
Sphingolipid Signaling and Complement Activation in Glioblastoma: A Promising Avenue for Therapeutic Intervention


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
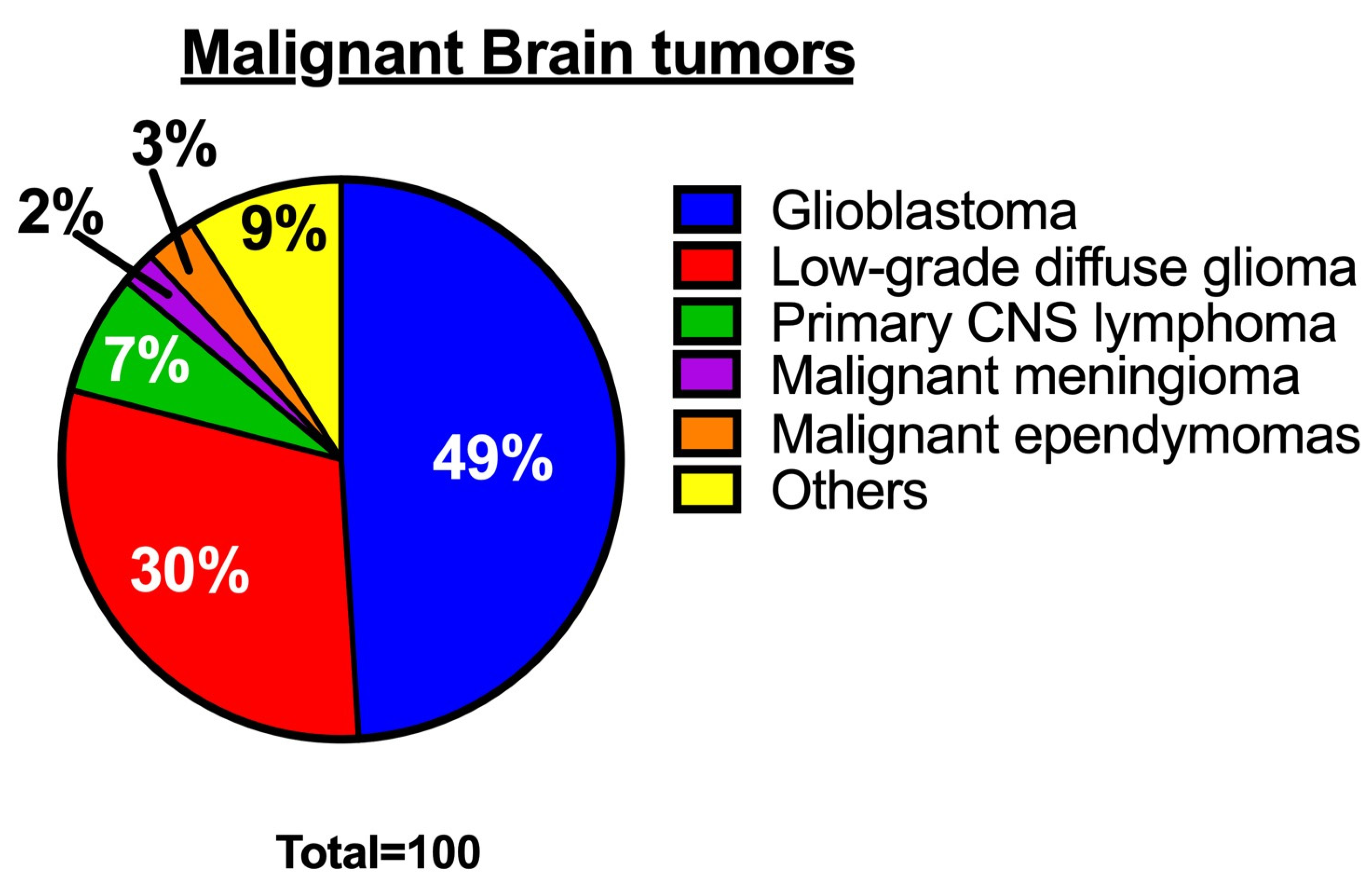
1. Introduction
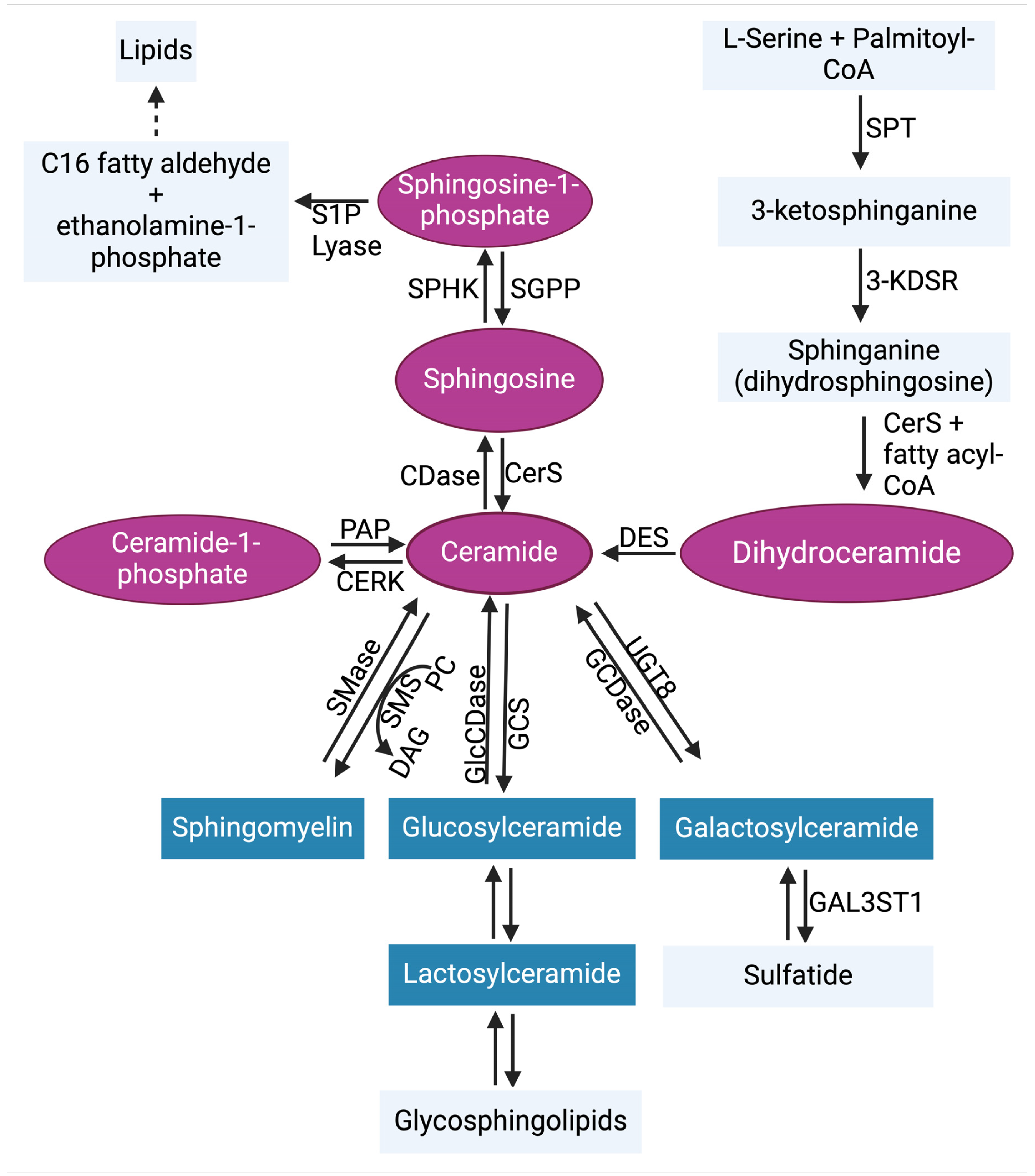
2. Sphingolipid Metabolism in Glioblastoma
2.1. Ceramides
2.2. Sphingosine-1-Phosphate (S1P) Signaling
3. Complement Signaling in Glioblastoma
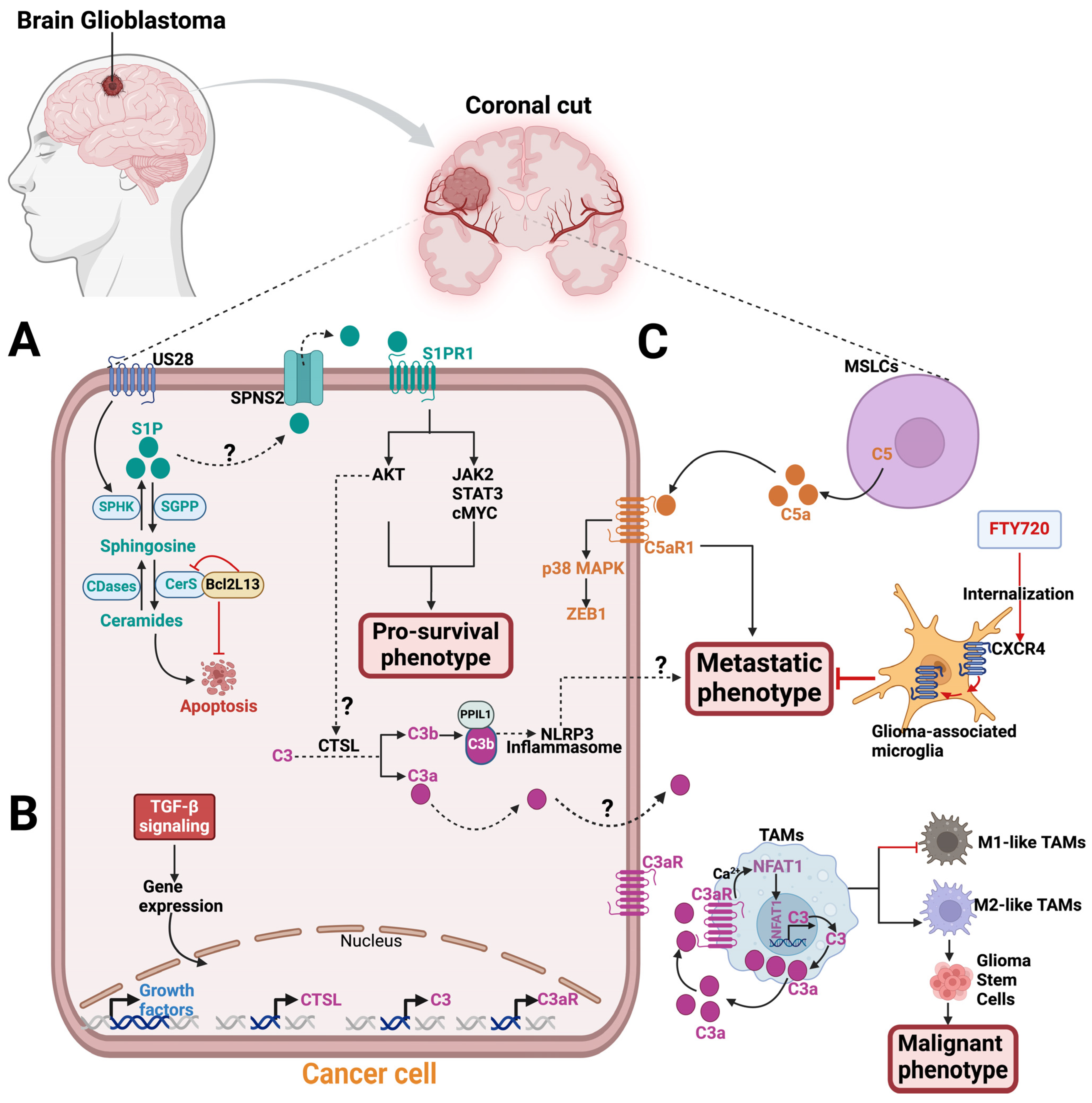
4. Crosstalk between Sphingolipid Metabolism and Complement Signaling
5. Targeting Sphingolipids and the Complement System in Glioblastoma
6. Conclusions and Future Directions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schaff, L.R.; Mellinghoff, I.K. Glioblastoma and other primary brain malignancies in adults: A review. JAMA 2023, 329, 574–587. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group, Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K. Effect of tumor-treating fields plus maintenance temozolomide vs. maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef]
- Ah-Pine, F.; Khettab, M.; Bedoui, Y.; Slama, Y.; Daniel, M.; Doray, B.; Gasque, P. On the origin and development of glioblastoma: Multifaceted role of perivascular mesenchymal stromal cells. Acta Neuropathol. Commun. 2023, 11, 104. [Google Scholar] [CrossRef]
- Kim, H.J.; Park, J.W.; Lee, J.H. Genetic architectures and cell-of-origin in glioblastoma. Front. Oncol. 2021, 10, 615400. [Google Scholar] [CrossRef]
- Greenwald, A.C.; Darnell, N.G.; Hoefflin, R.; Simkin, D.; Mount, C.W.; Castro, L.N.G.; Harnik, Y.; Dumont, S.; Hirsch, D.; Nomura, M. Integrative spatial analysis reveals a multi-layered organization of glioblastoma. Cell 2024, 187, 2485–2501.e2426. [Google Scholar] [CrossRef]
- Wang, W.; Li, T.; Cheng, Y.; Li, F.; Qi, S.; Mao, M.; Wu, J.; Liu, Q.; Zhang, X.; Li, X. Identification of hypoxic macrophages in glioblastoma with therapeutic potential for vasculature normalization. Cancer Cell 2024, 42, 815–832.e12. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2014–2018. Neuro-Oncology 2021, 23, iii1–iii105. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.; Pfister, S.M.; Reifenberger, G. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Hamad, A.; Yusubalieva, G.M.; Baklaushev, V.P.; Chumakov, P.M.; Lipatova, A.V. Recent developments in glioblastoma therapy: Oncolytic viruses and emerging future strategies. Viruses 2023, 15, 547. [Google Scholar] [CrossRef]
- Brat, D.J.; Aldape, K.; Colman, H.; Holland, E.C.; Louis, D.N.; Jenkins, R.B.; Kleinschmidt-DeMasters, B.; Perry, A.; Reifenberger, G.; Stupp, R. cIMPACT-NOW update 3: Recommended diagnostic criteria for “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV”. Acta Neuropathol. 2018, 136, 805–810. [Google Scholar] [CrossRef]
- Valentinis, L.; Tuniz, F.; Valent, F.; Mucchiut, M.; Little, D.; Skrap, M.; Bergonzi, P.; Zanchin, G. Headache attributed to intracranial tumours: A prospective cohort study. Cephalalgia 2010, 30, 389–398. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2015–2019. Neuro-Oncology 2022, 24, v1–v95. [Google Scholar] [CrossRef]
- Chen, H.; Judkins, J.; Thomas, C.; Wu, M.; Khoury, L.; Benjamin, C.G.; Pacione, D.; Golfinos, J.G.; Kumthekar, P.; Ghamsari, F. Mutant IDH1 and seizures in patients with glioma. Neurology 2017, 88, 1805–1813. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; Van Den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Walker, M.D.; Green, S.B.; Byar, D.P.; Alexander, E., Jr.; Batzdorf, U.; Brooks, W.H.; Hunt, W.E.; MacCarty, C.S.; Mahaley, M.S., Jr.; Mealey, J., Jr. Randomized comparisons of radiotherapy and nitrosoureas for the treatment of malignant glioma after surgery. N. Engl. J. Med. 1980, 303, 1323–1329. [Google Scholar] [CrossRef]
- Brown, T.J.; Brennan, M.C.; Li, M.; Church, E.W.; Brandmeir, N.J.; Rakszawski, K.L.; Patel, A.S.; Rizk, E.B.; Suki, D.; Sawaya, R. Association of the extent of resection with survival in glioblastoma: A systematic review and meta-analysis. JAMA Oncol. 2016, 2, 1460–1469. [Google Scholar] [CrossRef]
- Molinaro, A.M.; Hervey-Jumper, S.; Morshed, R.A.; Young, J.; Han, S.J.; Chunduru, P.; Zhang, Y.; Phillips, J.J.; Shai, A.; Lafontaine, M. Association of maximal extent of resection of contrast-enhanced and non–contrast-enhanced tumor with survival within molecular subgroups of patients with newly diagnosed glioblastoma. JAMA Oncol. 2020, 6, 495–503. [Google Scholar] [CrossRef]
- Niyazi, M.; Brada, M.; Chalmers, A.J.; Combs, S.E.; Erridge, S.C.; Fiorentino, A.; Grosu, A.L.; Lagerwaard, F.J.; Minniti, G.; Mirimanoff, R.-O. ESTRO-ACROP guideline “target delineation of glioblastomas”. Radiother. Oncol. 2016, 118, 35–42. [Google Scholar] [CrossRef]
- Wernicke, A.G.; Smith, A.W.; Taube, S.; Mehta, M.P. Glioblastoma: Radiation treatment margins, how small is large enough? Pract. Radiat. Oncol. 2016, 6, 298–305. [Google Scholar] [CrossRef]
- Minniti, G.; Niyazi, M.; Alongi, F.; Navarria, P.; Belka, C. Current status and recent advances in reirradiation of glioblastoma. Radiat. Oncol. 2021, 16, 36. [Google Scholar] [CrossRef]
- Ostermann, S.; Csajka, C.; Buclin, T.; Leyvraz, S.; Lejeune, F.; Decosterd, L.A.; Stupp, R. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin. Cancer Res. 2004, 10, 3728–3736. [Google Scholar] [CrossRef]
- Stupp, R.; Dietrich, P.-Y.; Kraljevic, S.O.; Pica, A.; Maillard, I.; Maeder, P.; Meuli, R.; Janzer, R.; Pizzolato, G.; Miralbell, R. Promising survival for patients with newly diagnosed glioblastoma multiforme treated with concomitant radiation plus temozolomide followed by adjuvant temozolomide. J. Clin. Oncol. 2002, 20, 1375–1382. [Google Scholar] [CrossRef]
- Hegi, M.E.; Genbrugge, E.; Gorlia, T.; Stupp, R.; Gilbert, M.R.; Chinot, O.L.; Nabors, L.B.; Jones, G.; Van Criekinge, W.; Straub, J. MGMT promoter methylation cutoff with safety margin for selecting glioblastoma patients into trials omitting temozolomide: A pooled analysis of four clinical trials. Clin. Cancer Res. 2019, 25, 1809–1816. [Google Scholar] [CrossRef]
- Tea, M.N.; Poonnoose, S.I.; Pitson, S.M. Targeting the sphingolipid system as a therapeutic direction for glioblastoma. Cancers 2020, 12, 111. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Mellinghoff, I.K.; Cloughesy, T.F. Balancing risk and efficiency in drug development for rare and challenging tumors: A new paradigm for glioma. J. Clin. Oncol. 2022, 40, 3510. [Google Scholar] [CrossRef]
- Omuro, A.; Brandes, A.A.; Carpentier, A.F.; Idbaih, A.; Reardon, D.A.; Cloughesy, T.; Sumrall, A.; Baehring, J.; van den Bent, M.; Bähr, O. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: An international randomized phase III trial. Neuro-Oncology 2023, 25, 123–134. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O. Effect of nivolumab vs. bevacizumab in patients with recurrent glioblastoma: The CheckMate 143 phase 3 randomized clinical trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Janneh, A.H.; Kassir, M.F.; Dwyer, C.J.; Chakraborty, P.; Pierce, J.S.; Flume, P.A.; Li, H.; Nadig, S.N.; Mehrotra, S.; Ogretmen, B. Alterations of lipid metabolism provide serologic biomarkers for the detection of asymptomatic versus symptomatic COVID-19 patients. Sci. Rep. 2021, 11, 14232. [Google Scholar] [CrossRef]
- Yang, D.; Wang, X.; Zhang, L.; Fang, Y.; Zheng, Q.; Liu, X.; Yu, W.; Chen, S.; Ying, J.; Hua, F. Lipid metabolism and storage in neuroglia: Role in brain development and neurodegenerative diseases. Cell Biosci. 2022, 12, 106. [Google Scholar] [CrossRef]
- Venkataraman, K.; Riebeling, C.; Bodennec, J.; Riezman, H.; Allegood, J.C.; Sullards, M.C.; Merrill, A.H.; Futerman, A.H. Upstream of growth and differentiation factor 1 (uog1), a mammalian homolog of the yeast Longevity Assurance Gene 1 (LAG1), regulatesN-Stearoyl-sphinganine (C18-(Dihydro) ceramide) synthesis in a fumonisin B1-independent manner in mammalian cells. J. Biol. Chem. 2002, 277, 35642–35649. [Google Scholar] [CrossRef]
- Bandhuvula, P.; Saba, J.D. Sphingosine-1-phosphate lyase in immunity and cancer: Silencing the siren. Trends Mol. Med. 2007, 13, 210–217. [Google Scholar] [CrossRef]
- Zamora-Pineda, J.; Kumar, A.; Suh, J.H.; Zhang, M.; Saba, J.D. Dendritic cell sphingosine-1-phosphate lyase regulates thymic egress. J. Exp. Med. 2016, 213, 2773–2791. [Google Scholar] [CrossRef]
- Bielawski, J.; Pierce, J.S.; Snider, J.; Rembiesa, B.; Szulc, Z.M.; Bielawska, A. Sphingolipid analysis by high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS). In Sphingolipids as Signaling and Regulatory Molecules; Springer: New York, NY, USA, 2010; pp. 46–59. [Google Scholar]
- Scherer, M.; Leuthäuser-Jaschinski, K.; Ecker, J.; Schmitz, G.; Liebisch, G. A rapid and quantitative LC-MS/MS method to profile sphingolipids. J. Lipid Res. 2010, 51, 2001–2011. [Google Scholar] [CrossRef]
- Haynes, C.A.; Allegood, J.C.; Park, H.; Sullards, M.C. Sphingolipidomics: Methods for the comprehensive analysis of sphingolipids. J. Chromatogr. B 2009, 877, 2696–2708. [Google Scholar] [CrossRef]
- Bielawska, A.; Perry, D.K.; Hannun, Y.A. Determination of ceramides and diglycerides by the diglyceride kinase assay. Anal. Biochem. 2001, 298, 141–150. [Google Scholar] [CrossRef]
- Edsall, L.C.; Spiegel, S. Enzymatic measurement of sphingosine 1-phosphate. Anal. Biochem. 1999, 272, 80–86. [Google Scholar] [CrossRef]
- Merrill, A.H., Jr.; Wang, E.; Mullins, R.E.; Jamison, W.C.L.; Nimkar, S.; Liotta, D.C. Quantitation of free sphingosine in liver by high-performance liquid chromatography. Anal. Biochem. 1988, 171, 373–381. [Google Scholar] [CrossRef]
- Caligan, T.B.; Peters, K.; Ou, J.; Wang, E.; Saba, J.; Merrill, A.H., Jr. A high-performance liquid chromatographic method to measure sphingosine 1-phosphate and related compounds from sphingosine kinase assays and other biological samples. Anal. Biochem. 2000, 281, 36–44. [Google Scholar] [CrossRef]
- Janneh, A.H.; Ogretmen, B. Targeting sphingolipid metabolism as a therapeutic strategy in cancer treatment. Cancers 2022, 14, 2183. [Google Scholar] [CrossRef]
- Ogretmen, B.; Hannun, Y.A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 2004, 4, 604–616. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Bartke, N.; Hannun, Y.A. Bioactive sphingolipids: Metabolism and function. J. Lipid Res. 2009, 50, S91–S96. [Google Scholar] [CrossRef]
- Janneh, A.H.; Atkinson, C.; Tomlinson, S.; Ogretmen, B. Sphingolipid metabolism and complement signaling in cancer progression. Trends Cancer 2023, 9, 782–787. [Google Scholar] [CrossRef]
- O’Brien, R.M.; Cannon, A.; Reynolds, J.V.; Lysaght, J.; Lynam-Lennon, N. Complement in tumourigenesis and the response to cancer therapy. Cancers 2021, 13, 1209. [Google Scholar] [CrossRef]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef]
- Klos, A.; Tenner, A.J.; Johswich, K.-O.; Ager, R.R.; Reis, E.S.; Köhl, J. The role of the anaphylatoxins in health and disease. Mol. Immunol. 2009, 46, 2753–2766. [Google Scholar] [CrossRef]
- Maffia, P.; Mauro, C.; Case, A.; Kemper, C. Canonical and non-canonical roles of complement in atherosclerosis. Nat. Rev. Cardiol. 2024, 1–19. [Google Scholar] [CrossRef]
- Forneris, F.; Wu, J.; Gros, P. The modular serine proteases of the complement cascade. Curr. Opin. Struct. Biol. 2012, 22, 333–341. [Google Scholar] [CrossRef]
- West, E.E.; Kemper, C. Complosome—The intracellular complement system. Nat. Rev. Nephrol. 2023, 19, 426–439. [Google Scholar] [CrossRef]
- Liszewski, M.K.; Kolev, M.; Le Friec, G.; Leung, M.; Bertram, P.G.; Fara, A.F.; Subias, M.; Pickering, M.C.; Drouet, C.; Meri, S. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity 2013, 39, 1143–1157. [Google Scholar] [CrossRef]
- Janneh, A.H.; Kassir, M.F.; Atilgan, F.C.; Lee, H.G.; Sheridan, M.; Oleinik, N.; Szulc, Z.; Voelkel-Johnson, C.; Nguyen, H.; Li, H. Crosstalk between pro-survival sphingolipid metabolism and complement signaling induces inflammasome-mediated tumor metastasis. Cell Rep. 2022, 41, 111742. [Google Scholar] [CrossRef]
- Ding, P.; Xu, Y.; Li, L.; Lv, X.; Li, L.; Chen, J.; Zhou, D.; Wang, X.; Wang, Q.; Zhang, W. Intracellular complement C5a/C5aR1 stabilizes β-catenin to promote colorectal tumorigenesis. Cell Rep. 2022, 39, 110851. [Google Scholar] [CrossRef]
- Frade, R.; Rodrigues-Lima, F.; Huang, S.; Xie, K.; Guillaume, N.; Bar-Eli, M. Procathepsin-L, a proteinase that cleaves human C3 (the third component of complement), confers high tumorigenic and metastatic properties to human melanoma cells. Cancer Res. 1998, 58, 2733–2736. [Google Scholar] [CrossRef]
- Jean, D.; Bar-Eli, M.; Huang, S.; Xie, K.; Rodrigues-Lima, F.; Hermann, J.; Frade, R. A cysteine proteinase, which cleaves human C3, the third component of complement, is involved in tumorigenicity and metastasis of human melanoma. Cancer Res. 1996, 56, 254–258. [Google Scholar]
- Markiewski, M.M.; DeAngelis, R.A.; Benencia, F.; Ricklin-Lichtsteiner, S.K.; Koutoulaki, A.; Gerard, C.; Coukos, G.; Lambris, J.D. Modulation of the antitumor immune response by complement. Nat. Immunol. 2008, 9, 1225–1235. [Google Scholar] [CrossRef]
- Roumenina, L.T.; Daugan, M.V.; Petitprez, F.; Sautès-Fridman, C.; Fridman, W.H. Context-dependent roles of complement in cancer. Nat. Rev. Cancer 2019, 19, 698–715. [Google Scholar] [CrossRef]
- Roumenina, L.T.; Daugan, M.V.; Noé, R.; Petitprez, F.; Vano, Y.A.; Sanchez-Salas, R.; Becht, E.; Meilleroux, J.; Clec’h, B.L.; Giraldo, N.A. Tumor Cells Hijack Macrophage-Produced Complement C1q to Promote Tumor Growth. Cancer Immunol. Res. 2019, 7, 1091–1105. [Google Scholar] [CrossRef]
- Song, W.-C. Crosstalk between complement and toll-like receptors. Toxicol. Pathol. 2012, 40, 174–182. [Google Scholar] [CrossRef]
- Amara, U.; Rittirsch, D.; Flierl, M.; Bruckner, U.; Klos, A.; Gebhard, F.; Lambris, J.D.; Huber-Lang, M. Interaction between the coagulation and complement system. In Current Topics in Complement II; Springer: New York, NY, USA, 2008; pp. 68–76. [Google Scholar]
- Kolev, M.; Friec, G.L.; Kemper, C. Complement—Tapping into new sites and effector systems. Nat. Rev. Immunol. 2014, 14, 811–820. [Google Scholar] [CrossRef]
- Arbore, G.; West, E.E.; Spolski, R.; Robertson, A.A.; Klos, A.; Rheinheimer, C.; Dutow, P.; Woodruff, T.M.; Yu, Z.X.; O’Neill, L.A. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science 2016, 352, aad1210. [Google Scholar] [CrossRef]
- King, B.C.; Kulak, K.; Krus, U.; Rosberg, R.; Golec, E.; Wozniak, K.; Gomez, M.F.; Zhang, E.; O’Connell, D.J.; Renström, E. Complement component C3 is highly expressed in human pancreatic islets and prevents β cell death via ATG16L1 interaction and autophagy regulation. Cell Metab. 2019, 29, 202–210.e6. [Google Scholar] [CrossRef]
- Jacobs, J.; Iranpour, R.; Behrooz, A.B.; da Silva Rosa, S.C.; Ghavami, S. The role of BCL2L13 in Glioblastoma: Turning a need into a target. Biochem. Cell Biol. 2023, 102, 2. [Google Scholar] [CrossRef]
- Taïb, B.; Aboussalah, A.M.; Moniruzzaman, M.; Chen, S.; Haughey, N.J.; Kim, S.F.; Ahima, R.S. Lipid accumulation and oxidation in glioblastoma multiforme. Sci. Rep. 2019, 9, 19593. [Google Scholar] [CrossRef]
- Garcia, J.H.; Akins, E.A.; Jain, S.; Wolf, K.J.; Zhang, J.; Choudhary, N.; Lad, M.; Shukla, P.; Rios, J.; Seo, K. Multiomic screening of invasive GBM cells reveals targetable transsulfuration pathway alterations. J. Clin. Investig. 2024, 134, e170397. [Google Scholar] [CrossRef]
- Senkal, C.E.; Salama, M.F.; Snider, A.J.; Allopenna, J.J.; Rana, N.A.; Koller, A.; Hannun, Y.A.; Obeid, L.M. Ceramide is metabolized to acylceramide and stored in lipid droplets. Cell Metab. 2017, 25, 686–697. [Google Scholar] [CrossRef]
- Sentelle, R.D.; Senkal, C.E.; Jiang, W.; Ponnusamy, S.; Gencer, S.; Panneer Selvam, S.; Ramshesh, V.K.; Peterson, Y.K.; Lemasters, J.J.; Szulc, Z.M. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 2012, 8, 831–838. [Google Scholar] [CrossRef]
- Obeid, L.M.; Linardic, C.M.; Karolak, L.A.; Hannun, Y.A. Programmed cell death induced by ceramide. Science 1993, 259, 1769–1771. [Google Scholar] [CrossRef]
- Bose, R.; Verheij, M.; Haimovitz-Friedman, A.; Scotto, K.; Fuks, Z.; Kolesnick, R. Ceramide synthase mediates daunorubicin-induced apoptosis: An alternative mechanism for generating death signals. Cell 1995, 82, 405–414. [Google Scholar] [CrossRef]
- Mullen, T.D.; Hannun, Y.A.; Obeid, L.M. Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem. J. 2012, 441, 789–802. [Google Scholar] [CrossRef]
- Pavoine, C.; Pecker, F. Sphingomyelinases: Their regulation and roles in cardiovascular pathophysiology. Cardiovasc. Res. 2009, 82, 175–183. [Google Scholar] [CrossRef]
- Haimovitz-Friedman, A.; Kan, C.-C.; Ehleiter, D.; Persaud, R.S.; Mcloughlin, M.; Fuks, Z.; Kolesnick, R.N. Ionizing radiation acts on cellular membranes to generate ceramide and initiate apoptosis. J. Exp. Med. 1994, 180, 525–535. [Google Scholar] [CrossRef]
- Vit, J.-P.; Rosselli, F. Role of the ceramide-signaling pathways in ionizing radiation-induced apoptosis. Oncogene 2003, 22, 8645–8652. [Google Scholar] [CrossRef]
- Grassmé, H.; Riethmüller, J.; Gulbins, E. Biological aspects of ceramide-enriched membrane domains. Prog. Lipid Res. 2007, 46, 161–170. [Google Scholar] [CrossRef]
- Riboni, L.; Campanella, R.; Bassi, R.; Villani, R.; Gaini, S.M.; Martinelli-Boneschi, F.; Viani, P.; Tettamanti, G. Ceramide levels are inversely associated with malignant progression of human glial tumors. Glia 2002, 39, 105–113. [Google Scholar] [CrossRef]
- Jensen, S.A.; Calvert, A.E.; Volpert, G.; Kouri, F.M.; Hurley, L.A.; Luciano, J.P.; Wu, Y.; Chalastanis, A.; Futerman, A.H.; Stegh, A.H. Bcl2L13 is a ceramide synthase inhibitor in glioblastoma. Proc. Natl. Acad. Sci. USA 2014, 111, 5682–5687. [Google Scholar] [CrossRef]
- Yacoub, A.; Hamed, H.A.; Allegood, J.; Mitchell, C.; Spiegel, S.; Lesniak, M.S.; Ogretmen, B.; Dash, R.; Sarkar, D.; Broaddus, W.C. PERK–Dependent Regulation of Ceramide Synthase 6 and Thioredoxin Play a Key Role in mda-7/IL-24–Induced Killing of Primary Human Glioblastoma Multiforme Cells. Cancer Res. 2010, 70, 1120–1129. [Google Scholar] [CrossRef]
- Hamed, H.A.; Yacoub, A.; Park, M.A.; Archer, K.; Das, S.K.; Sarkar, D.; Grant, S.; Fisher, P.B.; Dent, P. Histone deacetylase inhibitors interact with melanoma differentiation associated-7/interleukin-24 to kill primary human glioblastoma cells. Mol. Pharmacol. 2013, 84, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wen, L.; Zhu, F.; Wang, Y.; Xie, Q.; Chen, Z.; Li, Y. Overexpression of ceramide synthase 1 increases C18-ceramide and leads to lethal autophagy in human glioma. Oncotarget 2017, 8, 104022. [Google Scholar] [CrossRef]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 2003, 4, 397–407. [Google Scholar] [CrossRef]
- Wang, P.; Yuan, Y.; Lin, W.; Zhong, H.; Xu, K.; Qi, X. Roles of sphingosine-1-phosphate signaling in cancer. Cancer Cell Int. 2019, 19, 295. [Google Scholar] [CrossRef]
- Takabe, K.; Spiegel, S. Export of sphingosine-1-phosphate and cancer progression. J. Lipid Res. 2014, 55, 1839–1846. [Google Scholar] [CrossRef]
- Abuhusain, H.J.; Matin, A.; Qiao, Q.; Shen, H.; Kain, N.; Day, B.W.; Stringer, B.W.; Daniels, B.; Laaksonen, M.A.; Teo, C. A metabolic shift favoring sphingosine 1-phosphate at the expense of ceramide controls glioblastoma angiogenesis. J. Biol. Chem. 2013, 288, 37355–37364. [Google Scholar] [CrossRef]
- Bien-Möller, S.; Lange, S.; Holm, T.; Böhm, A.; Paland, H.; Küpper, J.; Herzog, S.; Weitmann, K.; Havemann, C.; Vogelgesang, S. Expression of S1P metabolizing enzymes and receptors correlate with survival time and regulate cell migration in glioblastoma multiforme. Oncotarget 2016, 7, 13031. [Google Scholar] [CrossRef]
- Quint, K.; Stiel, N.; Neureiter, D.; Schlicker, H.U.; Nimsky, C.; Ocker, M.; Strik, H.; Kolodziej, M.A. The role of sphingosine kinase isoforms and receptors S1P1, S1P2, S1P3, and S1P5 in primary, secondary, and recurrent glioblastomas. Tumor Biol. 2014, 35, 8979–8989. [Google Scholar] [CrossRef]
- Yoshida, Y.; Nakada, M.; Sugimoto, N.; Harada, T.; Hayashi, Y.; Kita, D.; Uchiyama, N.; Hayashi, Y.; Yachie, A.; Takuwa, Y. Sphingosine-1-phosphate receptor type 1 regulates glioma cell proliferation and correlates with patient survival. Int. J. Cancer 2010, 126, 2341–2352. [Google Scholar] [CrossRef]
- Strub, G.M.; Maceyka, M.; Hait, N.C.; Milstien, S.; Spiegel, S. Extracellular and intracellular actions of sphingosine-1-phosphate. In Sphingolipids as Signaling and Regulatory Molecules; Springer: New York, NY, USA, 2010; pp. 141–155. [Google Scholar]
- Bryan, L.; Paugh, B.S.; Kapitonov, D.; Wilczynska, K.M.; Alvarez, S.M.; Singh, S.K.; Milstien, S.; Spiegel, S.; Kordula, T. Sphingosine-1-phosphate and interleukin-1 independently regulate plasminogen activator inhibitor-1 and urokinase-type plasminogen activator receptor expression in glioblastoma cells: Implications for invasiveness. Mol. Cancer Res. 2008, 6, 1469–1477. [Google Scholar] [CrossRef]
- Hawkins, C.C.; Ali, T.; Ramanadham, S.; Hjelmeland, A.B. Sphingolipid metabolism in glioblastoma and metastatic brain tumors: A review of sphingomyelinases and sphingosine-1-phosphate. Biomolecules 2020, 10, 1357. [Google Scholar] [CrossRef]
- Rostami, N.; Nikkhoo, A.; Ajjoolabady, A.; Azizi, G.; Hojjat-Farsangi, M.; Ghalamfarsa, G.; Yousefi, B.; Yousefi, M.; Jadidi-Niaragh, F. S1PR1 as a novel promising therapeutic target in cancer therapy. Mol. Diagn. Ther. 2019, 23, 467–487. [Google Scholar] [CrossRef]
- Anu, B.; Namitha, N.; Harikumar, K. S1PR1 signaling in cancer: A current perspective. Adv. Protein Chem. Struct. Biol. 2021, 125, 259–274. [Google Scholar]
- Bergkamp, N.D.; van Senten, J.R.; Brink, H.J.; Bebelman, M.P.; van den Bor, J.; Çobanoğlu, T.S.; Dinkla, K.; Köster, J.; Klau, G.; Siderius, M. A virally encoded GPCR drives glioblastoma through feed-forward activation of the SK1-S1P1 signaling axis. Sci. Signal. 2023, 16, eade6737. [Google Scholar] [CrossRef]
- Arseni, L.; Sharma, R.; Mack, N.; Nagalla, D.; Ohl, S.; Hielscher, T.; Singhal, M.; Pilz, R.; Augustin, H.; Sandhoff, R. Sphingosine-1-phosphate recruits macrophages and microglia and induces a pro-tumorigenic phenotype that favors glioma progression. Cancers 2023, 15, 479. [Google Scholar] [CrossRef]
- Bien-Möller, S.; Chen, F.; Xiao, Y.; Köppe, H.; Jedlitschky, G.; Meyer, U.; Tolksdorf, C.; Grube, M.; Marx, S.; Tzvetkov, M.V. The Putative S1PR1 Modulator ACT-209905 Impairs Growth and Migration of Glioblastoma Cells In Vitro. Cancers 2023, 15, 4273. [Google Scholar] [CrossRef]
- Young, N.; Van Brocklyn, J.R. Roles of sphingosine-1-phosphate (S1P) receptors in malignant behavior of glioma cells. Differential effects of S1P2 on cell migration and invasiveness. Exp. Cell Res. 2007, 313, 1615–1627. [Google Scholar] [CrossRef]
- Young, N.; Pearl, D.K.; Van Brocklyn, J.R. Sphingosine-1-phosphate regulates glioblastoma cell invasiveness through the urokinase plasminogen activator system and CCN1/Cyr61. Mol. Cancer Res. 2009, 7, 23–32. [Google Scholar] [CrossRef]
- Bohlson, S.S.; Tenner, A.J. Complement in the brain: Contributions to neuroprotection, neuronal plasticity, and neuroinflammation. Annu. Rev. Immunol. 2023, 41, 431–452. [Google Scholar] [CrossRef]
- Lim, E.-J.; Kim, S.; Oh, Y.; Suh, Y.; Kaushik, N.; Lee, J.-H.; Lee, H.-J.; Kim, M.-J.; Park, M.-J.; Kim, R.-K. Crosstalk between GBM cells and mesenchymal stemlike cells promotes the invasiveness of GBM through the C5a/p38/ZEB1 axis. Neuro-Oncology 2020, 22, 1452–1462. [Google Scholar] [CrossRef]
- Broekman, M.L.; Maas, S.L.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef]
- Zhu, H.; Yu, X.; Zhang, S.; Shu, K. Targeting the complement pathway in malignant glioma microenvironments. Front. Cell Dev. Biol. 2021, 9, 657472. [Google Scholar] [CrossRef]
- Sharma, P.; Aaroe, A.; Liang, J.; Puduvalli, V.K. Tumor microenvironment in glioblastoma: Current and emerging concepts. Neuro-Oncol. Adv. 2023, 5, vdad009. [Google Scholar] [CrossRef]
- Bouwens, T.; Trouw, L.; Veerhuis, R.; Dirven, C.; Lamfers, M.; Al-Khawaja, H. Complement activation in Glioblastoma multiforme pathophysiology: Evidence from serum levels and presence of complement activation products in tumor tissue. J. Neuroimmunol. 2015, 278, 271–276. [Google Scholar] [CrossRef]
- Mangogna, A.; Belmonte, B.; Agostinis, C.; Zacchi, P.; Iacopino, D.G.; Martorana, A.; Rodolico, V.; Bonazza, D.; Zanconati, F.; Kishore, U. Prognostic implications of the complement protein C1q in gliomas. Front. Immunol. 2019, 10, 2366. [Google Scholar] [CrossRef]
- Mäkelä, K.; Helén, P.; Haapasalo, H.; Paavonen, T. Complement activation in astrocytomas: Deposition of C4d and patient outcome. BMC Cancer 2012, 12, 565. [Google Scholar] [CrossRef]
- Jonsson, K.F.; Liljedahl, E.; Osther, K.; Bengzon, J.; Skattum, L.M.; Redebrandt, H.N. Complement Components in Peripheral Blood from Adult Patients with IDH Wild-Type Glioblastoma. World Neurosurg. 2023, 177, e742–e747. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Ah-Pine, F.; Malaterre-Septembre, A.; Bedoui, Y.; Khettab, M.; Neal, J.W.; Freppel, S.; Gasque, P. Complement activation and up-regulated expression of anaphylatoxin C3a/C3aR in glioblastoma: Deciphering the links with TGF-β and VEGF. Cancers 2023, 15, 2647. [Google Scholar] [CrossRef]
- Zhang, Y.; Song, Y.; Wang, X.; Shi, M.; Lin, Y.; Tao, D.; Han, S. An NFAT1-C3a-C3aR Positive Feedback Loop in Tumor-Associated Macrophages Promotes a Glioma Stem Cell Malignant Phenotype. Cancer Immunol. Res. 2024, 12, 363–376. [Google Scholar] [CrossRef]
- Kim, S.-M.; Kang, S.-G.; Park, N.-R.; Mok, H.-S.; Huh, Y.-M.; Lee, S.-J.; Jeun, S.-S.; Hong, Y.-K.; Park, C.-K.; Lang, F.F. Presence of glioma stroma mesenchymal stem cells in a murine orthotopic glioma model. Child’s Nerv. Syst. 2011, 27, 911–922. [Google Scholar] [CrossRef]
- Ho, C.-M.; Chang, S.-F.; Hsiao, C.-C.; Chien, T.-Y.; Shih, D.T.-B. Isolation and characterization of stromal progenitor cells from ascites of patients with epithelial ovarian adenocarcinoma. J. Biomed. Sci. 2012, 19, 23. [Google Scholar] [CrossRef]
- Jang, C.; Cho, B.-K.; Hwang, S.H.; Shin, H.J.; Yoon, S.H. Leptomeningeal spread at the diagnosis of glioblastoma multiforme: A case report and literature review. Brain Tumor Res. Treat. 2022, 10, 183. [Google Scholar] [CrossRef]
- Birzu, C.; Tran, S.; Bielle, F.; Touat, M.; Mokhtari, K.; Younan, N.; Psimaras, D.; Hoang-Xuan, K.; Sanson, M.; Delattre, J.Y. Leptomeningeal spread in glioblastoma: Diagnostic and therapeutic challenges. Oncologist 2020, 25, e1763–e1776. [Google Scholar] [CrossRef]
- Autran, D.; Barrie, M.; Matta, M.; Monserrat, C.; Campello, C.; Petrirena, G.; Boucard, C.; Padovani, L.; Loundou, A.; Appay, R. Leptomeningeal gliomatosis: A single institution study of 31 patients. Anticancer Res. 2019, 39, 1035–1041. [Google Scholar] [CrossRef]
- Boire, A.; Zou, Y.; Shieh, J.; Macalinao, D.G.; Pentsova, E.; Massagué, J. Complement component 3 adapts the cerebrospinal fluid for leptomeningeal metastasis. Cell 2017, 168, 1101–1113.e13. [Google Scholar] [CrossRef]
- Nguyen, H.; Kuril, S.; Bastian, D.; Kim, J.; Zhang, M.; Vaena, S.G.; Dany, M.; Dai, M.; Heinrichs, J.L.; Daenthanasanmak, A. Complement C3a and C5a receptors promote GVHD by suppressing mitophagy in recipient dendritic cells. JCI Insight 2018, 3, 121697. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Kim, C.; Wu, W.; Shin, D.M.; Bryndza, E.; Kucia, M.; Ratajczak, J. The role of innate immunity in trafficking of hematopoietic stem cells—An emerging link between activation of complement cascade and chemotactic gradients of bioactive sphingolipids. In Current Topics in Innate Immunity II; Springer: New York, NY, USA, 2012; pp. 37–54. [Google Scholar]
- Kim, C.H.; Wu, W.; Wysoczynski, M.; Abdel-Latif, A.; Sunkara, M.; Morris, A.; Kucia, M.; Ratajczak, J.; Ratajczak, M.Z. Conditioning for hematopoietic transplantation activates the complement cascade and induces a proteolytic environment in bone marrow: A novel role for bioactive lipids and soluble C5b-C9 as homing factors. Leukemia 2012, 26, 106–116. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Lee, H.; Wysoczynski, M.; Wan, W.; Marlicz, W.; Laughlin, M.J.; Kucia, M.; Janowska-Wieczorek, A.; Ratajczak, J. Novel insight into stem cell mobilization-plasma sphingosine-1-phosphate is a major chemoattractant that directs the egress of hematopoietic stem progenitor cells from the bone marrow and its level in peripheral blood increases during mobilization due to activation of complement cascade/membrane attack complex. Leukemia 2010, 24, 976–985. [Google Scholar]
- Ratajczak, M.Z.; Kucia, M. Hematopoiesis and innate immunity: An inseparable couple for good and bad times, bound together by an hormetic relationship. Leukemia 2022, 36, 23–32. [Google Scholar] [CrossRef]
- Lei, Y.-C.; Lu, C.-L.; Chen, L.; Ge, K.; Yang, L.-L.; Li, W.; Wu, Y.-H. C5a/C5aR pathway is essential for up-regulating SphK1 expression through p38-MAPK activation in acute liver failure. World J. Gastroenterol. 2016, 22, 10148. [Google Scholar] [CrossRef]
- Bachmaier, K.; Guzman, E.; Kawamura, T.; Gao, X.; Malik, A.B. Sphingosine kinase 1 mediation of expression of the anaphylatoxin receptor C5L2 dampens the inflammatory response to endotoxin. PLoS ONE 2012, 7, e30742. [Google Scholar] [CrossRef]
- Rao, R.P.; Yuan, C.; Allegood, J.C.; Rawat, S.S.; Edwards, M.B.; Wang, X.; Merrill, A.H., Jr.; Acharya, U.; Acharya, J.K. Ceramide transfer protein function is essential for normal oxidative stress response and lifespan. Proc. Natl. Acad. Sci. USA 2007, 104, 11364–11369. [Google Scholar] [CrossRef]
- Bode, G.H.; Losen, M.; Buurman, W.A.; Veerhuis, R.; Molenaar, P.C.; Steinbusch, H.W.; De Baets, M.H.; Daha, M.R.; Martinez-Martinez, P. Complement activation by ceramide transporter proteins. J. Immunol. 2014, 192, 1154–1161. [Google Scholar] [CrossRef]
- Rosenbloom, B.E.; Weinreb, N.J.; Zimran, A.; Kacena, K.A.; Charrow, J.; Ward, E. Gaucher disease and cancer incidence: A study from the Gaucher Registry. Blood 2005, 105, 4569–4572. [Google Scholar] [CrossRef]
- Pandey, M.K.; Burrow, T.A.; Rani, R.; Martin, L.J.; Witte, D.; Setchell, K.D.; Mckay, M.A.; Magnusen, A.F.; Zhang, W.; Liou, B. Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease. Nature 2017, 543, 108–112. [Google Scholar] [CrossRef]
- Liang, J.; Nagahashi, M.; Kim, E.Y.; Harikumar, K.B.; Yamada, A.; Huang, W.-C.; Hait, N.C.; Allegood, J.C.; Price, M.M.; Avni, D. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 2013, 23, 107–120. [Google Scholar] [CrossRef]
- Cohen, J.A.; Barkhof, F.; Comi, G.; Hartung, H.-P.; Khatri, B.O.; Montalban, X.; Pelletier, J.; Capra, R.; Gallo, P.; Izquierdo, G. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 402–415. [Google Scholar] [CrossRef]
- Kappos, L.; Radue, E.-W.; O’Connor, P.; Polman, C.; Hohlfeld, R.; Calabresi, P.; Selmaj, K.; Agoropoulou, C.; Leyk, M.; Zhang-Auberson, L. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 387–401. [Google Scholar] [CrossRef]
- Chun, J.; Hartung, H.-P. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin. Neuropharmacol. 2010, 33, 91–101. [Google Scholar] [CrossRef]
- Guo, X.-D.; Ji, J.; Xue, T.-F.; Sun, Y.-Q.; Guo, R.-B.; Cheng, H.; Sun, X.-L. FTY720 exerts anti-glioma effects by regulating the glioma microenvironment through increased CXCR4 internalization by glioma-associated microglia. Front. Immunol. 2020, 11, 178. [Google Scholar] [CrossRef]
- Kolodziej, M.; Al Barim, B.; Nagl, J.; Weigand, M.; Uhl, E.; Uhle, F.; Di Fazio, P.; Schwarm, F.; Stein, M. Sphingosine-1-phosphate analogue FTY720 exhibits a potent anti-proliferative effect on glioblastoma cells. Int. J. Oncol. 2020, 57, 1039–1046. [Google Scholar] [CrossRef]
- Sonoda, Y.; Yamamoto, D.; Sakurai, S.; Hasegawa, M.; Aizu-Yokota, E.; Momoi, T.; Kasahara, T. FTY720, a novel immunosuppressive agent, induces apoptosis in human glioma cells. Biochem. Biophys. Res. Commun. 2001, 281, 282–288. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H.; Ding, K.; Xu, J. FTY720 induces autophagy-related apoptosis and necroptosis in human glioblastoma cells. Toxicol. Lett. 2015, 236, 43–59. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H.; Zhu, J.; Ding, K.; Xu, J. FTY720 reduces migration and invasion of human glioblastoma cell lines via inhibiting the PI3K/AKT/mTOR/p70S6K signaling pathway. Tumor Biol. 2014, 35, 10707–10714. [Google Scholar] [CrossRef]
- Estrada-Bernal, A.; Palanichamy, K.; Ray Chaudhury, A.; Van Brocklyn, J.R. Induction of brain tumor stem cell apoptosis by FTY720: A potential therapeutic agent for glioblastoma. Neuro-Oncology 2012, 14, 405–415. [Google Scholar] [CrossRef]
- Afzali, B.; Noris, M.; Lambrecht, B.N.; Kemper, C. The state of complement in COVID-19. Nat. Rev. Immunol. 2022, 22, 77–84. [Google Scholar] [CrossRef]
- Mastaglio, S.; Ruggeri, A.; Risitano, A.M.; Angelillo, P.; Yancopoulou, D.; Mastellos, D.C.; Huber-Lang, M.; Piemontese, S.; Assanelli, A.; Garlanda, C. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin. Immunol. 2020, 215, 108450. [Google Scholar] [CrossRef]
- Mastellos, D.C.; da Silva, B.G.P.; Fonseca, B.A.; Fonseca, N.P.; Auxiliadora-Martins, M.; Mastaglio, S.; Ruggeri, A.; Sironi, M.; Radermacher, P.; Chrysanthopoulou, A. Complement C3 vs. C5 inhibition in severe COVID-19: Early clinical findings reveal differential biological efficacy. Clin. Immunol. 2020, 220, 108598. [Google Scholar] [CrossRef]
- Companioni, O.; Mir, C.; Garcia-Mayea, Y.; LLeonart, M.E. Targeting sphingolipids for cancer therapy. Front. Oncol. 2021, 11, 745092. [Google Scholar] [CrossRef]
- Ryland, L.K.; Doshi, U.A.; Shanmugavelandy, S.S.; Fox, T.E.; Aliaga, C.; Broeg, K.; Baab, K.T.; Young, M.; Khan, O.; Haakenson, J.K. C6-ceramide nanoliposomes target the Warburg effect in chronic lymphocytic leukemia. PLoS ONE 2013, 8, e84648. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cai, H.; Ren, L.; Yang, Y.; Yang, H.; Liu, J.; Li, S.; Zhang, Y.; Zheng, X.; Tan, W. Sphingosine kinase 1 promotes growth of glioblastoma by increasing inflammation mediated by the NF-κB/IL-6/STAT3 and JNK/PTX3 pathways. Acta Pharm. Sin. B 2022, 12, 4390–4406. [Google Scholar] [CrossRef] [PubMed]
- Sousa, N.; Geiß, C.; Bindila, L.; Lieberwirth, I.; Kim, E.; Régnier-Vigouroux, A. Targeting sphingolipid metabolism with the sphingosine kinase inhibitor SKI-II overcomes hypoxia-induced chemotherapy resistance in glioblastoma cells: Effects on cell death, self-renewal, and invasion. BMC Cancer 2023, 23, 762. [Google Scholar] [CrossRef] [PubMed]
- Arias, M.A.; Cios, K.J.; Kacsoh, D.B.; Montgomery, B.E.; Song, J.J.; Patel, A.R.; Chobrutskiy, A.; Chobrutskiy, B.I.; Blanck, G. Electrostatic Complementarities of Glioblastoma-Resident T-Cell Receptors and Cancer Testis Antigens Linked to Poor Outcomes and High Levels of Sphingosine Kinase-2 Expression. Biology 2023, 12, 575. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, H.A.; Tea, M.N.; Zebol, J.R.; Gliddon, B.L.; Stefanidis, C.; Moretti, P.A.; Pitman, M.R.; Costabile, M.; Kular, J.; Stringer, B.W. Cytoplasmic dynein regulates the subcellular localization of sphingosine kinase 2 to elicit tumor-suppressive functions in glioblastoma. Oncogene 2019, 38, 1151–1165. [Google Scholar] [CrossRef] [PubMed]
- Sordillo, L.A.; Sordillo, P.P.; Helson, L. Sphingosine kinase inhibitors as maintenance therapy of glioblastoma after ceramide-induced response. Anticancer Res. 2016, 36, 2085–2095. [Google Scholar]
- Van Brocklyn, J.R.; Jackson, C.A.; Pearl, D.K.; Kotur, M.S.; Snyder, P.J.; Prior, T.W. Sphingosine kinase-1 expression correlates with poor survival of patients with glioblastoma multiforme: Roles of sphingosine kinase isoforms in growth of glioblastoma cell lines. J. Neuropathol. Exp. Neurol. 2005, 64, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Kapitonov, D.; Allegood, J.C.; Mitchell, C.; Hait, N.C.; Almenara, J.A.; Adams, J.K.; Zipkin, R.E.; Dent, P.; Kordula, T.; Milstien, S. Targeting sphingosine kinase 1 inhibits Akt signaling, induces apoptosis, and suppresses growth of human glioblastoma cells and xenografts. Cancer Res. 2009, 69, 6915–6923. [Google Scholar] [CrossRef]
- Bektas, M.; Johnson, S.P.; Poe, W.E.; Bigner, D.D.; Friedman, H.S. A sphingosine kinase inhibitor induces cell death in temozolomide resistant glioblastoma cells. Cancer Chemother. Pharmacol. 2009, 64, 1053–1058. [Google Scholar] [CrossRef]
- Levičar, N.; Dewey, R.A.; Daley, E.; Bates, T.E.; Davies, D.; Kos, J.; Pilkington, G.J.; Lah, T.T. Selective suppression of cathepsin L by antisense cDNA impairs human brain tumor cell invasion in vitro and promotes apoptosis. Cancer Gene Ther. 2003, 10, 141–151. [Google Scholar] [CrossRef]
- Xiong, Y.; Ji, W.; Fei, Y.; Zhao, Y.; Wang, L.; Wang, W.; Han, M.; Tan, C.; Fei, X.; Huang, Q. Cathepsin L is involved in X-ray-induced invasion and migration of human glioma U251 cells. Cell. Signal. 2017, 29, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Zhang, C.; Chen, H.; Ren, M.; Liu, X. Cathepsins trigger cell death and regulate radioresistance in glioblastoma. Cells 2022, 11, 4108. [Google Scholar] [CrossRef]
- Dong, Q.; Li, Q.; Duan, L.; Wang, H.; Yan, Y.; Yin, H.; Niu, L.; Zhang, H.; Wang, B.; Yuan, G. Expressions and significances of CTSL, the target of COVID-19 on GBM. J. Cancer Res. Clin. Oncol. 2022, 148, 599–608. [Google Scholar] [CrossRef]
- Rao, J.S. Molecular mechanisms of glioma invasiveness: The role of proteases. Nat. Rev. Cancer 2003, 3, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Lu, B.; Gao, B.; Shi, Y.; Xu, J.; Yang, R.; Xu, B.; Ding, P. NLRP3 promotes glioma cell proliferation and invasion via the interleukin-1β/NF-κB p65 signals. Oncol. Res. 2019, 27, 557–564. [Google Scholar] [CrossRef]
- Sim, J.; Park, J.; Moon, J.-S.; Lim, J. Dysregulation of inflammasome activation in glioma. Cell Commun. Signal. 2023, 21, 239. [Google Scholar] [CrossRef]
- Rolim, G.B.; Lima, A.J.P.D.; dos Santos Cardoso, V.I.; de Fátima Machado Soares, É.; Nunes, D.N.; Barros, H.C.S.; Leite, A.B.; Alexandre-Moreira, M.S.; Duarte, A.W.F.; de Sales Marques, C. Can inflammasomes promote the pathophysiology of glioblastoma multiforme? A view about the potential of the anti-inflammasome therapy as pharmacological target. Crit. Rev. Oncol. Hematol. 2022, 172, 103641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Peng, Q.; Tang, Y.; Wang, C.; Wang, S.; Yu, D.; Hou, S.; Wang, Y.; Zhang, L.; Lin, N. Resveratrol ameliorates glioblastoma inflammatory response by reducing NLRP3 inflammasome activation through inhibition of the JAK2/STAT3 pathway. J. Cancer Res. Clin. Oncol. 2024, 150, 168. [Google Scholar] [CrossRef]
- Asgari, E.; Le Friec, G.; Yamamoto, H.; Perucha, E.; Sacks, S.S.; Köhl, J.; Cook, H.T.; Kemper, C. C3a modulates IL-1β secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood J. Am. Soc. Hematol. 2013, 122, 3473–3481. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janneh, A.H. Sphingolipid Signaling and Complement Activation in Glioblastoma: A Promising Avenue for Therapeutic Intervention. BioChem 2024, 4, 126-143. https://doi.org/10.3390/biochem4020007
Janneh AH. Sphingolipid Signaling and Complement Activation in Glioblastoma: A Promising Avenue for Therapeutic Intervention. BioChem. 2024; 4(2):126-143. https://doi.org/10.3390/biochem4020007
Chicago/Turabian StyleJanneh, Alhaji H. 2024. "Sphingolipid Signaling and Complement Activation in Glioblastoma: A Promising Avenue for Therapeutic Intervention" BioChem 4, no. 2: 126-143. https://doi.org/10.3390/biochem4020007
APA StyleJanneh, A. H. (2024). Sphingolipid Signaling and Complement Activation in Glioblastoma: A Promising Avenue for Therapeutic Intervention. BioChem, 4(2), 126-143. https://doi.org/10.3390/biochem4020007