
Beyond Docetaxel: Targeting Resistance Pathways in Prostate Cancer Treatment


Abstract
1. Introduction
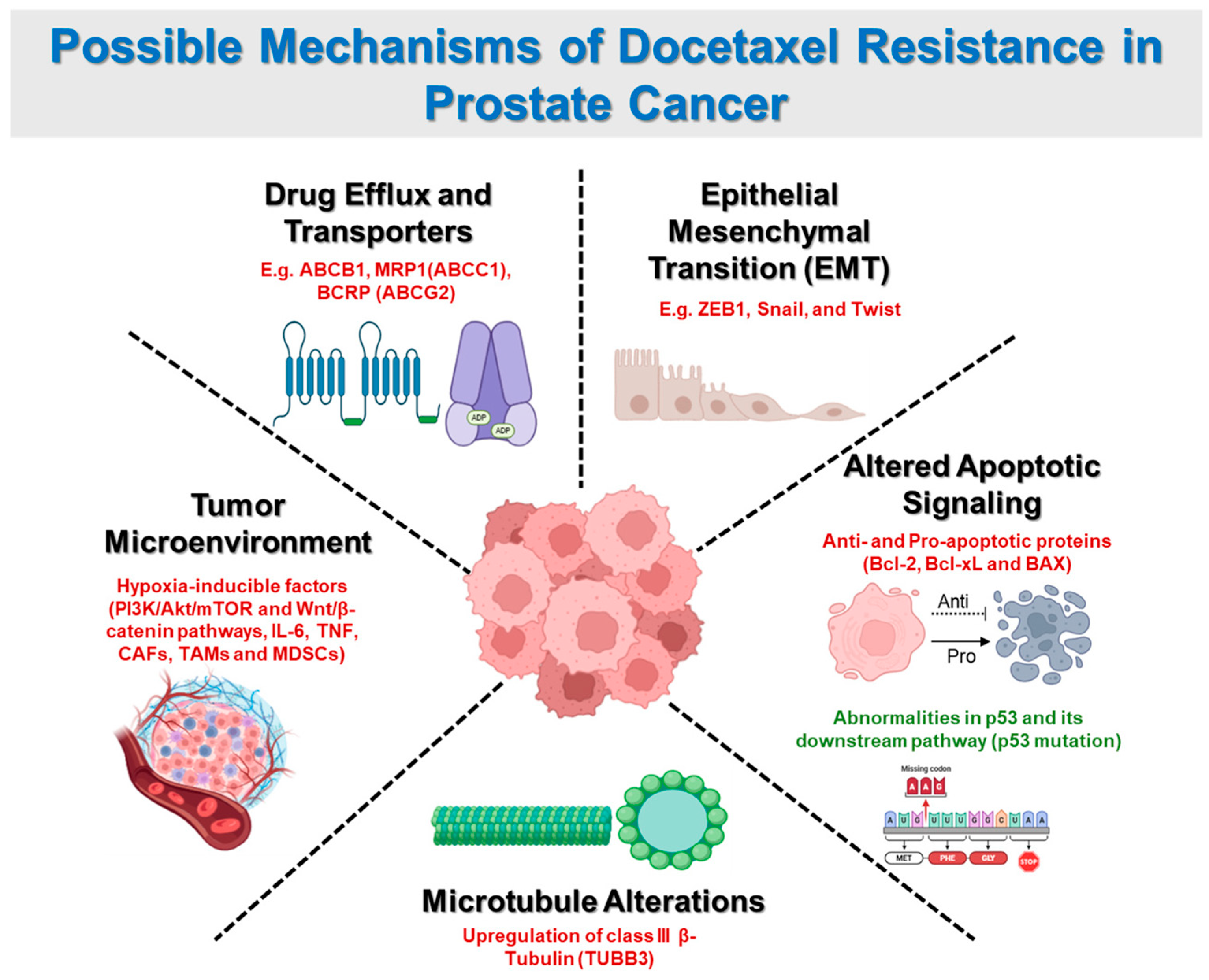
2. Mechanisms of Docetaxel Resistance
2.1. Drug Efflux and Transporters
2.2. Epithelial-Mesenchymal Transition (EMT)
2.3. Altered Apoptotic Signaling
2.4. Microtubule Alterations
2.5. Tumor Microenvironment Influence
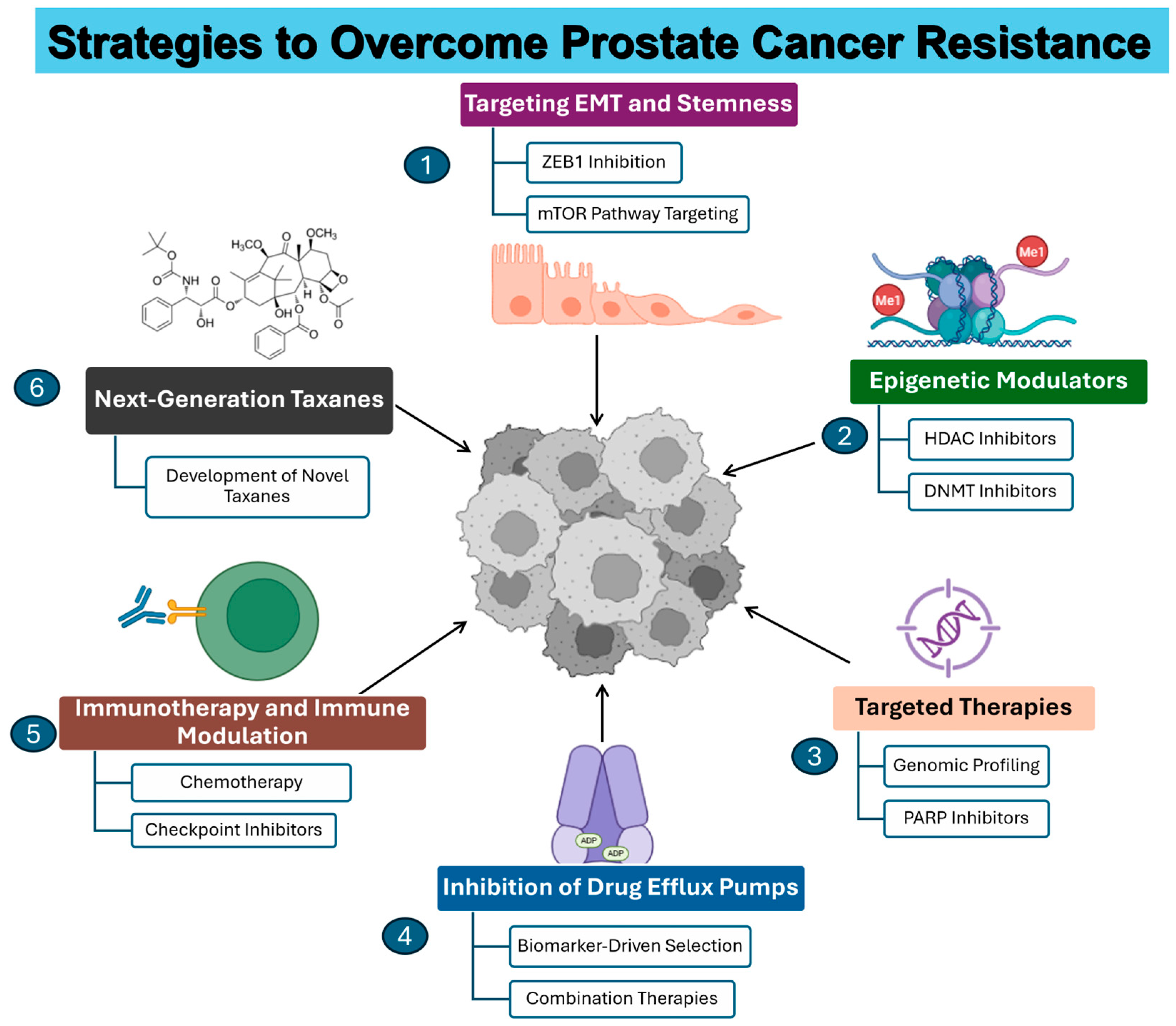
3. Emerging Therapies and Combination Strategies
3.1. Targeting EMT and Stemness
3.2. Inhibition of Drug Efflux Pumps
3.3. Epigenetic Modulators
3.4. Immunotherapy and Immune Modulation
3.5. Targeted Therapies
4. Clinical Implications and Future Directions
5. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| mCRPC | Metastatic castration-resistant prostate cancer |
| AR | Androgen receptor |
| FGF2 | fibroblast growth factor 2 |
| ABC | ATP-binding cassette |
| P-gp | P-glycoprotein |
| BCRP | Breast cancer resistance protein |
| MDR | Multidrug resistance |
| MRP | Multidrug resistance proteins |
| EMT | Epithelial-mesenchymal transition |
| MOMP | Mitochondrial outer membrane permeabilization |
| TUBB3 | Class III β-tubulin |
| HIFs | Hypoxia-inducible factors |
| CAFs | Cancer-associated fibroblasts |
| TAMs | Tumor-associated macrophages |
| MDSCs | Myeloid-derived suppressor cells |
| CSCs | Cancer stem cells |
| ADT | Androgen deprivation therapy |
| DNMT | DNA methyltransferases |
| HDAC | Histone deacetylases |
References
- Adekiya, T.A.; Moore, M.; Thomas, M.; Lake, G.; Hudson, T.; Adesina, S.K. Preparation, Optimization, and In-Vitro Evaluation of Brusatol- and Docetaxel-Loaded Nanoparticles for the Treatment of Prostate Cancer. Pharmaceutics 2024, 16, 114. [Google Scholar] [CrossRef]
- Adekiya, T.A.; Owoseni, O. Emerging Frontiers in Nanomedicine Targeted Therapy for Prostate Cancer. Cancer Treat. Res. Commun. 2023, 37, 100778. [Google Scholar] [CrossRef] [PubMed]
- Owoseni, O.B.; Adekiya, T.A.; Akinboye, E.S.; Adesina, S.K. Development of a Prostate-Specific Antigen Targeted Dual Drug Conjugate for Prostate Cancer Therapy. ACS Omega 2025, 10, 17611–17625. [Google Scholar] [CrossRef]
- Bracarda, S.; Logothetis, C.; Sternberg, C.N.; Oudard, S. Current and Emerging Treatment Modalities for Metastatic Castration-resistant Prostate Cancer. BJU Int. 2011, 107 (Suppl. S2), 13–20. [Google Scholar] [CrossRef]
- Adekiya, T.A.; Hudson, T.; Bakare, O.; Ameyaw, E.E.; Adebayo, A.; Olajubutu, O.; Adesina, S.K. PSMA-Targeted Combination Brusatol and Docetaxel Nanotherapeutics for the Treatment of Prostate Cancer. Biomed. Pharmacother. 2024, 177, 117125. [Google Scholar] [CrossRef]
- Cheng, B.; Li, L.; Wu, Y.; Luo, T.; Tang, C.; Wang, Q.; Zhou, Q.; Wu, J.; Lai, Y.; Zhu, D.; et al. Correction: The Key Cellular Senescence Related Molecule RRM2 Regulates Prostate Cancer Progression and Resistance to Docetaxel Treatment. Cell Biosci. 2024, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Su, Y.; Zhang, Y.; Han, B.; Liu, H.; Wang, X. Endothelial Cells Promote Docetaxel Resistance of Prostate Cancer Cells by Inducing ERG Expression and Activating Akt/mTOR Signaling Pathway. Front. Oncol. 2020, 10, 584505. [Google Scholar] [CrossRef] [PubMed]
- Sekino, Y.; Teishima, J. Molecular Mechanisms of Docetaxel Resistance in Prostate Cancer. Cancer Drug Resist. 2020, 3, 676–685. [Google Scholar] [CrossRef]
- Galletti, G.; Matov, A.; Beltran, H.; Fontugne, J.; Miguel Mosquera, J.; Cheung, C.; MacDonald, T.Y.; Sung, M.; O’Toole, S.; Kench, J.G.; et al. ERG Induces Taxane Resistance in Castration-Resistant Prostate Cancer. Nat. Commun. 2014, 5, 5548. [Google Scholar] [CrossRef]
- Sweeney, C.J.; Chen, Y.-H.; Carducci, M.; Liu, G.; Jarrard, D.F.; Eisenberger, M.; Wong, Y.-N.; Hahn, N.; Kohli, M.; Cooney, M.M.; et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer. N. Engl. J. Med. 2015, 373, 737–746. [Google Scholar] [CrossRef]
- Leonard, G.D.; Fojo, T.; Bates, S.E. The Role of ABC Transporters in Clinical Practice. Oncologist 2003, 8, 411–424. [Google Scholar] [CrossRef]
- Xia, C.Q.; Smith, P.G. Drug Efflux Transporters and Multidrug Resistance in Acute Leukemia: Therapeutic Impact and Novel Approaches to Mediation. Mol. Pharmacol. 2012, 82, 1008–1021. [Google Scholar] [CrossRef]
- Choi, Y.; Yu, A.-M. ABC Transporters in Multidrug Resistance and Pharmacokinetics, and Strategies for Drug Development. Curr. Pharm. Des. 2014, 20, 793–807. [Google Scholar] [CrossRef]
- Rajagopal, A.; Simon, S.M. Subcellular Localization and Activity of Multidrug Resistance Proteins. Mol. Biol. Cell 2003, 14, 3389–3399. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Ma, Q.; Ye, L.; Piao, G. The Traditional Medicine and Modern Medicine from Natural Products. Molecules 2016, 21, 559. [Google Scholar] [CrossRef]
- Cort, A.; Ozben, T. Natural Product Modulators to Overcome Multidrug Resistance In Cancer. Nutr. Cancer 2015, 67, 411–423. [Google Scholar] [CrossRef]
- Lombard, A.P.; Liu, C.; Armstrong, C.M.; Cucchiara, V.; Gu, X.; Lou, W.; Evans, C.P.; Gao, A.C. ABCB1 Mediates Cabazitaxel–Docetaxel Cross-Resistance in Advanced Prostate Cancer. Mol. Cancer Ther. 2017, 16, 2257–2266. [Google Scholar] [CrossRef]
- Tang, S.C.; Kort, A.; Cheung, K.L.; Rosing, H.; Fukami, T.; Durmus, S.; Wagenaar, E.; Hendrikx, J.J.M.A.; Nakajima, M.; Van Vlijmen, B.J.M.; et al. P-Glycoprotein, CYP3A, and Plasma Carboxylesterase Determine Brain Disposition and Oral Availability of the Novel Taxane Cabazitaxel (Jevtana) in Mice. Mol. Pharm. 2015, 12, 3714–3723. [Google Scholar] [CrossRef] [PubMed]
- Lombard, A.P.; Lou, W.; Armstrong, C.M.; D’Abronzo, L.S.; Ning, S.; Evans, C.P.; Gao, A.C. Activation of the ABCB1 Amplicon in Docetaxel- and Cabazitaxel-Resistant Prostate Cancer Cells. Mol. Cancer Ther. 2021, 20, 2061–2070. [Google Scholar] [CrossRef]
- Duran, G.E.; Wang, Y.C.; Francisco, E.B.; Rose, J.C.; Martinez, F.J.; Coller, J.; Brassard, D.; Vrignaud, P.; Sikic, B.I. Mechanisms of Resistance to Cabazitaxel. Mol. Cancer Ther. 2015, 14, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Buck, S.A.J.; Van Hemelryk, A.; De Ridder, C.; Stuurman, D.; Erkens-Schulze, S.; Van ’T Geloof, S.; Teubel, W.J.; Koolen, S.L.W.; Martens-Uzunova, E.S.; Van Royen, M.E.; et al. Darolutamide Added to Docetaxel Augments Antitumor Effect in Models of Prostate Cancer through Cell Cycle Arrest at the G1–S Transition. Mol. Cancer Ther. 2024, 23, 711–720. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Morgado-Diaz, J.A.; Wagner, M.S.; Sousa-Squiavinato, A.C.M.; de-Freitas-Junior, J.C.M.; De Araújo, W.M.; Tessmann, J.W.; Rocha, M.R. Epithelial-Mesenchymal Transition in Metastatic Colorectal Cancer. In Gastrointestinal Cancers; Morgado-Diaz, J.A., Ed.; Cellular and Molecular Oncobiology Program, Cellular Dynamic and Structure Group, National Cancer Institute-INCA, Rio de Janeiro, Brazil; Exon Publications: Brisbane, AU, Australia, 2022; pp. 25–42. [Google Scholar] [CrossRef]
- Marín-Aguilera, M.; Codony-Servat, J.; Reig, Ò.; Lozano, J.J.; Fernández, P.L.; Pereira, M.V.; Jiménez, N.; Donovan, M.; Puig, P.; Mengual, L.; et al. Epithelial-to-Mesenchymal Transition Mediates Docetaxel Resistance and High Risk of Relapse in Prostate Cancer. Mol. Cancer Ther. 2014, 13, 1270–1284. [Google Scholar] [CrossRef]
- Sánchez-Tilló, E.; Liu, Y.; De Barrios, O.; Siles, L.; Fanlo, L.; Cuatrecasas, M.; Darling, D.S.; Dean, D.C.; Castells, A.; Postigo, A. EMT-Activating Transcription Factors in Cancer: Beyond EMT and Tumor Invasiveness. Cell Mol. Life Sci. 2012, 69, 3429–3456. [Google Scholar] [CrossRef]
- Beach, S.; Tang, H.; Park, S.; Dhillon, A.S.; Keller, E.T.; Kolch, W.; Yeung, K.C. Snail Is a Repressor of RKIP Transcription in Metastatic Prostate Cancer Cells. Oncogene 2008, 27, 2243–2248. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.N.; Odero-Marah, V.A. The Role of Snail in Prostate Cancer. Cell Adhes. Migr. 2012, 6, 433–441. [Google Scholar] [CrossRef]
- Ong, M.S.; Deng, S.; Halim, C.E.; Cai, W.; Tan, T.Z.; Huang, R.Y.-J.; Sethi, G.; Hooi, S.C.; Kumar, A.P.; Yap, C.T. Cytoskeletal Proteins in Cancer and Intracellular Stress: A Therapeutic Perspective. Cancers 2020, 12, 238. [Google Scholar] [CrossRef]
- Gajula, R.P.; Chettiar, S.T.; Williams, R.D.; Thiyagarajan, S.; Kato, Y.; Aziz, K.; Wang, R.; Gandhi, N.; Wild, A.T.; Vesuna, F.; et al. The Twist Box Domain Is Required for Twist1-Induced Prostate Cancer Metastasis. Mol. Cancer Res. 2013, 11, 1387–1400. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Sun, Y.; Liang, X.; Gu, X.; Ning, J.; Xu, Y.; Chen, S.; Pan, L. Emerging New Therapeutic Antibody Derivatives for Cancer Treatment. Signal Transduct. Target. Ther. 2022, 7, 39. [Google Scholar] [CrossRef]
- Chen, Y.; Lu, X.; Montoya-Durango, D.E.; Liu, Y.-H.; Dean, K.C.; Darling, D.S.; Kaplan, H.J.; Dean, D.C.; Gao, L.; Liu, Y. ZEB1 Regulates Multiple Oncogenic Components Involved in Uveal Melanoma Progression. Sci. Rep. 2017, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Hanrahan, K.; O’Neill, A.; Prencipe, M.; Bugler, J.; Murphy, L.; Fabre, A.; Puhr, M.; Culig, Z.; Murphy, K.; Watson, R.W. The Role of Epithelial–Mesenchymal Transition Drivers ZEB 1 and ZEB 2 in Mediating Docetaxel-resistant Prostate Cancer. Mol. Oncol. 2017, 11, 251–265. [Google Scholar] [CrossRef]
- Wade, C.; Kyprianou, N. Profiling Prostate Cancer Therapeutic Resistance. Int. J. Mol. Sci. 2018, 19, 904. [Google Scholar] [CrossRef]
- Georgakopoulos-Soares, I.; Chartoumpekis, D.V.; Kyriazopoulou, V.; Zaravinos, A. EMT Factors and Metabolic Pathways in Cancer. Front. Oncol. 2020, 10, 499. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Wei, Z.; Yang, W.; Huang, J.; Yang, Y.; Wang, J. The Role of BCL-2 Family Proteins in Regulating Apoptosis and Cancer Therapy. Front. Oncol. 2022, 12, 985363. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhu, Y.; Lou, W.; Nadiminty, N.; Chen, X.; Zhou, Q.; Shi, X.B.; deVere White, R.W.; Gao, A.C. Functional P53 Determines Docetaxel Sensitivity in Prostate Cancer Cells. Prostate 2013, 73, 418–427. [Google Scholar] [CrossRef]
- Gimenez-Bonafe, P.; Tortosa, A.; Perez-Tomas, R. Overcoming Drug Resistance by Enhancing Apoptosis of Tumor Cells. Curr. Cancer Drug Targets 2009, 9, 320–340. [Google Scholar] [CrossRef]
- He, L.-Y. Silencing Notch-1 Induces Apoptosis and Increases the Chemosensitivity of Prostate Cancer Cells to Docetaxel through Bcl-2 and Bax. Oncol. Lett. 2012, 3, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.N.; Engelman, J.A.; Faber, A.C. The BCL2 Family: Key Mediators of the Apoptotic Response to Targeted Anticancer Therapeutics. Cancer Discov. 2015, 5, 475–487. [Google Scholar] [CrossRef]
- Tian, X.; Srinivasan, P.R.; Tajiknia, V.; Sanchez Sevilla Uruchurtu, A.F.; Seyhan, A.A.; Carneiro, B.A.; De La Cruz, A.; Pinho-Schwermann, M.; George, A.; Zhao, S.; et al. Targeting Apoptotic Pathways for Cancer Therapy. J. Clin. Investig. 2024, 134, e179570. [Google Scholar] [CrossRef]
- Muller, P.A.J.; Vousden, K.H. Mutant P53 in Cancer: New Functions and Therapeutic Opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef]
- Nyquist, M.D.; Corella, A.; Coleman, I.; De Sarkar, N.; Kaipainen, A.; Ha, G.; Gulati, R.; Ang, L.; Chatterjee, P.; Lucas, J.; et al. Combined TP53 and RB1 Loss Promotes Prostate Cancer Resistance to a Spectrum of Therapeutics and Confers Vulnerability to Replication Stress. Cell Rep. 2020, 31, 107669. [Google Scholar] [CrossRef]
- Zhou, C.K.; Young, D.; Yeboah, E.D.; Coburn, S.B.; Tettey, Y.; Biritwum, R.B.; Adjei, A.A.; Tay, E.; Niwa, S.; Truelove, A.; et al. TMPRSS2:ERG Gene Fusions in Prostate Cancer of West African Men and a Meta-Analysis of Racial Differences. Am. J. Epidemiol. 2017, 186, 1352–1361. [Google Scholar] [CrossRef]
- Ngoi, N.Y.L.; Choong, C.; Lee, J.; Bellot, G.; Wong, A.L.; Goh, B.C.; Pervaiz, S. Targeting Mitochondrial Apoptosis to Overcome Treatment Resistance in Cancer. Cancers 2020, 12, 574. [Google Scholar] [CrossRef]
- Perimbeti, S.; Jamroze, A.; Attwood, K.; Farmer, B.; Beumer, J.H.; Bies, R.; Levine, E.G.; Kirk, J.; Tang, D.; Chatta, G.S.; et al. Phase Ib Trial of Enzalutamide (Enza) with Venetoclax (Ven) in Metastatic Castration-Resistant Prostate Cancer (mCRPC). J. Clin. Oncol. 2023, 41 (Suppl. S6), 182. [Google Scholar] [CrossRef]
- Verrills, N.; Kavallaris, M. Improving the Targeting of Tubulin-Binding Agents: Lessons from Drug Resistance Studies. Curr. Pharm. Des. 2005, 11, 1719–1733. [Google Scholar] [CrossRef] [PubMed]
- Maahs, L.; Sanchez, B.E.; Gupta, N.; Van Harn, M.; Barrack, E.R.; Reddy, P.-V.; Hwang, C. Class III β-Tubulin Expression as a Predictor of Docetaxel-Resistance in Metastatic Castration-Resistant Prostate Cancer. PLoS ONE 2019, 14, e0222510. [Google Scholar] [CrossRef]
- Rushworth, L.K.; Hewit, K.; Munnings-Tomes, S.; Somani, S.; James, D.; Shanks, E.; Dufès, C.; Straube, A.; Patel, R.; Leung, H.Y. Repurposing Screen Identifies Mebendazole as a Clinical Candidate to Synergise with Docetaxel for Prostate Cancer Treatment. Br. J. Cancer 2020, 122, 517–527. [Google Scholar] [CrossRef]
- Mohamed, O.A.A.; Tesen, H.S.; Hany, M.; Sherif, A.; Abdelwahab, M.M.; Elnaggar, M.H. The Role of Hypoxia on Prostate Cancer Progression and Metastasis. Mol. Biol. Rep. 2023, 50, 3873–3884. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Lu, Y.; Roca, H.; Keller, J.M.; Zhang, J.; McCauley, L.K.; Keller, E.T. Immune Mediators in the Tumor Microenvironment of Prostate Cancer. Chin. J. Cancer 2017, 36, 29. [Google Scholar] [CrossRef]
- Kim, I.; Choi, S.; Yoo, S.; Lee, M.; Kim, I.-S. Cancer-Associated Fibroblasts in the Hypoxic Tumor Microenvironment. Cancers 2022, 14, 3321. [Google Scholar] [CrossRef] [PubMed]
- Kundu, M.; Butti, R.; Panda, V.K.; Malhotra, D.; Das, S.; Mitra, T.; Kapse, P.; Gosavi, S.W.; Kundu, G.C. Modulation of the Tumor Microenvironment and Mechanism of Immunotherapy-Based Drug Resistance in Breast Cancer. Mol. Cancer 2024, 23, 92. [Google Scholar] [CrossRef]
- Wegiel, B.; Vuerich, M.; Daneshmandi, S.; Seth, P. Metabolic Switch in the Tumor Microenvironment Determines Immune Responses to Anti-Cancer Therapy. Front. Oncol. 2018, 8, 284. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Z.; Ajani, J.A.; Song, S. Drug Resistance and Cancer Stem Cells. Cell Commun. Signal 2021, 19, 19. [Google Scholar] [CrossRef]
- Li, P.; Yang, R.; Gao, W.-Q. Contributions of Epithelial-Mesenchymal Transition and Cancer Stem Cells to the Development of Castration Resistance of Prostate Cancer. Mol. Cancer 2014, 13, 55. [Google Scholar] [CrossRef]
- Garg, M. Epithelial Plasticity and Cancer Stem Cells: Major Mechanisms of Cancer Pathogenesis and Therapy Resistance. World J. Stem Cells 2017, 9, 118. [Google Scholar] [CrossRef]
- Erin, N.; Grahovac, J.; Brozovic, A.; Efferth, T. Tumor Microenvironment and Epithelial Mesenchymal Transition as Targets to Overcome Tumor Multidrug Resistance. Drug Resist. Updates 2020, 53, 100715. [Google Scholar] [CrossRef]
- Chen, H.; Fang, S.; Zhu, X.; Liu, H. Cancer-Associated Fibroblasts and Prostate Cancer Stem Cells: Crosstalk Mechanisms and Implications for Disease Progression. Front. Cell Dev. Biol. 2024, 12, 1412337. [Google Scholar] [CrossRef]
- Becerril-Rico, J.; Alvarado-Ortiz, E.; Toledo-Guzmán, M.E.; Pelayo, R.; Ortiz-Sánchez, E. The Cross Talk between Gastric Cancer Stem Cells and the Immune Microenvironment: A Tumor-Promoting Factor. Stem Cell Res. Ther. 2021, 12, 498. [Google Scholar] [CrossRef] [PubMed]
- Dave, B.; Mittal, V.; Tan, N.M.; Chang, J.C. Epithelial-Mesenchymal Transition, Cancer Stem Cells and Treatment Resistance. Breast Cancer Res. 2012, 14, 202. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, J.; Zhang, Y.; Guo, Q. Drug resistance and tumor immune microenvironment: An overview of current understandings. Int. J. Oncol. 2024, 65, 96. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, C.; Bazan, F.; Kim, S.; Demarchi, M.; Chaigneau, L.; Thiery-Vuillemin, A.; Nguyen, T.; Cals, L.; Dobi, E.; Pivot, X. Cabazitaxel: A Novel Microtubule Inhibitor. Drugs 2011, 71, 1251–1258. [Google Scholar] [CrossRef]
- Lohiya, V.; Aragon-Ching, J.B.; Sonpavde, G. Role of Chemotherapy and Mechanisms of Resistance to Chemotherapy in Metastatic Castration-Resistant Prostate Cancer. Clin. Med. Insights Oncol. 2016, 10 (Suppl. S1), 57–66. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.K.; Pu, H.; Penticuff, J.C.; Cao, Z.; Horbinski, C.; Kyprianou, N. Multinucleation and Mesenchymal-to-Epithelial Transition Alleviate Resistance to Combined Cabazitaxel and Antiandrogen Therapy in Advanced Prostate Cancer. Cancer Res. 2016, 76, 912–926. [Google Scholar] [CrossRef]
- Mukhtar, E.; Adhami, V.M.; Siddiqui, I.A.; Verma, A.K.; Mukhtar, H. Fisetin Enhances Chemotherapeutic Effect of Cabazitaxel against Human Prostate Cancer Cells. Mol. Cancer Ther. 2016, 15, 2863–2874. [Google Scholar] [CrossRef] [PubMed]
- De Leeuw, R.; Berman-Booty, L.D.; Schiewer, M.J.; Ciment, S.J.; Den, R.B.; Dicker, A.P.; Kelly, W.K.; Trabulsi, E.J.; Lallas, C.D.; Gomella, L.G.; et al. Novel Actions of Next-Generation Taxanes Benefit Advanced Stages of Prostate Cancer. Clin. Cancer Res. 2015, 21, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Nouri, M.; Ratther, E.; Stylianou, N.; Nelson, C.C.; Hollier, B.G.; Williams, E.D. Androgen-Targeted Therapy-Induced Epithelial Mesenchymal Plasticity and Neuroendocrine Transdifferentiation in Prostate Cancer: An Opportunity for Intervention. Front. Oncol. 2014, 4, 370. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, S.; Yin, C.; Hu, S.; Liu, P. The Role of the mTOR Pathway in Breast Cancer Stem Cells (BCSCs): Mechanisms and Therapeutic Potentials. Stem Cell Res. Ther. 2025, 16, 156. [Google Scholar] [CrossRef]
- Dzobo, K.; Senthebane, D.A.; Ganz, C.; Thomford, N.E.; Wonkam, A.; Dandara, C. Advances in Therapeutic Targeting of Cancer Stem Cells within the Tumor Microenvironment: An Updated Review. Cells 2020, 9, 1896. [Google Scholar] [CrossRef]
- Lei, Z.-N.; Teng, Q.-X.; Koya, J.; Liu, Y.; Chen, Z.; Zeng, L.; Chen, Z.-S.; Fang, S.; Wang, J.; Liu, Y.; et al. The Correlation between Cancer Stem Cells and Epithelial-Mesenchymal Transition: Molecular Mechanisms and Significance in Cancer Theragnosis. Front. Immunol. 2024, 15, 1417201. [Google Scholar] [CrossRef]
- Nobili, S.; Landini, I.; Giglioni, B.; Mini, E. Pharmacological Strategies for Overcoming Multidrug Resistance. Curr. Drug Targets 2006, 7, 861–879. [Google Scholar] [CrossRef]
- Goebel, J.; Chmielewski, J.; Hrycyna, C.A. The Roles of the Human ATP-Binding Cassette Transporters P-Glycoprotein and ABCG2 in Multidrug Resistance in Cancer and at Endogenous Sites: Future Opportunities for Structure-Based Drug Design of Inhibitors. Cancer Drug Resist. 2021, 4, 784. [Google Scholar] [CrossRef]
- Hu, T.; Li, Z.; Gao, C.-Y.; Cho, C.H. Mechanisms of Drug Resistance in Colon Cancer and Its Therapeutic Strategies. World J. Gastroenterol. 2016, 22, 6876. [Google Scholar] [CrossRef]
- Rana, Z.; Diermeier, S.; Hanif, M.; Rosengren, R.J. Understanding Failure and Improving Treatment Using HDAC Inhibitors for Prostate Cancer. Biomedicines 2020, 8, 22. [Google Scholar] [CrossRef]
- Xu, P.; Hu, G.; Luo, C.; Liang, Z. DNA Methyltransferase Inhibitors: An Updated Patent Review (2012–2015). Expert. Opin. Ther. Pat. 2016, 26, 1017–1030. [Google Scholar] [CrossRef]
- Fandy, T. Development of DNA Methyltransferase Inhibitors for the Treatment of Neoplastic Diseases. Curr. Med. Chem. 2009, 16, 2075–2085. [Google Scholar] [CrossRef]
- Park, J.; Thomas, S.; Munster, P.N. Epigenetic Modulation with Histone Deacetylase Inhibitors in Combination with Immunotherapy. Epigenomics 2015, 7, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Abdelfatah, E.; Kerner, Z.; Nanda, N.; Ahuja, N. Epigenetic Therapy in Gastrointestinal Cancer: The Right Combination. Ther. Adv. Gastroenterol. 2016, 9, 560–579. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Festuccia, C.; Marampon, F.; Popov, V.M.; Pestell, R.G.; Zani, B.M.; Tombolini, V. Biological Rationale for the Use of DNA Methyltransferase Inhibitors as New Strategy for Modulation of Tumor Response to Chemotherapy and Radiation. Mol. Cancer 2010, 9, 305. [Google Scholar] [CrossRef] [PubMed]
- Vendetti, F.P.; Rudin, C.M. Epigenetic Therapy in Non-Small-Cell Lung Cancer: Targeting DNA Methyltransferases and Histone Deacetylases. Expert Opin. Biol. Ther. 2013, 13, 1273–1285. [Google Scholar] [CrossRef]
- Lee, L.; Gupta, M.; Sahasranaman, S. Immune Checkpoint Inhibitors: An Introduction to the Next-generation Cancer Immunotherapy. J. Clin. Pharma. 2016, 56, 157–169. [Google Scholar] [CrossRef]
- Venkatachalam, S.; McFarland, T.R.; Agarwal, N.; Swami, U. Immune Checkpoint Inhibitors in Prostate Cancer. Cancers 2021, 13, 2187. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, W.; Li, N.; Neri, S.; Sharma, A.; Jiang, W.; Lin, S.H. Combining Immunotherapy and Radiotherapy for Cancer Treatment: Current Challenges and Future Directions. Front. Pharmacol. 2018, 9, 185. [Google Scholar] [CrossRef]
- Liu, Y.L.; Zamarin, D. Combination Immune Checkpoint Blockade Strategies to Maximize Immune Response in Gynecological Cancers. Curr. Oncol. Rep. 2018, 20, 94. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Piulats, J.M.; Gross-Goupil, M.; Goh, J.; Ojamaa, K.; Hoimes, C.J.; Vaishampayan, U.; Berger, R.; Sezer, A.; Alanko, T.; et al. Pembrolizumab for Treatment-Refractory Metastatic Castration-Resistant Prostate Cancer: Multicohort, Open-Label Phase II KEYNOTE-199 Study. J. Clin. Oncol. 2020, 38, 395–405. [Google Scholar] [CrossRef]
- Ruiz De Porras, V.; Pardo, J.C.; Notario, L.; Etxaniz, O.; Font, A. Immune Checkpoint Inhibitors: A Promising Treatment Option for Metastatic Castration-Resistant Prostate Cancer? Int. J. Mol. Sci. 2021, 22, 4712. [Google Scholar] [CrossRef]
- Mogensen, T.H. Genetic Susceptibility to Viral Disease in Humans. Clin. Microbiol. Infect. 2022, 28, 1411–1416. [Google Scholar] [CrossRef] [PubMed]
- Govers, C.; Calder, P.C.; Savelkoul, H.F.J.; Albers, R.; Van Neerven, R.J.J. Ingestion, Immunity, and Infection: Nutrition and Viral Respiratory Tract Infections. Front. Immunol. 2022, 13, 841532. [Google Scholar] [CrossRef] [PubMed]
- Collet, L.; Hanvic, B.; Turinetto, M.; Treilleux, I.; Chopin, N.; Le Saux, O.; Ray-Coquard, I. BRCA1/2 Alterations and Reversion Mutations in the Area of PARP Inhibitors in High Grade Ovarian Cancer: State of the Art and Forthcoming Challenges. Front. Oncol. 2024, 14, 1354427. [Google Scholar] [CrossRef] [PubMed]
- Janysek, D.C.; Kim, J.; Duijf, P.H.G.; Dray, E. Clinical Use and Mechanisms of Resistance for PARP Inhibitors in Homologous Recombination-Deficient Cancers. Transl. Oncol. 2021, 14, 101012. [Google Scholar] [CrossRef] [PubMed]
- Smatti, M.K.; Al Thani, A.A.; Yassine, H.M. Viral-Induced Enhanced Disease Illness. Front. Microbiol. 2018, 9, 2991. [Google Scholar] [CrossRef]
- Kitazawa, H.; Villena, J. Modulation of Respiratory TLR3-Anti-Viral Response by Probiotic Microorganisms: Lessons Learned from Lactobacillus Rhamnosus CRL1505. Front. Immunol. 2014, 5, 201. [Google Scholar] [CrossRef]
- Shornick, L.P.; Wells, A.G.; Zhang, Y.; Patel, A.C.; Huang, G.; Takami, K.; Sosa, M.; Shukla, N.A.; Agapov, E.; Holtzman, M.J. Airway Epithelial versus Immune Cell Stat1 Function for Innate Defense against Respiratory Viral Infection. J. Immunol. 2008, 180, 3319–3328. [Google Scholar] [CrossRef]
- Mullane, S.A.; Van Allen, E.M. Precision Medicine for Advanced Prostate Cancer. Curr. Opin. Urol. 2016, 26, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Al-Daffaie, F.M.; Al-Mudhafar, S.F.; Alhomsi, A.; Tarazi, H.; Almehdi, A.M.; El-Huneidi, W.; Abu-Gharbieh, E.; Bustanji, Y.; Alqudah, M.A.Y.; Abuhelwa, A.Y.; et al. Metabolomics and Proteomics in Prostate Cancer Research: Overview, Analytical Techniques, Data Analysis, and Recent Clinical Applications. Int. J. Mol. Sci. 2024, 25, 5071. [Google Scholar] [CrossRef] [PubMed]
- Hoseini, S.H.; Enayati, P.; Nazari, M.; Babakhanzadeh, E.; Rastgoo, M.; Sohrabi, N.B. Biomarker Profile of Colorectal Cancer: Current Findings and Future Perspective. J. Gastrointest. Cancer 2024, 55, 497–510. [Google Scholar] [CrossRef]
- Halabi, S.; Dutta, S.; Tangen, C.M.; Rosenthal, M.; Petrylak, D.P.; Thompson, I.M.; Chi, K.N.; De Bono, J.S.; Araujo, J.C.; Logothetis, C.; et al. Clinical Outcomes in Men of Diverse Ethnic Backgrounds with Metastatic Castration-Resistant Prostate Cancer. Ann. Oncol. 2020, 31, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-Z.; Wang, Z.-J.; De, W.; Zheng, M.; Xu, W.-Z.; Wu, H.-F.; Armstrong, A.; Zhu, J.-G. Targeting AXL Overcomes Resistance to Docetaxel Therapy in Advanced Prostate Cancer. Oncotarget 2017, 8, 41064–41077. [Google Scholar] [CrossRef]
- Kahn, B.; Collazo, J.; Kyprianou, N. Androgen Receptor as a Driver of Therapeutic Resistance in Advanced Prostate Cancer. Int. J. Biol. Sci. 2014, 10, 588–595. [Google Scholar] [CrossRef]
- Ning, S.; Armstrong, C.M.; Xing, E.; Leslie, A.R.; Gao, R.Y.; Sharifi, M.; Schaaf, Z.A.; Lou, W.; Han, X.; Xu, D.H.; et al. LX1 Dual Targets AR Variants and AKR1C3 in Advanced Prostate Cancer Therapy. Cancer Res. 2024, 84, 3617–3628. [Google Scholar] [CrossRef] [PubMed]
- Morrison, G.J.; Goldkorn, A. Development and Application of Liquid Biopsies in Metastatic Prostate Cancer. Curr. Oncol. Rep. 2018, 20, 35. [Google Scholar] [CrossRef]
- Chowdhury-Paulino, I.M.; Ericsson, C.; Vince, R.; Spratt, D.E.; George, D.J.; Mucci, L.A. Racial Disparities in Prostate Cancer among Black Men: Epidemiology and Outcomes. Prostate Cancer Prostatic Dis. 2022, 25, 397–402. [Google Scholar] [CrossRef]
- Pietro, G.D.; Chornokur, G.; Kumar, N.B.; Davis, C.; Park, J.Y. Racial Differences in the Diagnosis and Treatment of Prostate Cancer. Int. Neurourol. J. 2016, 20 (Suppl. S2), S112–S119. [Google Scholar] [CrossRef]
- Singh, R. Molecular Basis for Prostate Cancer Racial Disparities. Front. Biosci. 2017, 22, 428–450. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Agent | Type | Mechanism of Action | Status |
|---|---|---|---|
| Cabazitaxel | Second-gen taxane | Binds β-tubulin, overcomes P-gp-mediated efflux | FDA approved |
| Enzalutamide | AR antagonist | Inhibits AR nuclear translocation | FDA approved |
| Abiraterone acetate | CYP17 inhibitor | Blocks androgen biosynthesis | FDA approved |
| Olaparib | PARP inhibitor | Exploits DNA repair defects (e.g., BRCA mutations) | FDA approved |
| Ipilimumab + Nivolumab | Immune checkpoint blockade | Targets CTLA-4 and PD-1 | Phase III trials |
| AZD5363 (Capivasertib) | AKT inhibitor | Blocks PI3K/AKT pathway | Phase I trials |
| AVB-S6-500 batiraxcept | AXL inhibitor | Reverse EMT, decreasing the expression of mesenchymal markers and increasing the expression of epithelial markers like E-cadherin | Phase I/II trials |
| BET inhibitors (e.g., ZEN-3694) | Epigenetic modulators | Inhibit transcriptional reprogramming | Phase I/II trials |
| Sipuleucel-T | Therapeutic cancer vaccine | Activation of patient’s immune system to target prostatic acid phosphatase (PAP) | FDA Approved |
| Radium-223 | Radiopharmaceutical | The emitted high-energy alpha particles induce DNA double-strand breaks that might be irreparable and lead to cell death in nearby exposed tumor cells, osteoblasts, and osteoclasts. | FDA Approved |
| Niraparib (Akeega) combined with abiraterone acetate and prednisone | PARP inhibitor | Exploits DNA repair defects (e.g., BRCA mutations) | FDA approved |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adekiya, T.A. Beyond Docetaxel: Targeting Resistance Pathways in Prostate Cancer Treatment. BioChem 2025, 5, 24. https://doi.org/10.3390/biochem5030024
Adekiya TA. Beyond Docetaxel: Targeting Resistance Pathways in Prostate Cancer Treatment. BioChem. 2025; 5(3):24. https://doi.org/10.3390/biochem5030024
Chicago/Turabian StyleAdekiya, Tayo Alex. 2025. "Beyond Docetaxel: Targeting Resistance Pathways in Prostate Cancer Treatment" BioChem 5, no. 3: 24. https://doi.org/10.3390/biochem5030024
APA StyleAdekiya, T. A. (2025). Beyond Docetaxel: Targeting Resistance Pathways in Prostate Cancer Treatment. BioChem, 5(3), 24. https://doi.org/10.3390/biochem5030024