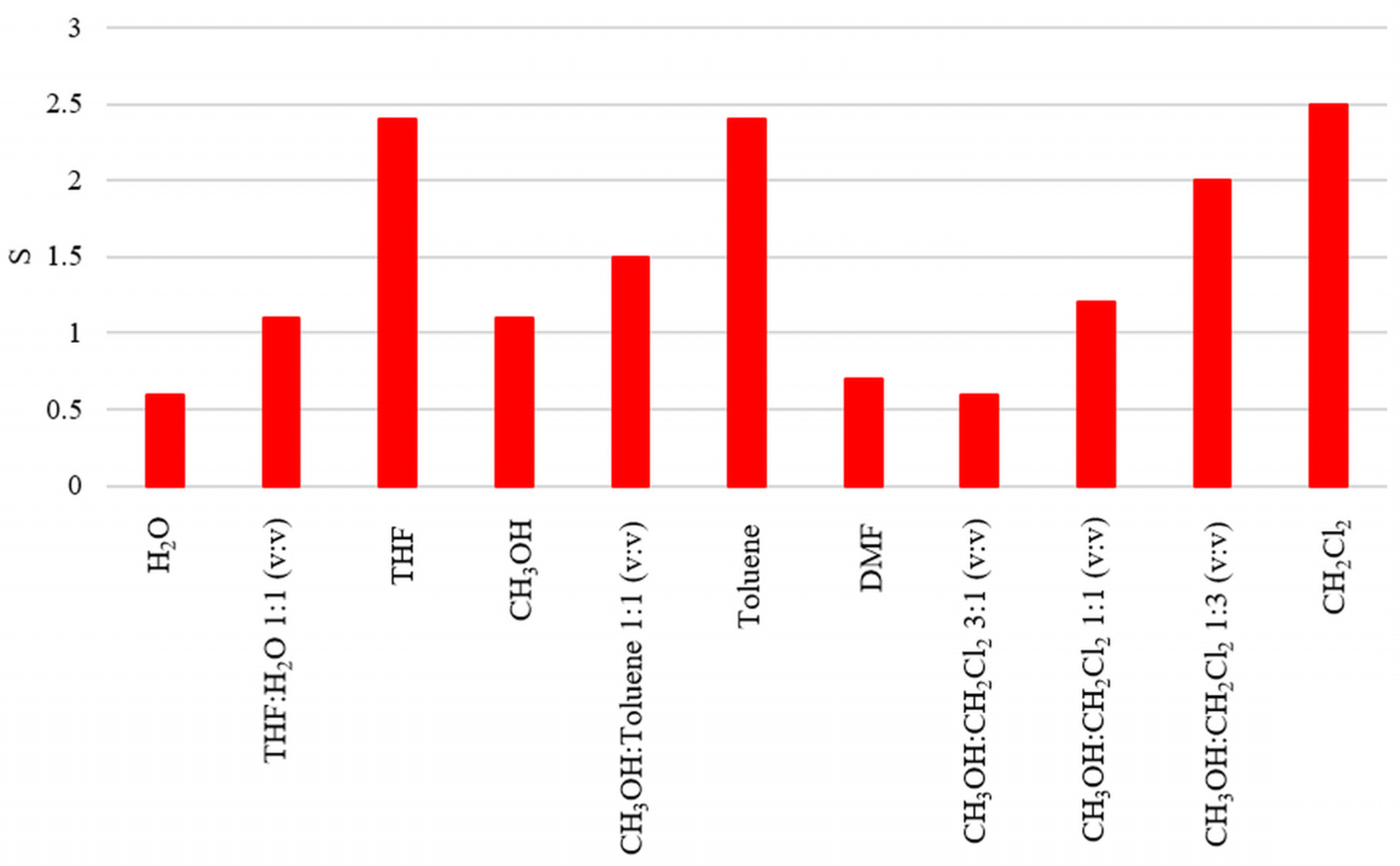
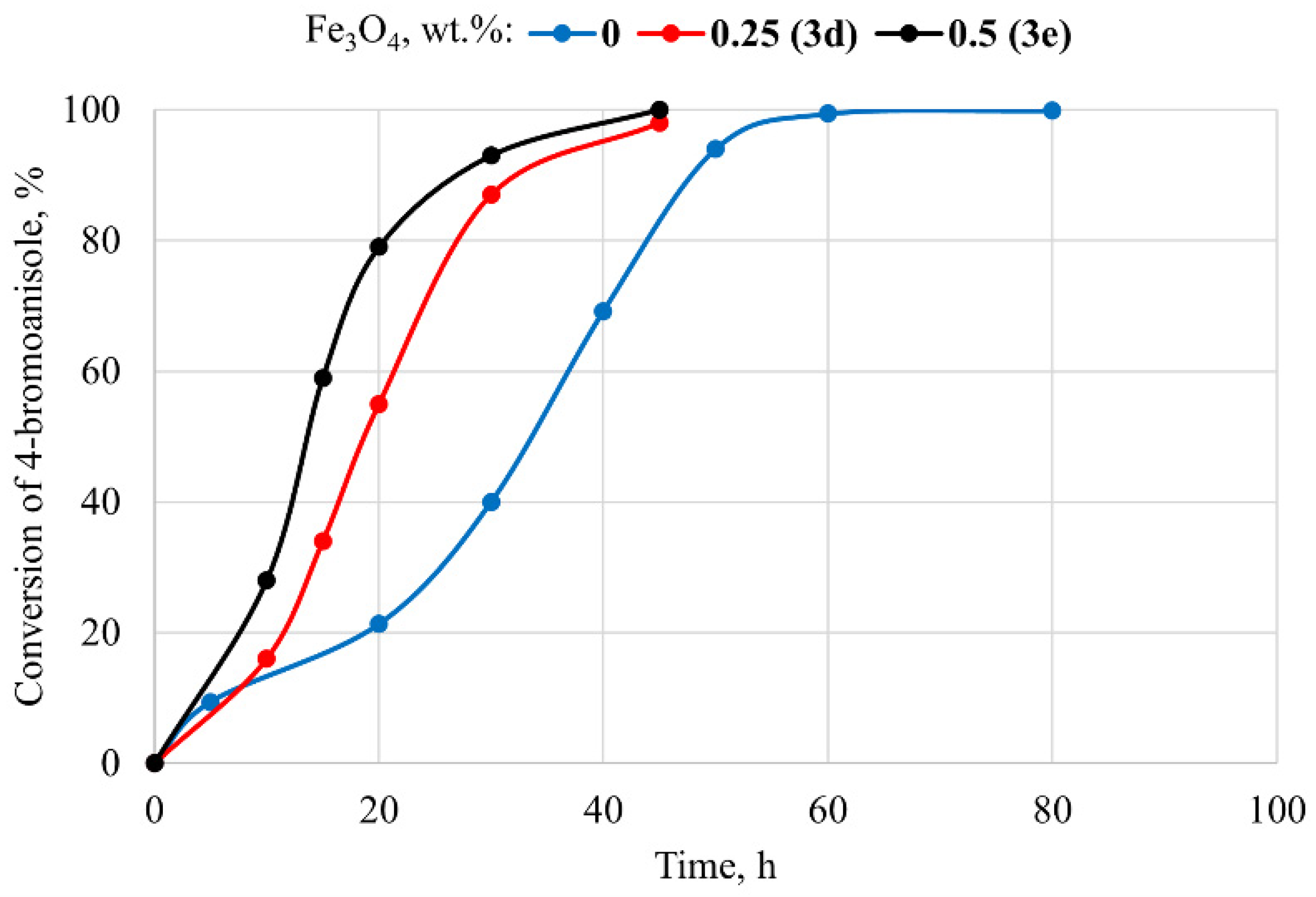
3.2. Attempts of Chemical Modification and Application of Composite Microparticles
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yudin, A.K. Aziridines and Epoxides in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Xiao, J.; Lu, Q.; Cong, H.; Shen, Y.; Yu, B. Microporous poly(glycidyl methacrylate-co-ethylene glycol dimethyl acrylate) microspheres: Synthesis, functionalization, and applications. Polym. Chem. 2021, 12, 6050–6070. [Google Scholar] [CrossRef]
- Vera, M.; Fodor, C.; Garcia, Y.; Pereira, E.; Loos, K.; Rivas, B.L. Multienzymatic immobilization of laccases on polymeric microspheres: A strategy to expand the maximum catalytic, Efficiency. J. Appl. Polym. Sci. 2020, 137, 49562. [Google Scholar] [CrossRef]
- Vasiliu, S.; Lungan, M.-A.; Racovita, S.; Popa, M. Porous microparticles based on methacrylic copolymers and gellan as drug delivery systems. Polym. Int. 2020, 69, 1066–1080. [Google Scholar] [CrossRef]
- Akbari, K.; Moghadam, P.N.; Behrouzi, M.; Fareghi, A.R. Synthesis of three-dimensional hydrogels based on poly(glycidyl methacrylate-alt-maleic anhydride): Characterization and study of furosemide drug release. Arab. J. Chem. 2020, 13, 8723–8733. [Google Scholar] [CrossRef]
- Horák, D.; Švec, F.; Bleha, M.; Kálal, J. Reactive polymers, XXXV. The effect of polymerization conditions on the specific surface area of macroporous copolymers from glycidylmethacrylate-ethylenedimethacrylate. Angew. Makromol. Chem. 1981, 95, 109–115. [Google Scholar] [CrossRef]
- Nastasović, A.; Marković, B.; Suručić, L.; Onjia, A. Methacrylate-based polymeric sorbents for recovery of metals from aqueous solutions. Metals 2022, 12, 814. [Google Scholar] [CrossRef]
- Drake, R.; Dunn, R.; Sherrington, D.C.; Thomson, S.J. Polymethacrylate and polystyrene-based resin-supported Pt catalysts in room temperature, solvent-less, oct-l-ene hydrosilylations using trichlorosilane and methyldichlorosilane. J. Molec. Catal. A Chem. 2001, 177, 49–69. [Google Scholar] [CrossRef]
- Bukowska, A.; Bukowski, W.; Bester, K.; Hus, K. Polymer supported copper(II) amine-imine complexes in the C-N and A3 coupling reactions. Appl. Organometal. Chem. 2017, 31, 3847. [Google Scholar] [CrossRef]
- Fofana, M.; Kuma, M.S. Use of a magnetic fluid-based process for coal separations. Miner. Met. Rev. 1997, 38, 35–40. [Google Scholar]
- Hatami, M.; Mohammadi-Rezaei, S.; Tahari, M.; Jing, D. Recent developments in magneto-hydrodynamic Fe3O4 nanofluids for different molecular applications: A review study. J. Molec. Liq. 2018, 250, 244–258. [Google Scholar] [CrossRef]
- Khollam, Y.B.; Dhage, S.R.; Potdar, H.S.; Deshpande, S.B.; Bakare, P.P.; Kulkarni, S.D.; Date, S.K. Microwave hydrothermal preparation of submicron-sized spherical magnetite (Fe3O4) powders. J. Magn. Magn. Mater. 2002, 56, 571–577. [Google Scholar] [CrossRef]
- Ali, A.; Shah, T.; Ullah, R.; Zhou, P.; Guo, M.; Ovais, M.; Tan, Z.; Rui, Y. Review on recent progress in magnetic nanoparticles: Synthesis, characterization, and diverse applications. Front. Chem. 2021, 9, 629054. [Google Scholar] [CrossRef] [PubMed]
- Soltys, L.; Olkhovyy, O.; Tatarchuk, T.; Naushad, M. Green synthesis of metal and metal oxide nanoparticles: Principles of green chemistry and raw materials. Magnetochemistry 2021, 7, 145. [Google Scholar] [CrossRef]
- Gambhir, R.P.; Rohiwal, S.S.; Tiwari, A.P. Multifunctional surface functionalized magnetic iron oxide nanoparticles for biomedical applications: A review. Appl. Surf. Sci. Adv. 2022, 11, 100303. [Google Scholar] [CrossRef]
- Ansari, K.; Ahmad, R.; Tanweer, M.S.; Azam, I. Magnetic iron oxide nanoparticles as a tool for the advancement of biomedical and environmental application: A review. Biomed. Mater. Devices 2023, 2, 139–157. [Google Scholar] [CrossRef]
- Fatmawati, T.; Shiddiq, M.; Armynah, B.; Tahir, D. Synthesis methods of Fe3O4 nanoparticles for biomedical applications. Chem. Eng. Technol. 2023, 46, 2356–2366. [Google Scholar] [CrossRef]
- Iota, M.A.; Cursaru, L.M.; Schiopu, A.G.; Tudor, I.A.; Motoc, A.M.; Piticescu, R.M. Fe3O4 Core–shell nanostructures with anticancer and antibacterial properties: A Mini-review. Processes 2023, 11, 1882. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Ma, S.; He, X.; Guo, C.; Liang, Z.; Hu, Y.; Wei, Y.; Lian, X.; Huang, D. Progress in cancer therapy with functionalized Fe3O4 nanomaterials. Front. Mater. Sci. 2023, 17, 230658. [Google Scholar] [CrossRef]
- Lu, A.H.; Salabas, E.L.; Schüth, F. Magnetic nanoparticles: Synthesis, protection, functionalization, and application. Angew. Chem. Int. Ed. 2007, 46, 1222–1244. [Google Scholar] [CrossRef]
- Laurent, S.; Forge, D.; Port, M.; Roch, A.; Robic, C.; Elst, L.V.; Muller, R.N. Magnetic iron oxide nanoparticles: Synthesis, stabilization, vectorization, physicochemical characterizations, and biological applications. Chem. Rev. 2008, 108, 2064–2110. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.D.; Tran, H.V.; Xu, S.; Lee, T.R. Fe3O4 Nanoparticles: Structures, synthesis, magnetic properties, surface functionalization, and emerging applications. Appl. Sci. 2021, 11, 11301. [Google Scholar] [CrossRef] [PubMed]
- Morais, P.C.; Garg, V.K.; Oliveira, A.C.; Silva, L.P.; Azevedo, R.B.; Silva, A.M.L.; Lima, E.C.D. Synthesis and characterization of size-controlled cobalt-ferrite-based ionic ferrofuids. J. Magn. Magn. Mat. 2001, 225, 37–40. [Google Scholar] [CrossRef]
- Shen, L.; Li, B.; Qiao, Y. Fe3O4 Nanoparticles in targeted drug/gene delivery systems. Materials 2018, 11, 324. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Niu, H.; Pan, Y.; Shi, Y.; Cai, Y. Chitosan-coated octadecyl-functionalized magnetite nanoparticles: Preparation and application in extraction of trace pollutants from environmental water samples. Anal. Chem. 2010, 82, 2363–2371. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Yan, Y.; Shuang, C.; Zhou, Q.; Ji, R.; Li, A. Biological magnetic ion exchange resin on advanced treatment of synthetic wastewater. Bioresour. Technol. 2023, 372, 128613. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Meng, X.; Lv, Z.; Jiang, T.; Liang, Q.; Shi, L.; Feng, J. Research progress on the removal of pesticides in water by Fe3O4-based adsorbents in the past decade: A review. Arab. J. Chem. 2024, 17, 105405. [Google Scholar] [CrossRef]
- Dong, X.; Zheng, Y.; Huang, Y.; Chen, X.; Jing, X. Synthesis and characterization of multifunctional poly(glycidyl methacrylate) microspheres and their use in cell separation. Anal. Biochem. 2010, 405, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.S.; Cong, Z.X.; Cao, J.B.; Ke, K.M.; Peng, Q.L.; Gao, J.; Yang, H.H.; Liu, G.; Chen, X. Multifunctional Fe3O4@polydopamine core&shell nanocomposites for intracellular mRNA detection and imaging-guided photothermal therapy. ACS Nano 2014, 8, 3876–3883. [Google Scholar]
- Wu, W.; Wu, Z.; Yu, T.; Jiang, C.; Kim, W.S. Recent progress on magnetic iron oxide nanoparticles: Synthesis, surface functional strategies and biomedical applications. Sci. Technol. Adv. Mater. 2015, 16, 023501. [Google Scholar] [CrossRef]
- Han, M.; Li, X.; Wang, X.; Liu, D.; Fu, S.; Xu, W.; Li, W.; Zhang, H. Preparation of polyhydroxyalkanoate-based magnetic microspheres for carbonyl reductase purification and immobilization. Int. J. Biol. Macromol. 2023, 253, 126814. [Google Scholar] [CrossRef]
- Wan, G.Z.; Ma, X.H.; Jin, L.; Chen, J. Fabrication of a magnetic porous organic polymer for α-glucosidase, immobilization and its application in inhibitor screening. Langmuir 2023, 39, 5239–5249. [Google Scholar] [CrossRef]
- Zhu, Y.; Stubbs, L.P.; Ho, F.; Liu, R.; Ship, C.P.; Maguire, J.A.; Hosmane, N.S. Magnetic nanocomposites: A new perspective in catalysis. ChemCatChem 2010, 2, 365–374. [Google Scholar] [CrossRef]
- Sharma, R.K.; Dutta, S.; Sharma, S.; Zboril, R.; Varma, R.S.; Gawande, M.B. Fe3O4 (iron oxide)-supported nanocatalysts: Synthesis, characterization and applications in coupling reactions. Green Chem. 2016, 18, 3184–3209. [Google Scholar] [CrossRef]
- Liu, R.; Guo, Y.; Odusote, G.; Qu, F.; Priestley, R.D. Core-shell Fe3O4 polydopamine nanoparticles serve multipurpose as drug carrier, catalyst support and carbon adsorbent. ACS Appl. Mater. Interfaces 2013, 5, 9167–9171. [Google Scholar] [CrossRef]
- Horák, D.; Babic, M.; Macková, H.; Benes, M.J. Preparation and properties of magnetic nano- and microsized particles for biological and environmental separations. J. Sep. Sci. 2007, 30, 1751–1772. [Google Scholar] [CrossRef] [PubMed]
- Wahajuddin, N.; Arora, S. Superparamagnetic iron oxide nanoparticles: Magnetic nanoplatforms as drug carriers. Int. J. Nanomed. 2012, 7, 3445–3471. [Google Scholar] [CrossRef]
- Bustamante-Torres, M.; Romero-Fierro, D.; Estrella-Nuñez, J.; Arcentales-Vera, B.; Chichande-Proaño, E.; Bucio, E. Polymeric Composite of Magnetite Iron Oxide Nanoparticles and Their Application in Biomedicine: A Review. Polymers 2022, 14, 752. [Google Scholar] [CrossRef]
- Gokmen, M.T.; Du Prez, F.E. Porous polymer particles—A comprehensive guide to synthesis, characterization, functionalization and applications. Prog. Polym. Sci. 2012, 37, 365–405. [Google Scholar] [CrossRef]
- Wang, C.; Yan, J.; Cui, X.; Cong, D.; Wang, H. Preparation and characterization of magnetic hollow PMMA nanospheres via in situ emulsion polymerization. Colloids Surf. A Physicochem. Eng. Asp. 2010, 363, 71–77. [Google Scholar] [CrossRef]
- Xu, S.; Ma, W.F.; You, L.J.; Li, J.M.; Guo, J.; Hu, J.J.; Wang, C.C. Toward designer magnetite/polystyrene colloidal composite microspheres with controllable nanostructures and desirable surface functionalities. Langmuir 2012, 28, 3271–3278. [Google Scholar] [CrossRef]
- Xu, H.; Cui, L.; Tong, N.; Gu, H. Development of high magnetization Fe3O4/polystyrene/silica nanospheres via combined miniemulsion/emulsion polymerization. J. Am. Chem. Soc. 2006, 128, 15582–15583. [Google Scholar] [CrossRef] [PubMed]
- Darwish, M.S.A.; Peuker, U.; Kunz, U. Bi-layer polymer-magnetite core/shell particles: Synthesis and characterization. J. Mater. Sci. 2011, 46, 2123–2134. [Google Scholar] [CrossRef]
- Hu, J.; Chen, M.; Wu, L. Organic-inorganic nanocomposites synthesized via miniemulsion polymerization. Polym. Chem. 2011, 2, 760–772. [Google Scholar] [CrossRef]
- Ma, Z.; Guan, Y.; Liu, H. Synthesis and characterization of micron-sized monodisperse superparamagnetic polymer particles with amino groups. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 3433–3439. [Google Scholar] [CrossRef]
- Zhang, J.; Yu, D.; Chen, W.; Xie, Y.; Wan, W.; Liang, H.; Min, C. Preparation of poly(styrene-glucidylmethacrylate)/Fe3O4 composite microsphere with high magnetite contents. J. Magn. Magn. Mater. 2009, 321, 572–577. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, C.; Li, Y.; Chen, Y.; Tong, Z. Facile fabrication of nanocomposite microspheres with polymer cores and magnetic shells by Pickering suspension polymerization. React. Funct. Polym. 2009, 69, 750–754. [Google Scholar] [CrossRef]
- Wang, L.; Tai, Y.; Gao, J.; Amer, W.A.; Ding, W.; Yu, H. Synthesis of Fe3O4@poly(methylmethacrylate-co-divinylbenzene) magnetic porous microspheres and their application in the separation of phenol from aqueous solutions. J. Colloid Interface Sci. 2011, 360, 731–738. [Google Scholar]
- Aksimentyeva, O.I.; Savchyn, V.P.; Dyakonov, V.P.; Piechota, S.; Horbenko, Y.Y.; Opainych, I.Y.; Demchenko, P.Y.; Popov, A.; Szymczak, H. Modification of Polymer-Magnetic Nanoparticles by Luminescent and Conducting Substances. Mol. Crys. Liq. 2014, 590, 35–42. [Google Scholar] [CrossRef]
- Lu, M.; Bai, S.; Yang, K.; Sun, Y. Synthesis and characterization of magnetic polymer microspheres with a core-shell structure. China Particuol. 2007, 5, 180–185. [Google Scholar] [CrossRef]
- Bukowska, A.; Bukowski, W.; Hus, K.; Depciuch, J.; Parlińska-Wojtan, M. Synthesis and characterization of new functionalized polymer-Fe3O4 nanocomposite particles. Express Polym. Lett. 2017, 11, 2–13. [Google Scholar] [CrossRef]
- Wang, L.; Yang, T.; Yang, J.; Li, Q.; Hao, D.; Yang, J.; Ma, G. In situ magnetization technique for synthesis of magnetic polymer microspheres. Powder Technol. 2013, 235, 1017–1024. [Google Scholar] [CrossRef]
- Rabelo, D.; Lima, E.C.D.; Reis, A.C.; Nunes, W.C.; Novak, M.A.; Garg, V.K.; Oliveira, A.C.; Morais, P.C. Preparation of Magnetite Nanoparticles in Mesoporous Copolymer Template. Nano Lett. 2001, 1, 105–108. [Google Scholar] [CrossRef]
- Liu, H.; Liu, X.; Xing, J.; Guan, Y.; Ma, Z.; Shan, G.; Yang, C. Preparation and characterization of superparamagnetic functional polymeric microparticles. China Particuol. 2003, 1, 76–79. [Google Scholar] [CrossRef]
- Bukowska, A.; Bukowski, W.; Noworól, J. Three-component terpolymers of glycidyl methacrylate with good swelling characteristics. J. Appl. Polym. Sci. 2007, 106, 3800–3807. [Google Scholar] [CrossRef]
- Bester, K.; Bukowska, A.; Bukowski, W. Palladium catalysts supported on amine-functionalized glycidyl methacrylate gel type terpolymers. Synthesis, characteristics and study of catalytic activity in Suzuki–Miyaura reactions. J. Molec. Catal. A Chem. 2013, 378, 124–134. [Google Scholar] [CrossRef]
- Duraczyńska, D.; Serwicka, E.M.; Drelinkiewicz, A.; Socha, R.P.; Zimowska, M.; Lityńska-Dobrzyńska, L.; Bukowska, A. Solvent and substituent effects in hydrogenation of aromatic ketones over Ru/polymer catalyst under very mild conditions. Molec. Catal. 2019, 470, 145–151. [Google Scholar] [CrossRef]
- Prymon, S.; Bukowska, A.; Bukowski, W.; Bester, K.; Hus, K.; Dychtoń, K.; Opiekun, Z. Effect of Fe3O4 particles on multi-hollow morphology of poly(HEMA-divinylbenzene-styrene) microspheres prepared by Pickering suspension polymerization. Express Polym. Lett. 2018, 12, 1026–1038. [Google Scholar] [CrossRef]
- Sherrington, D.C. Preparation, structure and morphology of polymer supports. Chem. Commun. 1998, 21, 2275–2286. [Google Scholar] [CrossRef]
- Zanini, M.; Marschelke, C.; Anachkov, S.E.; Marini, E.; Synytska, A.; Isa, L. Universal emulsion stabilization from the arrested adsorption of rough particles at liquid-liquid interfaces. Nat. Commun. 2017, 8, 15701. [Google Scholar] [CrossRef]
- Lan, Y.; Caciagli, A.; Guidetti, G.; Yu, Z.; Liu, J.; Johansen, V.E.; Kamp, M.; Abell, C.; Vignolini, S.; Scherman, O.A.; et al. Unexpected stability of aqueous dispersions of raspberry-like colloids. Nat. Commun. 2018, 9, 3614. [Google Scholar] [CrossRef]
- Binks, B.P. Particles as surfactants-similarities and differences. Curr. Opin. Colloid Interface Sci. 2002, 7, 21–41. [Google Scholar] [CrossRef]
- De Carvalho-Guimarães, F.B.; Correa, K.L.; de Souza, T.P.; Amado, J.R.R.; Ribeiro-Costa, R.M.; Silva-Júnior, J.O.C. A review of pickering emulsions: Perspectives and application. Pharmaceuticals 2022, 15, 141. [Google Scholar] [CrossRef]
- Pan, J.; Chen, J.; Wang, X.; Wang, Y.; Fan, J.-B. Pickering emulsion: From controllable fabrication to biomedical application. Interdiscip. Med. 2023, 1, e20230014. [Google Scholar] [CrossRef]
- Ortiz, D.G.; Pochat-Bohatier, C.; Cambedouzou, J.; Bechelany, M.; Miele, P. Current trends in pickering emulsions: Particle morphology and applications. Engineering 2020, 6, 468–482. [Google Scholar] [CrossRef]
- Bukowska, A.; Bukowski, W.; Bester, K.; Flaga, S. Linkage of the PAMAM type dendrimer with the gel type resin based on glycidyl methacrylate terpolymer as a method of preparation of the polymer support for the recyclable palladium catalyst for Suzuki–Miyaura cross-coupling reactions. RSC Adv. 2015, 5, 49036–49044. [Google Scholar] [CrossRef]
- Pretsch, E.; Bühlmann, P.; Badertscher, M. Structure Determination of Organic Compounds Tables of Spectral Data; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Martin, R.; Buchwald, S.L. Palladium-catalyzed Suzuki-Miyaura cross-coupling reactions employing dialkylbiaryl phosphine ligands. Acc. Chem. Res. 2008, 41, 1461–1473. [Google Scholar] [CrossRef]
- Torborga, C.; Beller, M. Recent applications of palladium-catalyzed coupling reactions in the pharmaceutical, agrochemical, and fine chemical industries. Adv. Synth. Catal. 2009, 351, 3027–3043. [Google Scholar] [CrossRef]
- Cho, S.H.; Kim, J.Y.; Kwak, J.; Chang, S. Recent advances in the transition metal-catalyzed twofold oxidative C–H bond activation strategy for C–C and C–N bond formation. Chem. Soc. Rev. 2011, 40, 5068–5083. [Google Scholar] [CrossRef]
- Emadi, R.; Nekoo, A.B.; Molaverdi, F.; Khorsandi, Z.; Sheibani, R.; Sadeghi-Aliabadi, H. Applications of palladium-catalyzed C–N crosscoupling reactions in pharmaceutical compounds. RSC Adv. 2023, 13, 18715–18733. [Google Scholar] [CrossRef] [PubMed]
- Seechurn, C.C.C.J.; Kitching, M.O.; Colacot, T.J.; Snieckus, V. Palladium-catalyzed cross-coupling: A historical contextual perspective to the 2010 Nobel Prize. Angew. Chem. Int. Ed. 2012, 51, 5062–5085. [Google Scholar] [CrossRef] [PubMed]
- Blaser, H.-U.; Indolese, A.; Naud, F.; Nettekoven, U.; Schnyder, A. Industrial R&D on catalytic C-C and C-N coupling reactions: A personal account on goals, approaches and results. Adv. Synth. Catal. 2004, 346, 1583–1598. [Google Scholar]
- Polshettiwar, V.; Len, C.; Fihri, A. Sustainable catalysts for cross-coupling reactions. Coord. Chem. Rev. 2009, 253, 2599–2626. [Google Scholar]
- Mora, M.; Jiménez-Sanchidrián, C.; Ruiz, J.R. Recent advances in the heterogeneous palladium-catalysed Suzuki cross-coupling reaction. Curr. Org. Chem. 2012, 16, 1128–1150. [Google Scholar] [CrossRef]
- Hussain, I.; Capricho, J.; Yawer, M.A. Synthesis of biaryls via ligand-free Suzuki–Miyaura cross-coupling reactions: A review of homogeneous and heterogeneous catalytic developments. Adv. Synth. Catal. 2016, 358, 3320–3349. [Google Scholar] [CrossRef]
- Lawrence, A.S.; Martin, N.; Sivakumar, B.; Cirujano, F.G.; Dhakshinamoorthy, A. Palladium-based metal organic frameworks as heterogeneous catalysts for C-C couplings. ChemCatChem 2022, 14, e202200403. [Google Scholar] [CrossRef]
- Martín, C.d.M.G.; García, J.I.H.; Bonardd, S.; Díaz, D.D. Lignin-based catalysts for C-C bond-forming reactions. Molecules 2023, 28, 3513. [Google Scholar] [CrossRef]
- Sheikh, S.; Nasseri, M.A.; Chahkandi, M.; Reiser, O.; Allahresani, A. Dendritic structured palladium complexes: Magnetically retrievable, highly efficient heterogeneous nanocatalyst for Suzuki and Heck cross-coupling reactions. RSC Adv. 2022, 12, 8833–8840. [Google Scholar] [CrossRef]
- Bester, K.; Bukowska, A.; Bukowski, W. Synthesis of gel-type imino-amino functionalized methacrylate-styrene terpolymers as supports for palladium catalysts for the Suzuki–Miyaura reaction. Appl. Catal. A General 2012, 443, 181–190. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Abbr. | Fe3O4, wt.% | Average Size, µm | Diameter Range, µm | Fraction, % | Fe Content | ||
|---|---|---|---|---|---|---|---|---|
| Calculated, wt.% | Determined, wt.% | Modification Degree, % | ||||||
| 1 | 1a | - | 211 | 100–448 | 100 | - | - | - |
| 2 | 1b | 0.05 | 272 | 142–632 | 100 | 0.036 | 0.013 | 36.0 |
| 3 | 1c | 0.10 | 328 | 159–710 | 100 | - | - | - |
| 4 | 1d | 0.25 | 386 | 159–1002 | 95.1 | 0.181 | 0.048 | 26.5 |
| 5 | 1e | 0.50 | 256 | 112–633 | 95.7 | 0.361 | 0.068 | 18.8 |
| 6 | 1f | 0.75 | 298 | 142–633 | 100 | 0.542 | 0.095 | 17.5 |
| 7 * | 1g | 0.50 | 247 | 100–564 | 100 | - | - | - |
| 8 ** | 1h | 0.50 | 173 | 50—448 | 88.8 | - | - | - |
| Polymerization Product | Epoxy Group Loading, mmol/ | Modification Product | Nitrogen Content, | Epoxy Group Conversion, % | |
|---|---|---|---|---|---|
| wt.% | mmol/g | ||||
| 1a | 1.33 | - | - | - | - |
| 1d | 1.25 | 2d | 4.99 | 3.56 | 36 |
| 1e | 1.29 | 2e | 5.74 | 4.10 | 41 |
 | |||||
|---|---|---|---|---|---|
| Entry | R | T °C | Catalyst 3e, mol% | Time, min | Conversion of ArBr, % |
| 1 | OCH3 | 70 | 0.1 | 150 | 98 |
| 2 | OCH3 | 70 | 0.25 | 60 | 100 |
| 3 | OCH3 | 70 | 0.5 | 45 | 100 |
| 4 | OCH3 | 25 | 0.5 | 360 | 88 |
| 5 | OCH3 | 50 | 0.5 | 120 | 96 |
| 6 | H | 70 | 0.5 | 20 | 100 |
| 7 | CHO | 70 | 0.5 | 10 | 100 |
| 8 | Naphtyl | 70 | 0.5 | 30 | 97 |
| 9 | F | 70 | 0.5 | 90 | 96 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bukowska, A.; Bester, K.; Flaga, S.; Bukowski, W. Reactive Polymer Composite Microparticles Based on Glycidyl Methacrylate and Magnetite Nanoparticles. Solids 2024, 5, 151-171. https://doi.org/10.3390/solids5010011
Bukowska A, Bester K, Flaga S, Bukowski W. Reactive Polymer Composite Microparticles Based on Glycidyl Methacrylate and Magnetite Nanoparticles. Solids. 2024; 5(1):151-171. https://doi.org/10.3390/solids5010011
Chicago/Turabian StyleBukowska, Agnieszka, Karol Bester, Sylwia Flaga, and Wiktor Bukowski. 2024. "Reactive Polymer Composite Microparticles Based on Glycidyl Methacrylate and Magnetite Nanoparticles" Solids 5, no. 1: 151-171. https://doi.org/10.3390/solids5010011
APA StyleBukowska, A., Bester, K., Flaga, S., & Bukowski, W. (2024). Reactive Polymer Composite Microparticles Based on Glycidyl Methacrylate and Magnetite Nanoparticles. Solids, 5(1), 151-171. https://doi.org/10.3390/solids5010011