
Formulation and In Vitro Characterization of Cellulose-Based Propranolol Hydrochloride Sustained Release Matrix Tablets


 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
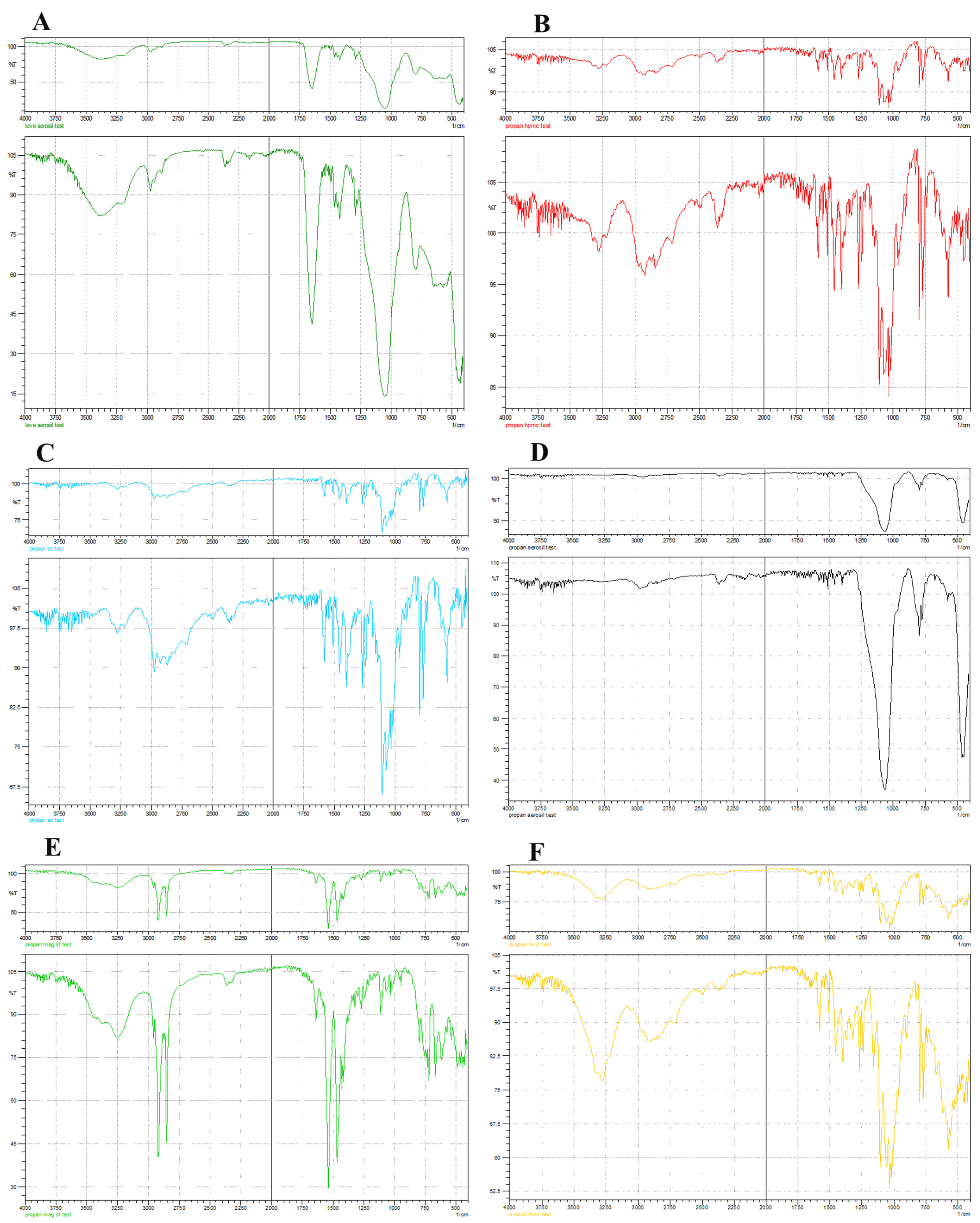
2.2. Drug Excipient Compatibility Study
2.3. Evaluation of Pre-Compression Properties of the Powder Mix
2.3.1. Bulk Density (Db)
2.3.2. Tapped Density (Dt)
2.3.3. Powder Flow Property Study
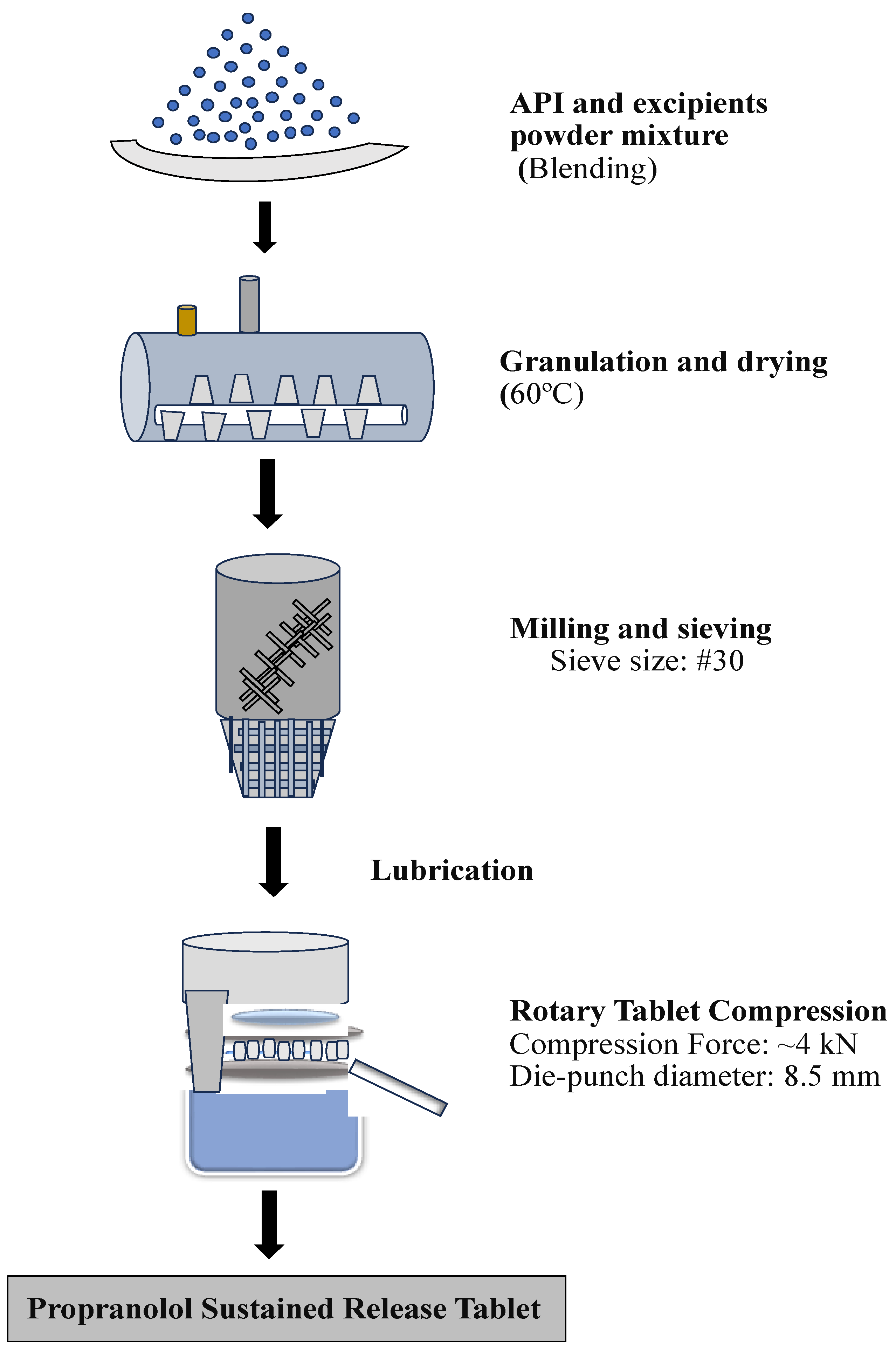
2.4. Formulation of Sustained-Release Matrix Tablets
2.5. Physical Characterization
2.5.1. Visual Inspection
2.5.2. Weight Variation Studies
2.5.3. Friability Test
2.5.4. Hardness Test
2.6. Drug Content
2.7. In Vitro Drug Release
2.8. Comparative Dissolution Profile Study
3. Results
3.1. Drug–Excipient Compatibility Study
3.2. Pre-Compression Analysis of Granules
3.3. Post-Compression Analysis of the Formulations
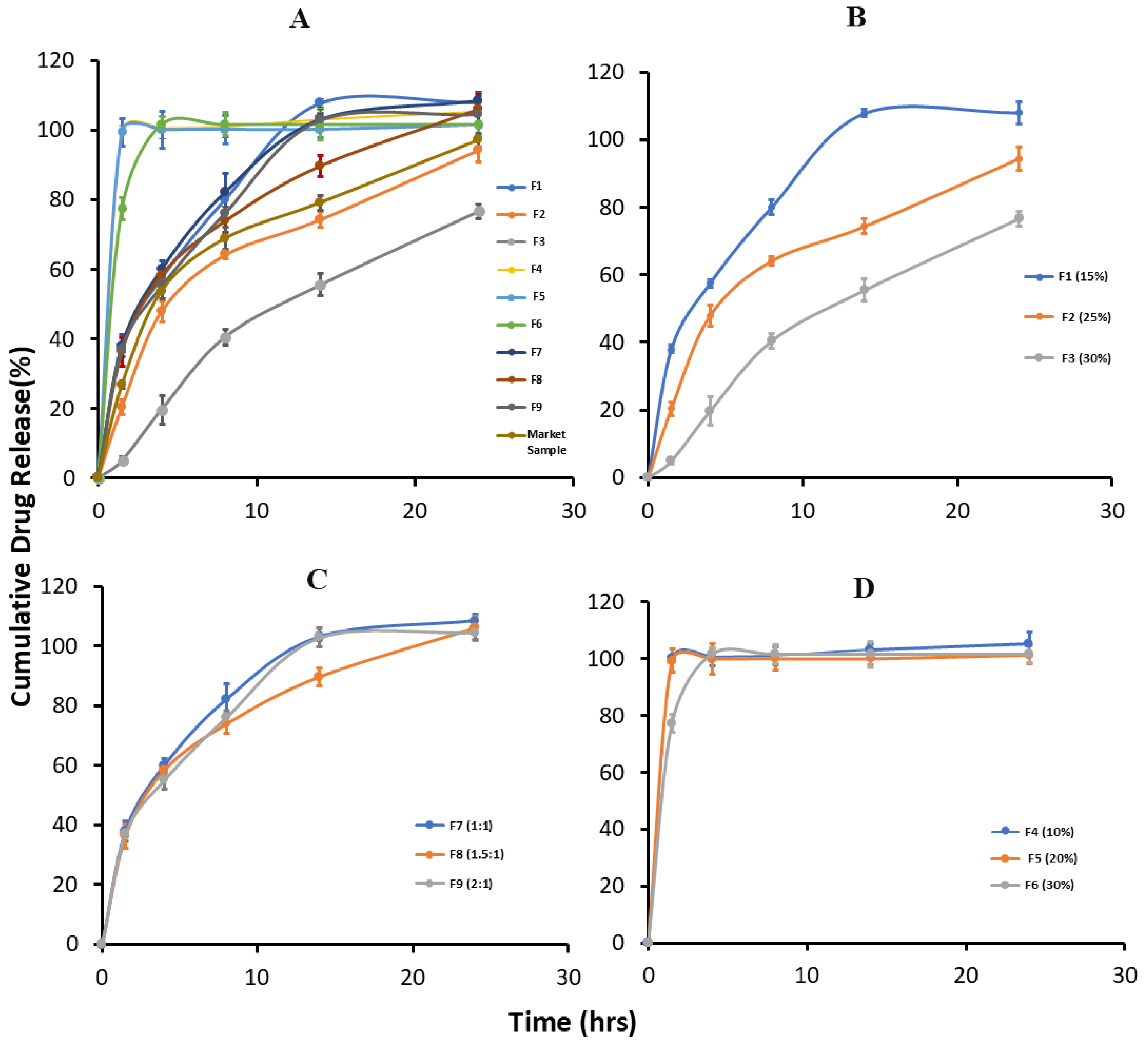
3.4. In Vitro Drug Release Study
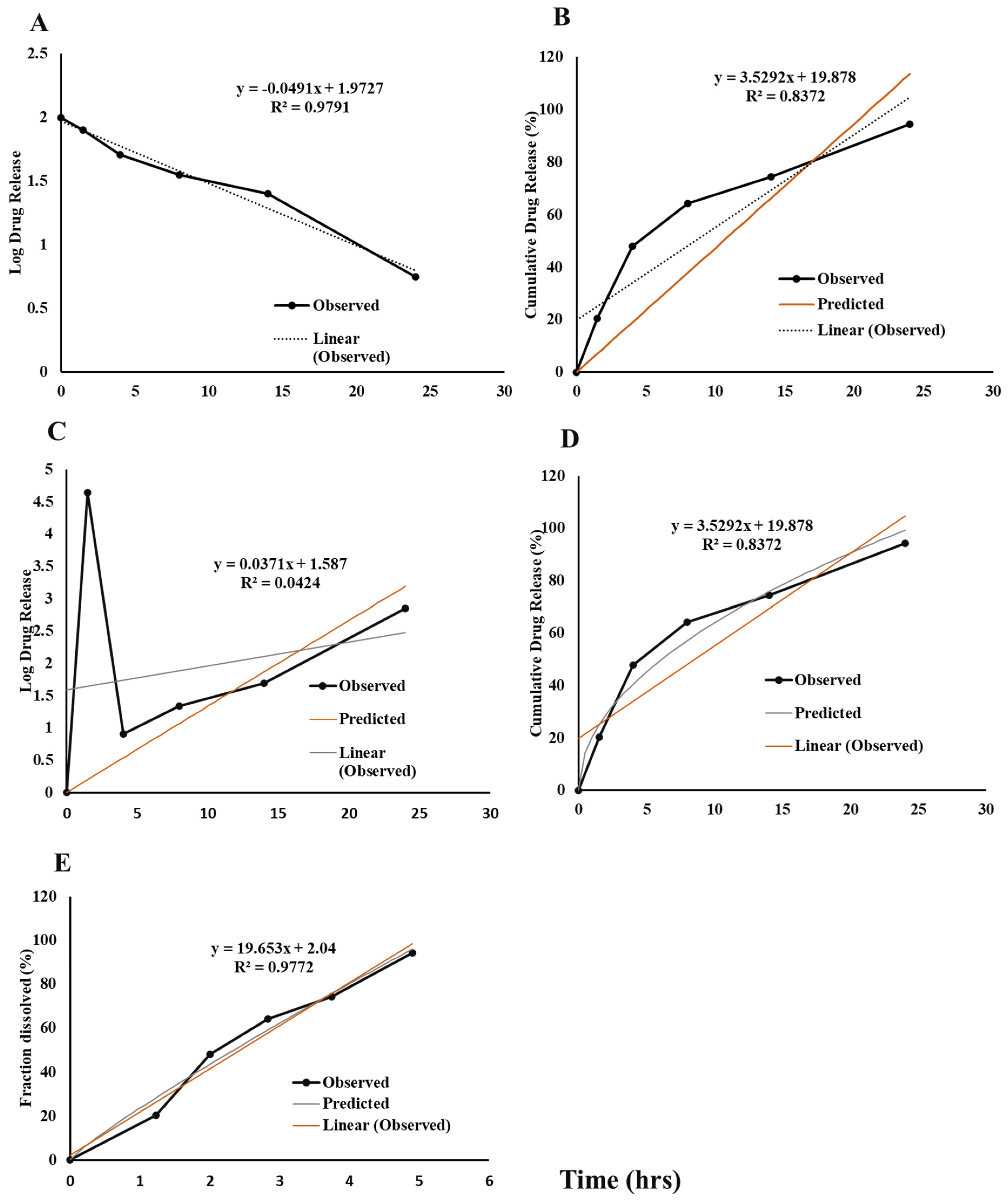
3.5. In Vitro Drug Release Kinetic Study
3.6. Comparative Dissolution Profile Study
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| EC | Ethylcellulose |
| HPMC | Hydroxypropyl methylcellulose |
| SR | Sustained Release |
| FT-IR | Fourier transform infrared spectroscopy |
| MCC | Microcrystalline cellulose |
| Db | Bulk density |
| Dt | Tapped density |
| USP | United State Pharmacopeia |
| HPLC | High-performance liquid chromatography |
| PVP | Polyvinyl pyrrolidone |
| API | Active Pharmaceutical Ingredient |
| DSC | Differential scanning calorimetry |
| CI | Carr’s index |
| HR | Hausner ratio |
References
- Stritzke, A.; Kabra, N.; Kaur, S.; Robertson, H.; Lodha, A. Oral propranolol in prevention of severe retinopathy of prematurity: A systematic review and meta-analysis. J. Perinatol. 2019, 39, 1584–1594. [Google Scholar] [CrossRef] [PubMed]
- Propranolol Hydrochloride. Available online: https://go.drugbank.com/salts/DBSALT000549 (accessed on 5 March 2025).
- Lampert, R.; Ickovics, J.R.; Viscoli, C.J.; Horwitz, R.I.; Lee, F.A. Effects of propranolol on recovery of heart rate variability following acute myocardial infarction and relation to outcome in the Beta-Blocker Heart Attack Trial. Am. J. Cardiol. 2003, 91, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Williams, F.N.; Herndon, D.N.; Kulp, G.A.; Jeschke, M.G. Propranolol decreases cardiac work in a dose-dependent manner in severely burned children. Surgery 2011, 149, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Chen, S.; Wang, Q.; Xiang, B.; Xu, Z.; Zhong, L.; Yang, K.; Lu, G.; Qiu, L. Intolerable side effects during propranolol therapy for infantile hemangioma: Frequency, risk factors and management. Sci. Rep. 2018, 8, 4264. [Google Scholar] [CrossRef]
- Acharya, P.; Baral, R.; Manandhar, S.; Shahi, P.; Gurung, B. Formulation and In-vitro Characterization of Fluoxetine Hydrochloride Loaded Fast Dissolving Oral Film Using HPMC 15CPS and HPMC K4M. J. Pharm. Drug Dev. 2021, 8, 101. [Google Scholar]
- Martinez, M.; Rathbone, M.; Burgess, D.; Huynh, M. In vitro and in vivo considerations associated with parenteral sustained release products: A review based upon information presented and points expressed at the 2007 Controlled Release Society Annual Meeting. J. Control. Release 2008, 129, 79–87. [Google Scholar] [CrossRef]
- Dash, A.; Cudworth, G., II. Therapeutic applications of implantable drug delivery systems. J. Pharmacol. Toxicol. Methods 1998, 40, 1–12. [Google Scholar] [CrossRef]
- Shen, J.; Burgess, D. Accelerated in-vitro release testing methods for extended-release parenteral dosage forms. J. Pharm. Pharmacol. 2012, 64, 986–996. [Google Scholar] [CrossRef]
- Nashed, N.; Lam, M.; Nokhodchi, A. A comprehensive overview of extended-release oral dosage forms manufactured through hot melt extrusion and its combination with 3D printing. Int. J. Pharm. 2021, 596, 120237. [Google Scholar] [CrossRef]
- Robinson, J.; Eriksen, S. Theoretical formulation of sustained-release dosage forms. J. Pharm. Sci. 1966, 55, 1254–1263. [Google Scholar]
- Almeida, A.; Possemiers, S.; Boone, M.; De Beer, T.; Quinten, T.; Van Hoorebeke, L.; Remon, J.P.; Vervaet, C. Ethylene vinyl acetate as matrix for oral sustained release dosage forms produced via hot-melt extrusion. Eur. J. Pharm. Biopharm. 2011, 77, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Tabasi, S.H.; Moolchandani, V.; Fahmy, R.; Hoag, S.W. Sustained release dosage forms dissolution behavior prediction: A study of matrix tablets using NIR spectroscopy. Int. J. Pharm. 2009, 382, 1–6. [Google Scholar] [CrossRef]
- Körner, A.; Piculell, L.; Iselau, F.; Wittgren, B.; Larsson, A. Influence of different polymer types on the overall release mechanism in hydrophilic matrix tablets. Molecules 2009, 14, 2699–2716. [Google Scholar] [CrossRef] [PubMed]
- Viridén, A.; Wittgren, B.; Andersson, T.; Larsson, A. The effect of chemical heterogeneity of HPMC on polymer release from matrix tablets. Eur. J. Pharm. Sci. 2009, 36, 392–400. [Google Scholar] [CrossRef]
- Giri, B.R.; Maniruzzaman, M. Fabrication of Sustained-Release Dosages Using Powder-Based Three-Dimensional (3D) Printing Technology. AAPS PharmSciTech 2022, 24, 4. [Google Scholar] [CrossRef]
- Giri, B.R.; Kwon, J.; Vo, A.Q.; Bhagurkar, A.M.; Bandari, S.; Kim, D.W. Hot-melt extruded amorphous solid dispersion for solubility, stability, and bioavailability enhancement of telmisartan. Pharmaceuticals 2021, 14, 73. [Google Scholar] [CrossRef]
- Pearnchob, N.; Siepmann, J.; Bodmeier, R. Pharmaceutical applications of shellac: Moisture-protective and taste-masking coatings and extended-release matrix tablets. Drug Dev. Ind. Pharm. 2003, 29, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Giri, B.R.; Poudel, S.; Kim, D.W. Cellulose and its derivatives for application in 3D printing of pharmaceuticals. J. Pharm. Investig. 2021, 51, 1–22. [Google Scholar] [CrossRef]
- Kwon, J.; Giri, B.R.; Song, E.S.; Bae, J.; Lee, J.; Kim, D.W. Spray-dried amorphous solid dispersions of atorvastatin calcium for improved supersaturation and oral bioavailability. Pharmaceutics 2019, 11, 461. [Google Scholar] [CrossRef]
- Viridén, A.; Wittgren, B.; Larsson, A. The consequence of the chemical composition of HPMC in matrix tablets on the release behavior of model drug substances having different solubility. Eur. J. Pharm. Biopharm. 2011, 77, 99–110. [Google Scholar] [CrossRef]
- Chavan, R.B.; Rathi, S.; Jyothi, V.G.S.; Shastri, N.R. Cellulose based polymers in development of amorphous solid dispersions. Asian J. Pharm. Sci. 2019, 14, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zuo, X.; Liang, H.; Zhu, T.; Zhong, Y.; Liu, J.; Nan, J. A heat-resistant poly (oxyphenylene benzimidazole)/ethyl cellulose blended polymer membrane for highly safe lithium-ion batteries. ACS Appl. Mater. Interfaces 2019, 12, 637–645. [Google Scholar] [CrossRef]
- Reddy, K.R.; Mutalik, S.; Reddy, S. Once-daily sustained-release matrix tablets of nicorandil: Formulation and in vitro evaluation. AAPS PharmSciTech 2003, 4, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Kuksal, A.; Tiwary, A.K.; Jain, N.K.; Jain, S. Formulation and in vitro, in vivo evaluation of extended-release matrix tablet of zidovudine: Influence of combination of hydrophilic and hydrophobic matrix formers. AAPS PharmSciTech 2006, 7, E1–E9. [Google Scholar] [CrossRef]
- Patil, H.; Tiwari, R.V.; Upadhye, S.B.; Vladyka, R.S.; Repka, M.A. Formulation and development of pH-independent/dependent sustained release matrix tablets of ondansetron HCl by a continuous twin-screw melt granulation process. Int. J. Pharm. 2015, 496, 33–41. [Google Scholar] [CrossRef]
- Mandal, S.; Basu, S.K.; Sa, B. Sustained release of a water-soluble drug from alginate matrix tablets prepared by wet granulation method. AAPS PharmSciTech 2009, 10, 1348–1356. [Google Scholar] [CrossRef]
- Hamza, Y.E.-S.; Aburahma, M.H. Design and in vitro evaluation of novel sustained-release matrix tablets for lornoxicam based on the combination of hydrophilic matrix formers and basic pH-modifiers. Pharm. Dev. Technol. 2010, 15, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Khaled, S.A.; Alexander, M.R.; Wildman, R.D.; Wallace, M.J.; Sharpe, S.; Yoo, J.; Roberts, C. 3D extrusion printing of high drug loading immediate release paracetamol tablets. Int. J. Pharm. 2018, 538, 223–230. [Google Scholar] [CrossRef]
- Abdelbary, G.A.; Tadros, M.I. Design and in vitro/in vivo evaluation of novel nicorandil extended release matrix tablets based on hydrophilic interpolymer complexes and a hydrophobic waxy polymer. Eur. J. Pharm. Biopharm. 2008, 69, 1019–1028. [Google Scholar] [CrossRef]
- Sahadevan, J.; Prabhakaran, R.; Vijay, J.; Gilhotra, R.M. Formulation and evaluation of cephalexin extended-release matrix tablets using hydroxy propyl methyl cellulose as rate-controlling polymer. J. Young Pharm. 2012, 4, 3–12. [Google Scholar] [CrossRef]
- Badshah, A.; Subhan, F.; Rauf, K.; Irfan Bukhari, N.; Shah, K.; Khan, S.; Ahmed, Z.; Khan, I. Development of controlled-release matrix tablet of risperidone: Influence of Methocel®- and Ethocel®-based novel polymeric blend on in vitro drug release and bioavailability. AAPS PharmSciTech 2011, 12, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, J.; Murthy, P.; Biswal, S.; Sahoo, S.; Mahapatra, A. Comparative study of propranolol hydrochloride release from matrix tablets with Kollidon® SR or hydroxy propyl methyl cellulose. AAPS PharmSciTech 2008, 9, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, S. Granulation techniques and technologies: Recent progresses. BioImpacts BI 2015, 5, 55. [Google Scholar] [CrossRef]
- De Simone, V.; Caccavo, D.; Dalmoro, A.; Lamberti, G.; D’amore, M.; Barba, A.A. Inside the phenomenological aspects of wet granulation: Role of process parameters. In Granularity in Materials Science; IntechOpen: Rijeka, Croatia, 2018; pp. 63–84. [Google Scholar]
- Mughal, M.A.; Iqbal, Z.; Neau, S.H. Guar gum, xanthan gum, and HPMC can define release mechanisms and sustain release of propranolol hydrochloride. AAPS PharmSciTech 2011, 12, 77–87. [Google Scholar] [CrossRef]
- El Nabarawi, M.A.; Teaima, M.H.; Abd El-Monem, R.A.; El Nabarawy, N.A.; Gaber, D.A. Formulation, release characteristics, and bioavailability study of gastroretentive floating matrix tablet and floating raft system of Mebeverine HCl. Drug Des. Dev. Ther. 2017, 2017, 1081–1093. [Google Scholar] [CrossRef]
- Tiwari, S.B.; Murthy, T.K.; Raveendra Pai, M.; Mehta, P.R.; Chowdary, P.B. Controlled release formulation of tramadol hydrochloride using hydrophilic and hydrophobic matrix system. AAPS PharmSciTech 2003, 4, 18–23. [Google Scholar] [CrossRef]
- Bose, A.; Wong, T.W.; Singh, N. Formulation development and optimization of sustained release matrix tablet of Itopride HCl by response surface methodology and its evaluation of release kinetics. Saudi Pharm. J. 2013, 21, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Peppas, N.A. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC). Adv. Drug Deliv. Rev. 2012, 64, 163–174. [Google Scholar] [CrossRef]
- Amalia, A.; Lestari, W.W.; Pratama, J.H.; Wibowo, F.R.; Larasati, L.; Saraswati, T.E. Modification of dry-gel synthesized MIL-100 (Fe) with carboxymethyl cellulose for curcumin slow-release. J. Polym. Res. 2022, 29, 487. [Google Scholar] [CrossRef]
- Costa, P.; Lobo, J.M. Comparison of dissolution profiles—Advancement of in vitro correlation method. Pharm. Sci. Technol. Today 2001, 4, 240–246. [Google Scholar]
- Prichard, B. Propranolol and beta-adrenergic receptor blocking drugs in the treatment of hypertension. Br. J. Clin. Pharmacol. 1982, 13, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Grewal, J.; Jyoti, K.; Jain, U.K.; Chandra, R.; Madan, J. Oral controlled and sustained drug delivery systems: Concepts, advances, preclinical, and clinical status. In Drug Targeting and Stimuli Sensitive Drug Delivery Systems; Elsevier: Amsterdam, The Netherlands, 2018; pp. 567–626. [Google Scholar]
- Dos Santos, J.; Da Silva, G.S.; Velho, M.C.; Beck, R.C.R. Eudragit®: A versatile family of polymers for hot melt extrusion and 3D printing processes in pharmaceutics. Pharmaceutics 2021, 13, 1424. [Google Scholar] [CrossRef] [PubMed]
- Kiortsis, S.; Kachrimanis, K.; Broussali, T.; Malamataris, S. Drug release from tableted wet granulations comprising cellulosic (HPMC or HPC) and hydrophobic component. Eur. J. Pharm. Biopharm. 2005, 59, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Bolívar-Monsalve, E.J.; Alvarez, M.M.; Hosseini, S.; Espinosa-Hernandez, M.A.; Ceballos-González, C.F.; Sanchez-Dominguez, M.; Shin, S.R.; Cecen, B.; Hassan, S.; Di Maio, E. Engineering bioactive synthetic polymers for biomedical applications: A review with emphasis on tissue engineering and controlled release. Mater. Adv. 2021, 2, 4447–4478. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulations | Propranolol | HPMC K100M | EC | PVP K30 | MCC PH101 | MgSt | Aerosil |
|---|---|---|---|---|---|---|---|
| F1 | 20 | 15 | - | 2.0 | 61.5 | 1.0 | 0.5 |
| F2 | 20 | 25 | - | 2.0 | 51.5 | 1.0 | 0.5 |
| F3 | 20 | 30 | - | 2.0 | 46.5 | 1.0 | 0.5 |
| F4 | 20 | - | 10.0 | 2.0 | 66.5 | 1.0 | 0.5 |
| F5 | 20 | - | 20.0 | 2.0 | 56.5 | 1.0 | 0.5 |
| F6 | 20 | - | 30.0 | 2.0 | 46.5 | 1.0 | 0.5 |
| F7 | 20 | 12.5 | 12.5 | 2.0 | 51.5 | 1.0 | 0.5 |
| F8 | 20 | 18.8 | 6.3 | 2.0 | 51.5 | 1.0 | 0.5 |
| F9 | 20 | 16.7 | 8.3 | 2.0 | 51.5 | 1.0 | 0.5 |
| Formulations | Angle of Repose (θ) | Carr’s Index (CI) | Hausner Ratio (HR) |
|---|---|---|---|
| F1 | 28.1 | 15.79 | 1.19 |
| F2 | 28.5 | 12.5 | 1.14 |
| F3 | 28.7 | 11.9 | 1.14 |
| F4 | 33.5 | 13.79 | 1.16 |
| F5 | 34.1 | 12.90 | 1.15 |
| F6 | 34.5 | 13.51 | 1.16 |
| F7 | 30.2 | 11.76 | 1.13 |
| F8 | 30.5 | 13.89 | 1.16 |
| F9 | 30.3 | 10.81 | 1.12 |
| Formulations | Average wt (mg) | Assay (%) | Hardness (Kp) | Friability (%) | Thickness (mm) |
|---|---|---|---|---|---|
| F1 | 200.45 ± 1.87 | 97.45 | 11.93 ± 1.24 | 0.18 | 3.81 ± 0.02 |
| F2 | 200.55 ± 1.70 | 98.36 | 12.34 ± 0.91 | 0.20 | 3.80 ± 0.02 |
| F3 | 200.90 ± 1.99 | 97.17 | 13.28 ± 1.14 | 0.15 | 3.80 ± 0.03 |
| F4 | 200.85 ± 1.95 | 96.36 | 12.84 ± 1.14 | 0.27 | 3.81 ± 0.02 |
| F5 | 200.55 ± 1.93 | 97.75 | 12.72 ± 1.08 | 0.30 | 3.81 ± 0.02 |
| F6 | 199.55 ± 1.57 | 98.65 | 12.26 ± 1.29 | 0.25 | 3.80 ± 0.02 |
| F7 | 201.25 ± 2.42 | 98.05 | 12.54 ± 0.98 | 0.28 | 3.82 ± 0.02 |
| F8 | 201.00 ± 2.27 | 97.80 | 12.29 ± 1.24 | 0.29 | 3.81 ± 0.02 |
| F9 | 200.75 ± 1.68 | 96.09 | 13.66 ± 1.08 | 0.32 | 3.81 ± 0.02 |
| Formulation | 1.5 h | 4 h | 8 h | 14 h | 24 h |
|---|---|---|---|---|---|
| F1 * | 38.02 ± 1.09 | 57.51 ± 1.08 | 80.07 ± 2.12 | 107.81 ± 1.14 | 107.95 ± 3.21 |
| F2 * | 20.40 ± 2.07 | 47.91 ± 3.02 | 64.11 ± 1.21 | 74.30 ± 2.21 | 94.30 ± 3.31 |
| F3 * | 05.00 ± 1.03 | 19.66 ± 4.15 | 40.53 ± 2.17 | 55.50 ± 3.17 | 76.67 ± 2.17 |
| F4 | 100.0 ± 1.16 | 100.60 ± 3.21 | 101.03 ± 3.21 | 103.10 ± 2.12 | 105.40 ± 4.15 |
| F5 | 99.33 ± 4.07 | 100.01 ± 5.32 | 100.12 ± 4.18 | 100.13 ± 2.31 | 101.66 ± 3.18 |
| F6 | 77.33 ± 3.15 | 101.66 ± 2.17 | 101.66 ± 3.31 | 101.66 ± 4.37 | 101.69 ± 3.14 |
| F7 * | 38.01 ± 3.24 | 60.03 ± 2.26 | 82.20 ± 5.21 | 103.15 ± 1.17 | 108.50 ± 2.19 |
| F8 * | 36.30 ± 4.02 | 58.33 ± 1.31 | 73.83 ± 3.24 | 89.66 ± 3.14 | 106.00 ± 4.17 |
| F9 * | 37.12 ± 1.12 | 55.07 ± 3.21 | 76.12 ± 2.15 | 102.87 ± 3.17 | 104.52 ± 2.36 |
| Formulation | Dissimilarity Factor (f1) | Similarity Factor (f2) |
|---|---|---|
| F1 | 40 | 20 |
| F2 | 8 | 64 |
| F3 | 39 | 29 |
| F4 | 57 | 18 |
| F5 | 53 | 19 |
| F6 | 49 | 22 |
| F7 | 20 | 42 |
| F8 | 12 | 54 |
| F9 | 15 | 45 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khadka, A.; Giri, B.R.; Baral, R.; Shakya, S.; Shrestha, A.K. Formulation and In Vitro Characterization of Cellulose-Based Propranolol Hydrochloride Sustained Release Matrix Tablets. BioChem 2025, 5, 14. https://doi.org/10.3390/biochem5020014
Khadka A, Giri BR, Baral R, Shakya S, Shrestha AK. Formulation and In Vitro Characterization of Cellulose-Based Propranolol Hydrochloride Sustained Release Matrix Tablets. BioChem. 2025; 5(2):14. https://doi.org/10.3390/biochem5020014
Chicago/Turabian StyleKhadka, Aashish, Bhupendra Raj Giri, Rishiram Baral, Shailendra Shakya, and Ashwinee Kumar Shrestha. 2025. "Formulation and In Vitro Characterization of Cellulose-Based Propranolol Hydrochloride Sustained Release Matrix Tablets" BioChem 5, no. 2: 14. https://doi.org/10.3390/biochem5020014
APA StyleKhadka, A., Giri, B. R., Baral, R., Shakya, S., & Shrestha, A. K. (2025). Formulation and In Vitro Characterization of Cellulose-Based Propranolol Hydrochloride Sustained Release Matrix Tablets. BioChem, 5(2), 14. https://doi.org/10.3390/biochem5020014