1. Introduction
SARS-CoV-2 is the etiologic agent responsible for coronavirus disease 2019 (COVID-19). About 80% of patients infected by SARS-CoV-2 are asymptomatic or present mild symptoms, but they can infect other people. On the other hand, some infected individuals which are symptomatic may present more severe symptoms which could also lead to death [
1]. Symptoms appear 2–14 days after the infection. Since the virus can affect both lungs, patients present signs and symptoms which are associated with viral pneumonia (such as fever, cough, sore throat, headache, fatigue, myalgia and dyspnoea). Loss of smell or taste and gastrointestinal disorders have also been reported in some cases [
1,
2].
The genome of SARS-CoV-2 is a positive-sense single-stranded RNA composed of 29,900 nucleotides. It encodes for 29 proteins which have different roles: infection, replication and virion assembly. Similarly to all coronaviruses, SARS-CoV-2 has crown-like spikes on its surface, the spike glycoprotein (S), presenting two functional subunits: S1 and S2. The S1 subunit is responsible for the binding to the host cell receptor, while the fusion of the viral membrane with the cell is ascribable to S2. In particular, the SARS-CoV-2 spike glycoprotein interacts with human angiotensin-converting enzyme 2 (ACE2) mediating the entry into the host cell. In more detail, the virus hijacks host cell receptors via its RBD (Receptor-binding domain) which is part of the S1 portion. This spike protein differs from the SARS and MERS ones by the presence of unique N- and O-linked glycosylation sites.
After the virus has entered the cell, viral RNA is released, and it is translated into polyprotein molecules by host translational machinery. At first, nonstructural proteins (NS) are encoded and activated; they form the RNA replicase–transcriptase complex which leads to the production of negative-sense RNAs. Then, structural proteins involved in virion assembly are produced: S1, S2, envelope (E), membrane (M) and nucleocapsid (N).
SARS-CoV-2 belongs to the hazard group 3 pathogens in many countries. Therefore, inactivation of the SARS-CoV-2 virus is a mandatory step in order to handle clinical samples and perform experiments under safe conditions. Hence, the choice of a suitable method compatible with an MS-based approach is crucial. Heat inactivation is able, similarly to other protocols, to block virus infectivity or reduce its viral load, as reported in [
3,
4,
5]. Moreover, organic solvents, such as ether, ethanol, chloroform, chlorine-containing or disinfectant agents and peroxyacetic acid have been shown to efficiently inactivate SARS-CoV-2 [
6].
Life science researchers are continuously taking advantage of the high contribution of mass spectrometry (MS)-based proteomic approaches able to improve the knowledge of the proteome. The dramatic SARS-CoV-2 pandemic has strongly motivated proteomics scientists to join efforts to better understand how the new virus affects patients and develop effective therapeutic approaches and vaccines [
7,
8]. MS-based proteomics is not dependent on prior knowledge of the disease, such as those based on affinity tools, and also allows untargeted experiments. Different MS-based proteomic approaches are indeed available and suitable for specific purposes. In particular, two different MS proteomic protocols could be chosen: targeted or untargeted. In the first strategy, MS can be focused only towards the detection of specific peptides deriving from SARS-CoV-2 proteins after enzymatic digestion, while in the second one, MS is used to collect as many signals (m/z) deriving from SARS-CoV-2 peptides as possible. Moreover, proteomics is a high-throughput technique that provides a huge amount of clinical and biological information from biological specimens. It should also be emphasised that proteogenomic approaches combining mass spectrometry and RNA sequencing data facilitate detailed characterization of novel nonsynonymous mutations and their impact on protein expression, post-translational modifications or phenotypic variations [
9]. Thus, MS-based proteomics could be an ideal technology useful in situations when quick responses are required.
Moreover, host proteome profiles of multiple biological fluids from patients affected by COVID-19 have been considered in order to understand its complex pathogenesis and the host immune response against the virus [
10]. Among them, blood (serum/plasma) is mainly investigated [
11]. For instance, sera and plasma of COVID-19 patients were evaluated by LC-MS/MS to highlight possible biomarkers of the COVID-19 infection [
7,
12,
13]. Messner et al. were able to associate some proteins with COVID-19 severity [
12]. Shen et al. observed differentially expressed sera proteins of COVID-19 patients not present in non-COVID-19 patients [
13]. Similarly, Overmyer et al. detected about 220 molecules (proteins, metabolites and lipids) correlating with COVID-19 and its severity [
14]. Saliva has also emerged as a useful tool for diagnosis of SARS-CoV-2: it has been shown that this biofluid allows directly detecting the virus, quantifying specific immunoglobulins produced against the virus and evaluating the innate immune response of the patient [
15,
16]. However, MS-based proteomics in saliva aimed at the study of human proteome signatures related to SARS-CoV-2 has not been reported yet.
Several laboratories have been working to develop mass spectrometry-based tools to discover SARS-CoV-2 proteins useful for diagnostics and research purposes [
17]. Ihling et al. were able to identify peptides originating from the SARS-CoV-2 nucleoprotein by analysing gargle solutions of COVID-19 patients. These authors developed a targeted MS-based method to specifically detect SARS-CoV-2 virus proteins in highly diluted samples [
18]. Another interesting study was realised by Bezstarosti et al.; by analysing the whole proteome of Vero cells infected with SARS-CoV-2 using a bottom-up proteomic approach, they were able to detect tryptic peptides of SARS-CoV-2 proteins with a level of sensitivity sufficient to reveal proteins theoretically deriving from approximately 10,000 SARS-CoV-2 particles [
17]. However, whether this low limit of detection (LOD) could allow MS-based approaches to play a possible role in the diagnostics of COVID-19 depending on the amount of virus particles present in the patient material (such as nasopharyngeal swabs, mucus, gargle solution) needs to be further investigated. Another challenging feature of the SARS-CoV-2 proteome is the presence of N- and O- glycosylation sites that need to be considered when the spike glycoprotein is used as a marker of positivity to the virus [
19].
In this study, we investigated the ability of a method based on nLC-ESI-MS/MS to simultaneously identify specific SARS-CoV-2 proteins and characterise the host human proteome both in saliva and plasma samples with the perspective of a future application in detecting possible variants of this virus and in clustering COVID-19 patients with different outcomes (mild or severe) in a single analysis.
2. Results and Discussion
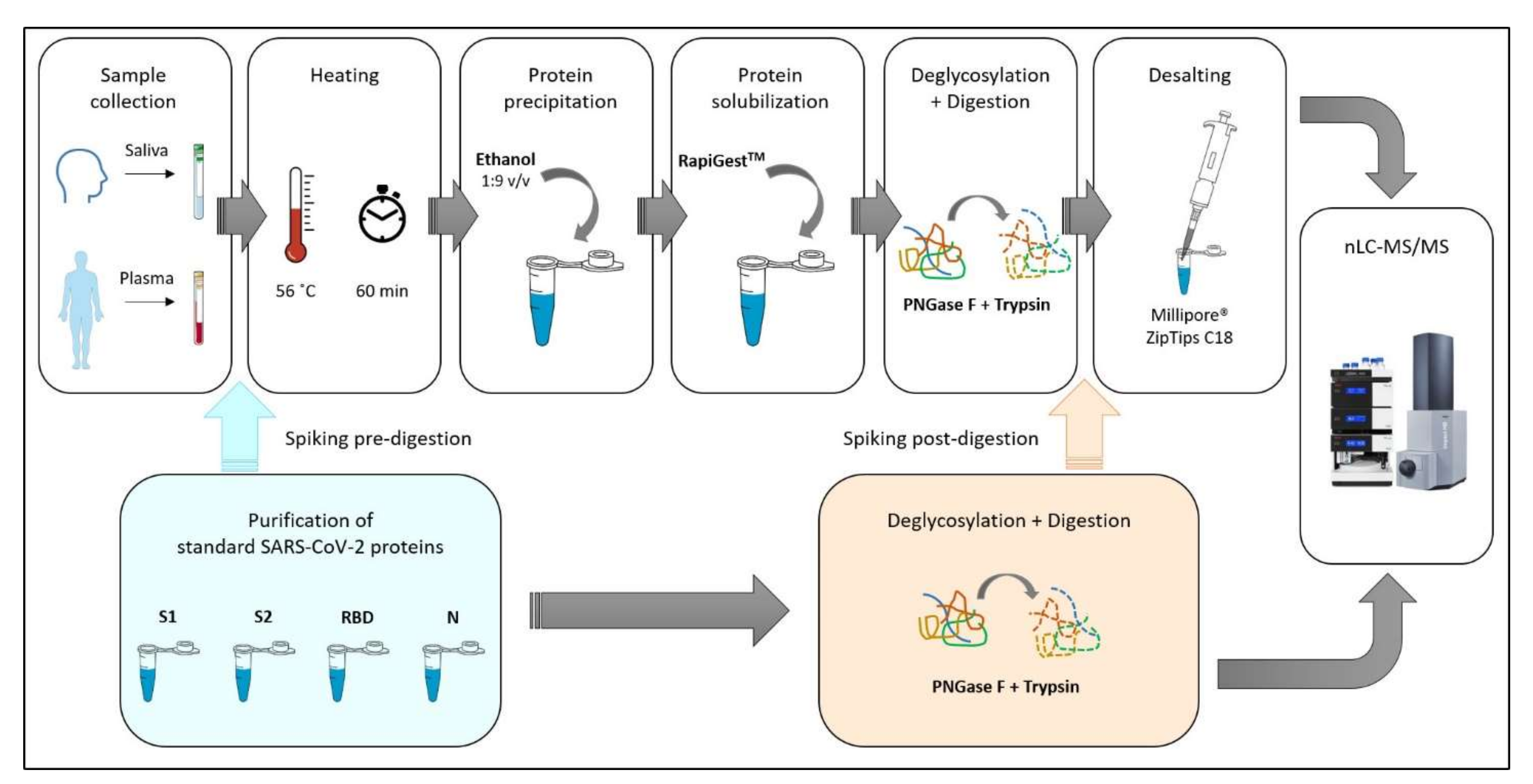
The experimental workflow was finalised to obtain a protocol able to simultaneously detect and characterise SARS-CoV-2 proteins and investigate the alterations of human saliva and plasma proteome of the patients affected by COVID-19. The work design was realised in order to have a completely safe manipulation of samples in each part of the processing, including by laboratories not equipped for biosafety risk 3.
To test the validity and the sensitivity of the method, four recombinant proteins highly expressed in SARS-CoV-2 (spike glycoprotein subunits S1 and S2, receptor-binding domain (RBD) and nucleoprotein N), were purified by Istituto Nazionale Genetica Molecolare (INGM) of Milan. As the first step, a standard solution of these proteins at a known concentration was first enzymatically digested and then injected into the nUHPLC Qq-TOF system to evaluate the limit of detection (LOD) independently from the biological matrix. Secondly, saliva and plasma spiked with different amounts of the four standard SARS-CoV-2 proteins (S1, S2, RBD, N) were injected and analysed to simultaneously investigate the limit of characterisation (LOC) of SARS-CoV-2 proteins and the human proteome. The step was conceived in order to verify the performance of the protocol simulating a real biofluid sample.
The experimental design, described in
Figure 1, was organised in different consequential steps with the following objectives: (i) establish the limit of detection with purified standard viral proteins and the maximum sequence coverage achievable; (ii) develop a method for safe manipulation of COVID-19 patient samples for proteomic applications; (iii) evaluate the matrix effect of the saliva/plasma proteome related to SARS-CoV-2 proteins identification; (iv) assess how the coverage percentage of viral proteins changes in correlation with their concentration (limit of characterization) and how it is influenced by sample manipulation; (v) evaluate the ability of the developed method to investigate the saliva and plasma proteome and its possible alterations in response to COVID-19.
2.1. Limit of Detection (LOD)
Most of the published methods rely on a targeted mass spectrometry approach, in which only selected ions of the SARS-CoV-2 proteins are monitored to improve the limit of detection [
18,
20,
21,
22]. On the contrary, we chose to set up a protocol based on an untargeted MS method, allowing the theoretical detection of all tryptic peptides, compared to the ones used by these authors, in which a multiple reaction monitoring (MRM) assay toward specific ions was used. One possible drawback of an untargeted method could be represented by the limited ability to detect proteins deriving from the virus if they are present in low amounts within complex biological specimens (such as saliva or blood). However, an untargeted approach has an attractive advantage of allowing a retrospective elaboration of the raw data for a possible detection of future viral variants since all the ions, and not only the selected ones, are acquired.
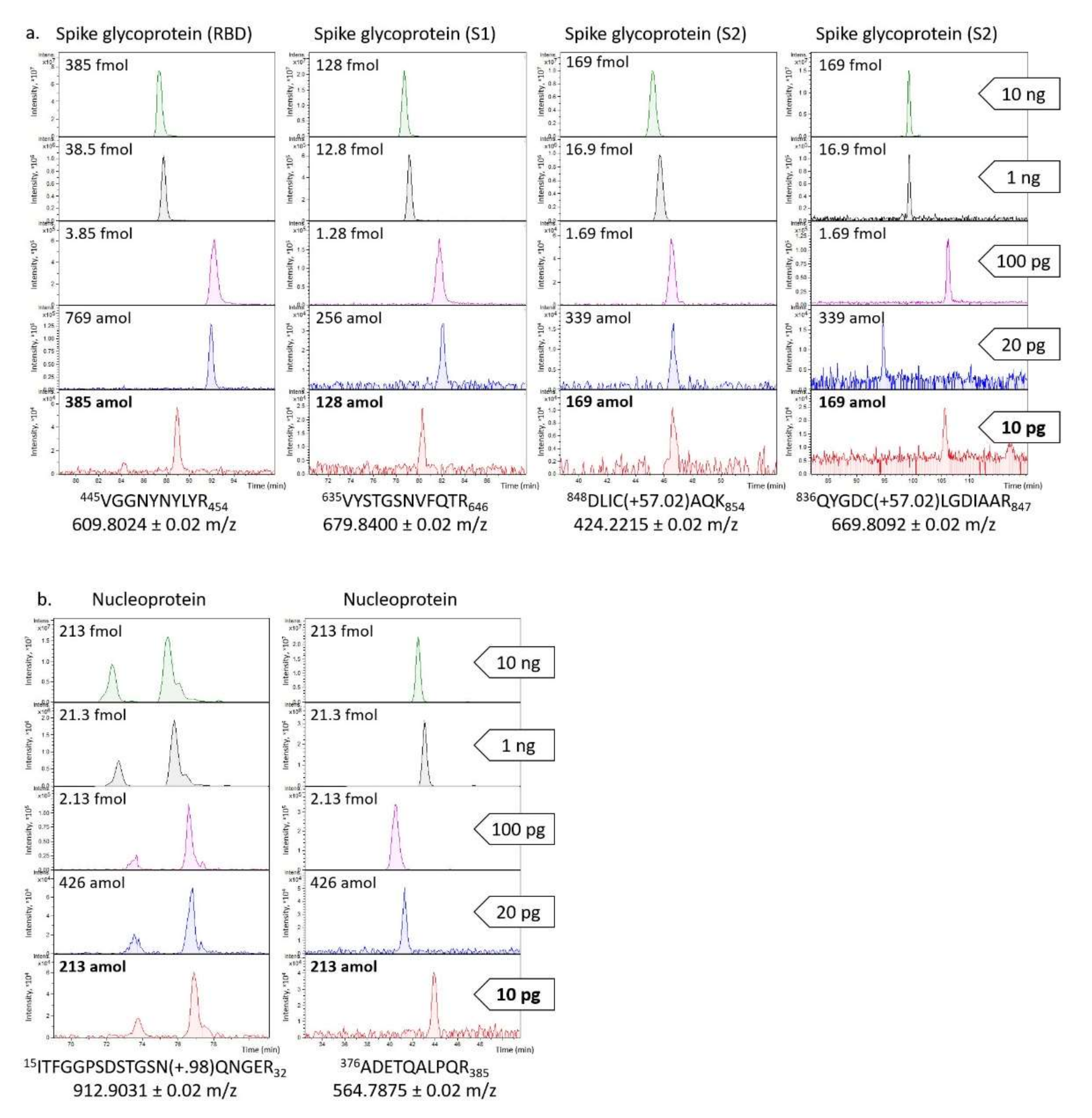
Therefore, to evaluate the limit of detection for the four viral proteins of the present untargeted method, serial dilutions of the deglycosylated and trypsinized mixture were prepared, and 10, 1, 0.1, 0.02, and 0.01 ng were analysed by nLC-ESI-MS/MS. Among the several tryptic peptides, two for the nucleoprotein (ITFGGPSDSTGSN(+0.98)QNGER and ADETQALPQR) and four for the spike glycoprotein (VGGNYNYLYR, VYSTGSNVFQTR, DLIC(+57.02)AQK and QYGDC(+57.02)LGDIAAR) were particularly intense (
Figure 2). Based on these signals, the LOD was estimated to be 10 pg (with a signal/noise ratio ≥ 3) for both proteins; it corresponds to 71 amol for the spike glycoprotein and 213 amol for the nucleoprotein. These findings are in line with those described by Van Puyvelde et al. who reported an instrumental LOD of 10 pg and 2 pg for the spike glycoprotein and the nucleoprotein, respectively [
21].
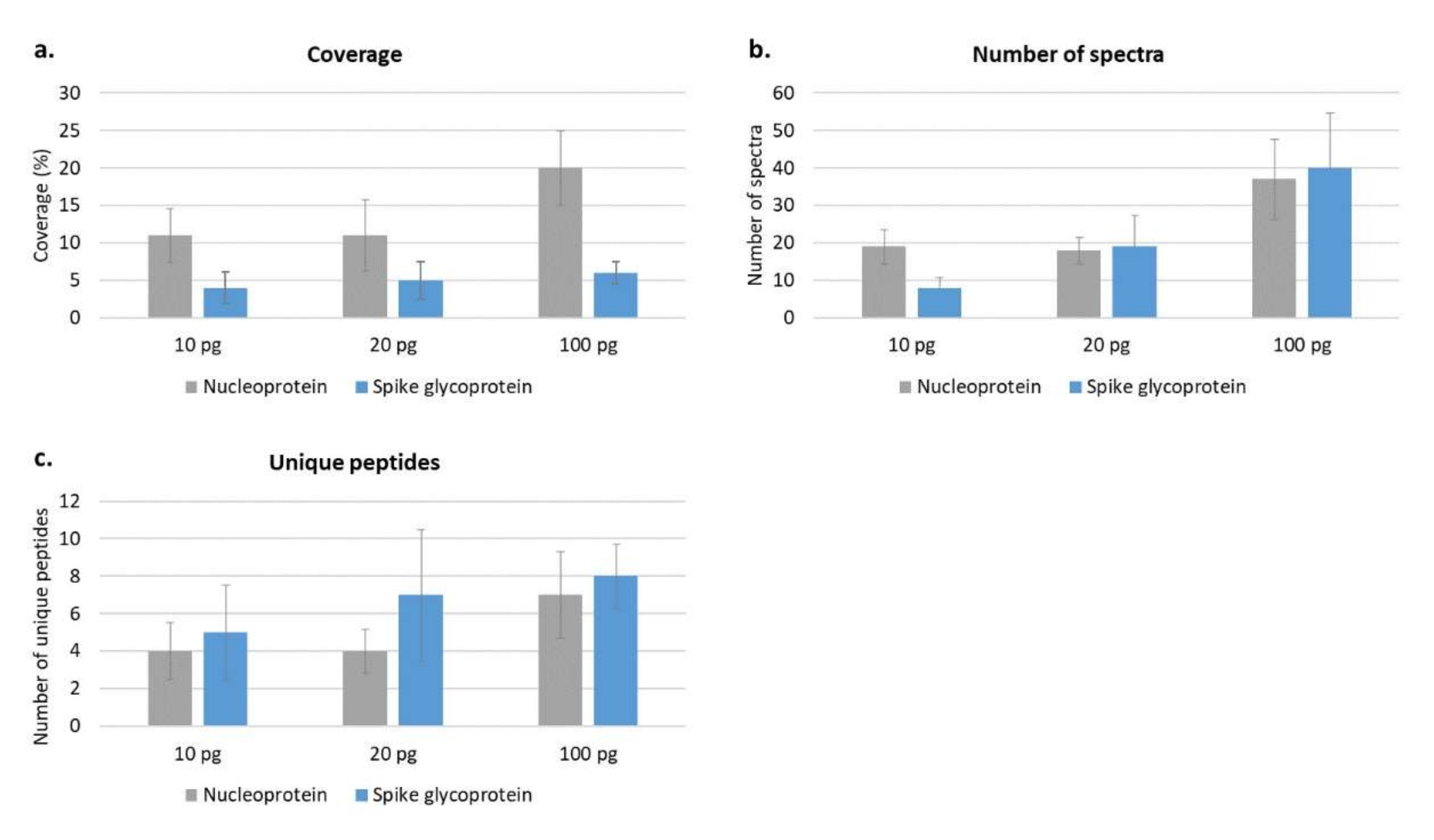
Then, to better evaluate the ability of the protocol to detect as much as possible of the amino acid sequence, the whole protein coverage was investigated at different amounts loaded into the nLC-MS/MS (from 10 ng to 10 pg). In particular, about 70% of the nucleoprotein sequence was detected at 10 ng, while this percentage decreased to 20% at 100 pg and to 11% at 10 pg. The spike glycoprotein showed a coverage of 30% at 10 ng, which decreased to 6% at 100 pg, to 5% at 20 pg and 4% at 10 pg (
Figure 3 and
Figure 4). It was observed that the nucleoprotein was identified more efficiently compared with the spike glycoprotein, especially in terms of sequence coverage percentage (
Figure 3a); this might be partly attributed to the known high level of N- and O-glycosylation brought by the spike glycoprotein [
19].
2.2. Virus Inactivation
For the safe manipulation of biological specimens, the virus had to be inactivated by heating and using an organic solvent. These approaches were chosen as inactivation methods in order to guarantee safe conditions and the minimum impact; they also did not interfere with sample processing and they did not compromise mass spectrometry analysis. Two protocols (A and B) were evaluated initially in saliva: in particular, the first one was adapted from a method already used in proteomic studies focused on SARS-CoV-2 in gargle samples, while the second one was slightly modified to improve protein recovery and make the strategy fitting for both biofluids [
18]. Protocol A consisted in an initial strong heating step at 70 ℃ for 30 min [
5] and consequent protein precipitation with acetone (1:4;
v/v) [
18,
21]; Protocol B was based on (i) the use of a mass friendly detergent for improving idrolisation quality, (ii) a milder heating at 56 ℃ for 1 h sufficient for viral inactivation [
4,
5] but with a lower extent of sample coagulation for plasma, (iii) followed by protein precipitation with ethanol (1:9;
v/v) for further enhancing the safety level of sample manipulation [
23]. PNGase F was used for both protocols to possibly improve the amino acid sequence coverage. Saliva was spiked with 1 µg of each standard viral protein and then processed with both protocols before LC-MS analysis. Protocol A resulted in a coverage of 23% for the spike glycoprotein and 28% for the nucleoprotein and allowed identifying about 255 different human protein isoforms, while Protocol B showed a slight improvement in detecting the spike glycoprotein (coverage, 28%) and the nucleoprotein (coverage, 33%), but a consistent increase in identification power nearly doubling the number of identified human proteins (554). Moreover, this last protocol also showed a greater applicability to plasma since the high temperature of inactivation used in Protocol A was observed to be associated with considerable “gelification” of the sample that prevented effective processing. According to these results, Protocol B emerged as a suitable method for virus inactivation and sample preparation for LC-MS/MS proteomic analysis both for saliva and plasma specimens.
2.3. Limits of Viral Protein Characterisation (LOC) and the Matrix Effect
The limit of characterisation was estimated as the minimum amount of protein spiked into a biofluid which is required to gather information about the amino acid sequence.
To this end, 100 µL of plasma and saliva were inactivated, deglycosylated and trypsin-digested as described in Protocol B. Then, different amounts of deglycosylated and enzymatically digested purified SARS-CoV-2 proteins were added to already processed saliva and plasma samples before MS analysis. In particular, 10, 100 and 1000 pg of each purified viral protein (S1, S2, RBD and N) spiked into 10 µL of saliva and 33, 330 and 3300 pg spiked into 10 µL of plasma were injected and run by means of nLC-MS/MS. To avoid false-positive results, only the SARS-CoV-2 proteins identified with at least one unique peptide were considered, and a database combining the human, SARS-CoV and SARS-CoV-2 proteomes was used.
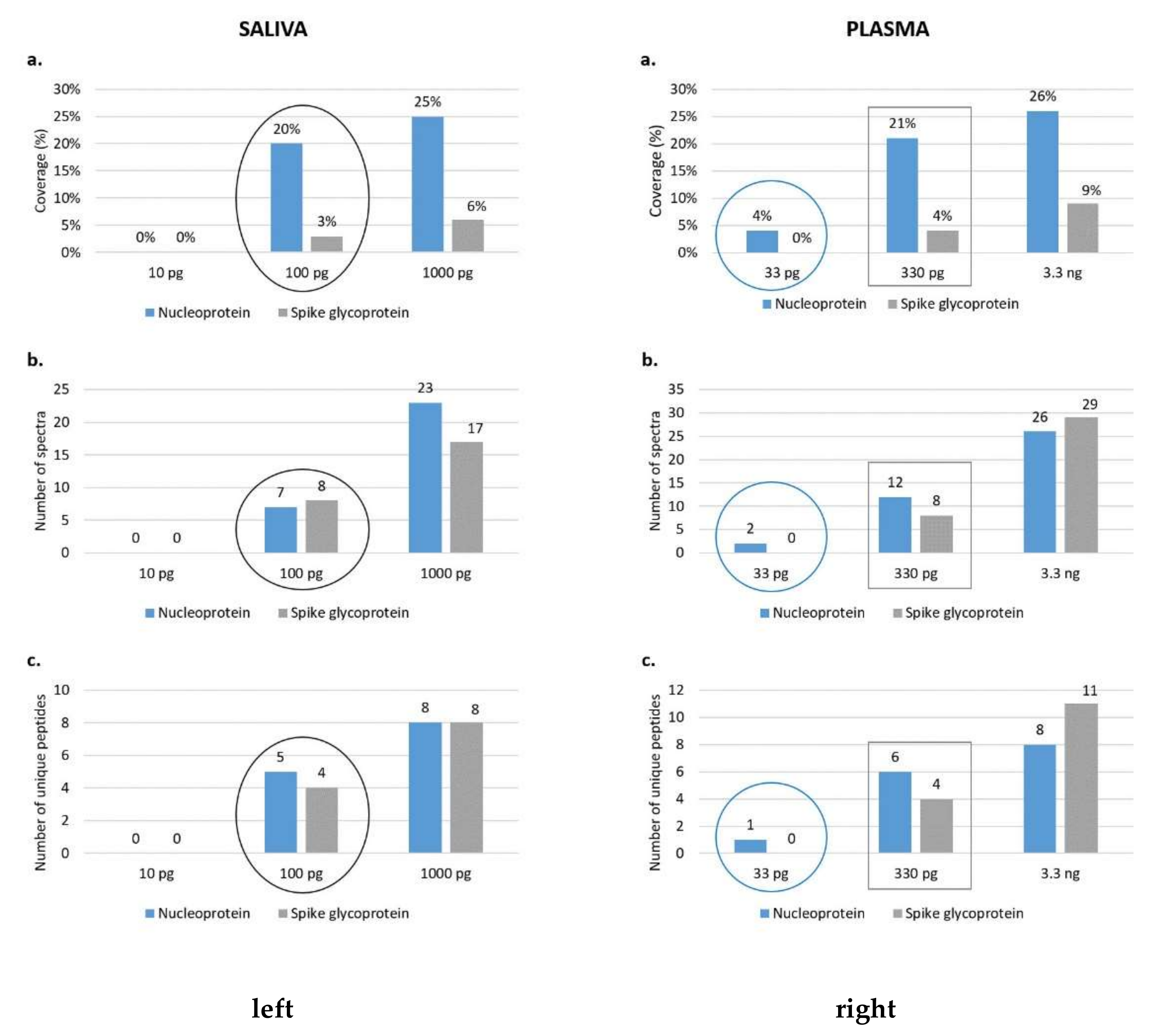
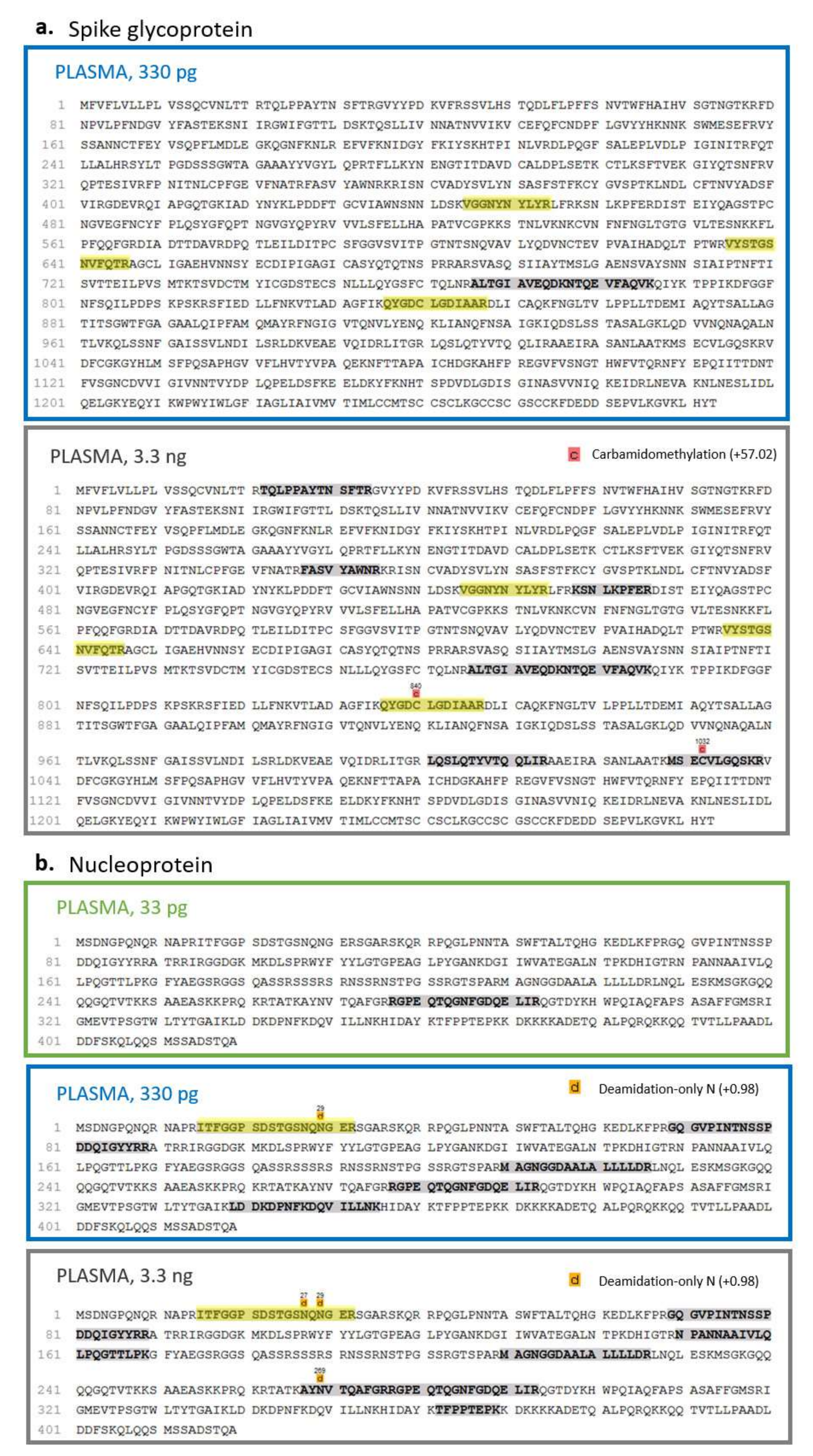
A coverage of 20% and 3% of the nucleoprotein and the spike glycoprotein, respectively, was observed by injecting 100 pg of viral standards spiked into saliva, while the viral proteins were not identified at lower quantities (
Figure 5 and
Figure 6). On the other hand, in the plasma biofluid, the LOC for the nucleoprotein was 33 pg, resulting in a sequence coverage of 4%; conversely, the spike glycoprotein was identified only after the injection of 330 pg of S1, S2 and RBD, reaching a sequence coverage of 4% (
Figure 5 and
Figure 7).
Therefore, our limit of characterization of SARS-CoV-2 proteins was lower in saliva samples than in plasma ones if we consider the spike glycoprotein. Conversely, the detection of N seemed to perform better in plasma even though the detection at 33 pg in plasma is quite low if compared to the one found at 100 pg in saliva (
Figure 5). This difference in recovery, especially related to the S protein, is likely to be attributed to a massive matrix effect, particularly evident in plasma, where an extremely high dynamic range of the proteome may mask the presence of viral proteins.
2.4. Sample Processing Effect on Method Sensitivity
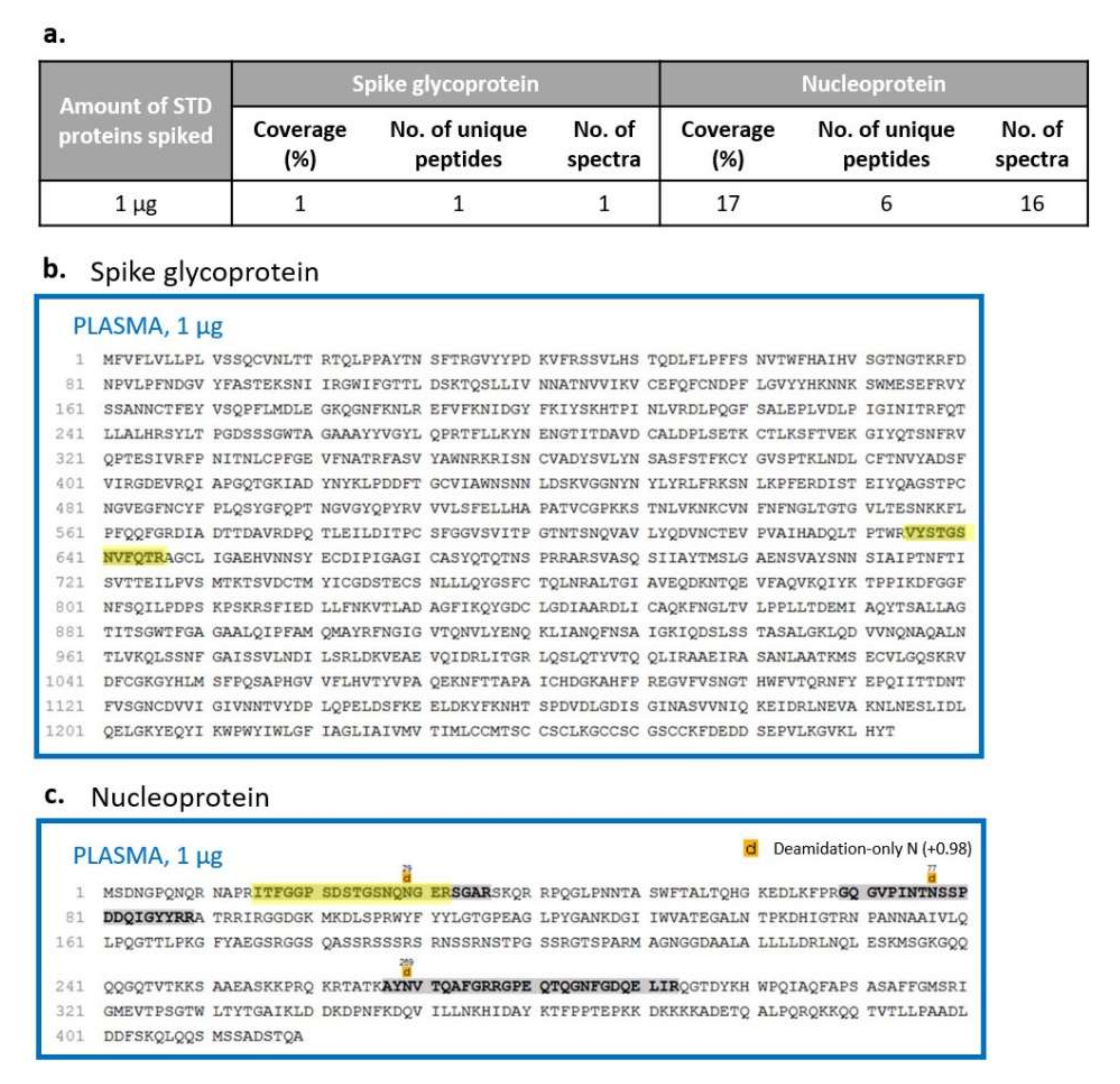
In order to simulate a real biological sample and evaluate the effect of the manipulation and processing on viral detection and characterisation beyond the component of matrix interference, we spiked saliva and plasma samples with different amounts of purified viral proteins before viral inactivation and enzymatic digestion. To avoid false-positive results, only identification with at least one unique peptide was considered, using a database combining the human, SARS-CoV and SARS-CoV-2 proteomes. Similarly to the previous results and as expected from the differences in the dynamic range of the two biofluids, the matrix effect was higher in plasma than in saliva samples. Indeed, the spike glycoprotein and the nucleoprotein were detectable when adding at least 1 µg of each purified viral protein in plasma, with a coverage of 1% for the spike glycoprotein and 17% for the nucleoprotein (
Figure 8). On the other hand, in saliva, the detection and characterisation limits were reached using a lower amount of each standard: in more detail, spiking 100 ng and 1 µg, we obtained a coverage of 10% and 28% for the spike glycoprotein and of 11% and 33% for the nucleoprotein, respectively (
Figure 9).
Then, in order to measure the impact of the sample preparation process on our sensitivity, independently from the matrix effect, the data obtained from plasma and saliva spiked before and after digestion were compared.
Considering a protein concentration of approximately 60 mg/mL in plasma [
24] and assuming an efficiency of 100% of sample digestion and processing, we estimated to inject 330 pg of each viral protein, spiking 1 µg of each one before digestion, corresponding to a ratio of 1:6000 between each purified viral protein and human proteins. Comparing this outcome with the one determined by adding the standards just before nLC-MS/MS analysis, a wider recovery of the S and N proteins was noted in the last condition, suggesting a loss of information during sample processing. In particular, the S protein was identified with four unique peptides and 4% of protein coverage if the purified standards were added at the end of sample processing (
Figure 5 and
Figure 7) while its detection decreased to one unique peptide and 1% of coverage if viral proteins were spiked at the beginning of the preparation (
Figure 8a,b). A similar behaviour was observed for the N protein (
Figure 5 and
Figure 7b) where the number of unique peptides was equal between the two conditions, while the percentage of coverage decreased if viral standards were added before sample preparation (17% compared to 21%) (
Figure 8a,b).
On the other hand, the high heterogeneity of protein amount and low protein solubility after precipitation in saliva samples makes the calculation of the theoretical SARS-CoV-2 protein quantity injected in LC-MS quite challenging. A rough estimation of the total protein concentration was conducted using 280 nm absorbance. Based on it, a comparison was conducted between the results obtained by spiking viral standard proteins before (
Figure 9) and after manipulation (
Figure 5 and
Figure 6). In particular, the quantity of 100 ng of standards for the first condition and the saliva spiked after digestion with 1 ng were compared considering a ratio of about 1:2000–1:4000 between each purified viral protein and human proteins. Similarly to plasma results, the nucleoprotein in saliva samples was recognised with a lower sequence coverage determined as 11% if added before any sample manipulation and as 25% if spiked before LC-MS analysis. Instead, spike glycoprotein identification remained almost unchanged considering the number of unique peptides and the sequence coverage.
2.5. Saliva and Plasma Human Proteome
Several authors optimised mass spectrometry-based protocols to investigate the proteome of biofluids and determine the changes in response to COVID-19 infection, exploring the physiopathology of the disease and the molecular basis of the difference in patient response. In particular, Overmyer et al. performed untargeted and targeted multiomics (proteomics, metabolomics and lipidomics) by means of high-resolution MS coupled either with gas chromatography (GC) or liquid chromatography (LC) systems on not-depleted plasma samples of 128 COVID-19-positive (
n = 102) and -negative (
n = 26) patients [
14]. This approach identified more than 500 proteins, about 650 lipids, 110 metabolites and about 2800 unidentified small molecules. Based on this protocol, they studied COVID-19 physiopathology, pinpointing the up- and downregulated proteins and pathways characterising specific molecular profiles of the patients in the cohort. In another interesting study performed by Messner et al., serum and plasma samples of thirty-one COVID-19 patients grouped on the basis of WHO severity grade (between 3 and 7 WHO scores) were evaluated [
12]. They developed an ultrafast proteomics method for the discovery of clinical classifiers based on protein expression signatures that allowed the identification of 27 potential biomarkers associated with COVID-19 severity. These alterations are involved in complement factors, coagulation system, inflammatory modulators and proinflammatory signalling. Finally, Park et al. implemented an ultrahigh-resolution LC-MS method based on BoxCar acquisition, and they performed plasma proteome profiling of three mild and five severe COVID-19 patients [
25]. In this study, the host response to SARS-CoV-2 infection resulted in 91 differentially expressed proteins depending on disease severity, and the proteins related to neutrophil activation, blood coagulation, proinflammatory factors and complement activation were mostly significant.
One of most desirable goals that can be reached by mass spectrometry-based methods is likely to have the simultaneous identification of the proteome of biofluids, as well as the detection of SARS-CoV-2 proteins in a single analysis, in order to gain information both on the viral molecular profile and its effect on the human proteome at the systemic level. To this end, we set and applied an untargeted protocol on plasma and saliva that permitted both detecting SARS-CoV-2 proteins spiked in plasma or saliva, as previously shown, and qualitatively and quantitatively evaluating the proteome of these biofluids. In particular, concerning the molecular modifications induced to the human counterpart, we evaluated the proteome identified in plasma/saliva from healthy samples with and without SARS-CoV-2 proteins through the application of this protocol in order to have an indication of the identification power of this approach, explore the consistency with the current knowledge and investigate its potential for future SARS-CoV-2-related studies.
Concerning plasma samples, our strategy resulted in the identification of 324 proteins in total (
Supplementary Table S1) and an average of 1387 unique peptides per run. The use of PNGase F enabled us to detect 98 plasma proteins containing at least one potential N-glycosylation site, corresponding to 30.2% of the total identified proteins. Then, to evaluate how representative the identified proteins might be for the study of COVID-19, a further characterisation of the proteome was carried out and compared with the data available in the literature. The results showed that the more represented protein families belong to defense/immunity proteins (PC00090) and protein-binding activity modulators (PC00095) covering the 16.5% and 11.2% of the total identifications, respectively. Moreover, functional investigation highlighted two pathways that are likely to be enriched in our dataset, including complement cascade (HSA-166658) and innate immune system (HSA-168249). These data seem to be in line with those reported in a similar proteomic analysis present in the literature regarding COVID-19 studies in human plasma [
13].
On the other hand, a mass spectrometry-based protocol to investigate the whole salivary proteome is more critical to develop. Despite the fact that this body fluid is very attractive for disease diagnosis due to different reasons (e.g., easy, economic and noninvasive collection), its use in clinical proteomic applications is strongly limited by high variability (inter- and intraindividual), biological rhythm influence (e.g., circadian rhythm) and several possible polymorphisms [
26].
Using our untargeted mass spectrometry-based method, in addition to SARS-CoV-2 proteins, we were able to identify an average of 2141 unique peptides per run and 868 proteins in total; among them, 136 were characterised by at least one potential N-linked glycosylation (15.7% of total proteins) (
Supplementary Table S2). As expected, approximately 30% of the total salivary identifications were serum-like proteins (e.g., albumin and immunoglobulins) [
27]. Moreover, in line with the data reported by Castagnola et al., we observed different protein families typical of salivary specimens, including basic, acidic and glycosylated proline-rich proteins (e.g., PRPC_HUMAN, SPR2A_HUMAN, SPR2B_HUMAN, SPRR3_HUMAN, PRP1_HUMAN, PRB2_HUMAN), amylases (AMY1A_HUMAN), cystatins (e.g., CYTA_HUMAN, CYTB_HUMAN, CYTC_HUMAN, CYTD_HUMAN, CYTT_HUMAN, CYTN_HUMAN), mucins (e.g., MUC2_HUMAN, MUC21_HUMAN, MUC5B_HUMAN) and statherins (e.g., STAT_HUMAN) [
28]. Functional analysis on our saliva sample proteome suggested the enrichment of some of the identified proteins, mainly in pathways related to the defense/immunity protein (PC00090), the immunoglobulin (PC00123), the translational protein (PC00263) and the protease inhibitor (PC00191), whose expression could provide useful information for the investigation of the mechanisms of pathogenesis and response to SARS-CoV-2. Indeed, recent proteomic studies of nasopharyngeal swabs in COVID-19 patients highlighted biological processes and molecular actors that we observed with the implemented protocol, such as a consistent alteration of the innate immune response, including the complement activation and the interferon-mediated immune response, components of protein synthesis, and in the regulation of proteosomal subunits [
29]. These data are likely to confirm the intriguing potential of saliva as a noninvasive biofluid for disease investigation in addition to SARS-CoV-2 protein detection.
3. Conclusions
In this work, a mass spectrometry-based method was developed, which, through an easy sample preparation workflow, allowed for safe manipulation of COVID-19 specimens in laboratories without biosafety level 3 (BSL3) equipment. Moreover, the application of an untargeted approach enabled the simultaneous identification of SARS-CoV-2 proteins and characterisation of alterations in the host human proteome from different biofluids.
Firstly, the limit of detection of the method was evaluated by analysing purified SARS-CoV-2 protein standards (S1, S2, RBD and N), and it resulted in 10 pg for the spike glycoprotein and the nucleoprotein. Then, the limit of characterisation (LOC) was measured in two biofluids, saliva and plasma, spiked with different amounts of purified and digested SARS-CoV-2 proteins before LC-MS. A LOC of 100 pg for both proteins in saliva and of 330 pg and 33 pg for the spike glycoprotein and the nucleoprotein, respectively, in plasma was observed. The standards were also spiked before sample manipulation to simulate a real sample, and the effect of matrix interference and sample processing was estimated.
Furthermore, a preliminary proteomic analysis was performed on saliva and plasma samples, identifying about 800 and 300 human proteins, respectively. Among them, several key effectors and pathways that are known to be altered in COVID-19 patients were observed.
The approach can be highly relevant for further application in COVID-19 patients, allowing SARS-CoV-2 proteins and the human proteome to be explored in saliva and plasma samples, and for retrospective studies aimed at investigating possible virus variants and stratifying patients with different outcomes.
4. Materials and Methods
4.1. Materials
SARS-CoV-2 recombinant proteins S1, S2, RBD and N were produced and provided by Istituto Nazionale Genetica Molecolare (INGM) of Milan. RapiGestTM SF was from Waters Corporation, Milford.
4.2. SARS-CoV-2 Protein Deglycosylation
Five µg of each viral recombinant protein (S1, S2, RBD, N) were diluted in 50 mM ammonium bicarbonate (NH4HCO3, Sigma-Aldrich, ≥99.0%) buffer solution to reach the final volume of 100 µL. In order to remove N-linked oligosaccharides which could interfere with the following steps, N-Glycosidase F (Roche Diagnostics, Mannehim, Germany) was added to SARS-CoV-2 standard proteins with a ratio of 1 U of enzyme over 25 µg of proteins and incubated overnight at 37 ℃.
4.3. Biological Fluid Collection
Saliva samples were collected from healthy volunteers working in the proteomic unit, while blood samples were collected from healthy subjects at San Gerardo Hospital (Monza, Italy). All the subjects had signed an informed consent form prior to sample donation, and analyses were carried out in compliance with the Declaration of Helsinki. Study protocols and procedures were approved by the local ethics committee (Comitato Etico Azienda Ospedaliera San Gerardo, Monza, Italy). Plasma samples were collected in Vacutainer® K3E tubes containing EDTA (Becton Dickinson Italia S.p.A.), centrifuged at 3700 rpm for 10 min and then stocked at −80 ℃.
4.4. Sample Inactivation and Deglycosylation of Biological Fluids
To allow for sample manipulation in safe conditions, an inactivation method for the virus in biological fluids was developed; 100 µL of each biological fluid sample were collected and underwent a process of heat inactivation and protein precipitation. Two protocols were evaluated. Protocol A: the samples were heated for 30 min at 70 ℃ and then incubated in freezing acetone (Honeywell, ≥99.8%, Offenbach, Germany), 4:1 acetone/sample (v/v), to allow for protein precipitation. Protocol B: the samples were heated for 60 min at 56 ℃ and then incubated in freezing ethanol (Honeywell, ≥99.8%, Offenbach, Germany), 9:1 ethanol/sample (v/v), to allow for protein precipitation and to increase the safety level. Then, the samples were centrifuged, the supernatant was discarded, while the pellet was left to air-dry and resuspended in 50 mM ammonium bicarbonate buffer solution. In Protocol B, to enhance enzymatic digestion, RapiGestTM SF Surfactant was added to the final concentration of 0.1%. After protein quantification with a Nanodrop spectrophotometer (NanoDrop OneC, Thermo scientific, Wilmington, DE, USA), N-Glycosidase F (Roche Diagnostics, Mannehim, Germany) was added to the samples (8 U/100 µL and 10 U/100 µL of the enzyme for the saliva and plasma samples, respectively), which were incubated overnight at 37 ℃.
4.5. Protein Digestion
Both SARS-CoV-2 standard proteins and biological samples were treated with DL-Dithiothreitol (DTT) (Sigma-Aldrich, St. Louis, MO, USA, ≥99.5%) at the final concentration of 20–25 mM for the viral proteins and saliva and of 40 mM for the plasma samples and incubated for 45 min at 56 ℃. Then, iodoacetamide (IAA) (Sigma-Aldrich, St. Louis, MO, USA) at the final concentration of 25–30 mM was added, and the samples were left at room temperature for 30 min for the carbamidomethylation reaction.
The proteins were enzymatically digested by adding trypsin (trypsin from the porcine pancreas, Sigma-Aldrich, St. Louis, MO, USA) at the enzyme/protein mass ratio of 1:25 for SARS-CoV-2 standard proteins, while 4 μg/100 µL and 8 μg/100 µL of trypsin were added to the saliva and plasma samples, respectively. Then, all the samples were incubated overnight at 37 ℃. The enzymatic reaction was stopped by adding formic acid (FA) (LiChropur®, Merck KGaA, Darmstadt, Germany) to reach an acidic pH (<2). Sample volume was reduced by means of a vacuum centrifugal evaporator (Hetovac, Savant); then, dried peptides were resuspended in 50 µL of loading pump phase A (H2O:ACN:TFA 98:2:0.1). The saliva and plasma protein content was obtained with a Nanodrop spectrophotometer followed by a desalting step using Ziptip™ µ-C18 Pipette Tips (Merck Millipore Ltd.). The peptides were eluted with a solution of 80% acetonitrile and 0.1% formic acid, dried by means of a vacuum centrifugal evaporator (Hetovac) and resuspended in 50 µL of loading pump phase A.
4.6. Mass Spectrometry Analysis
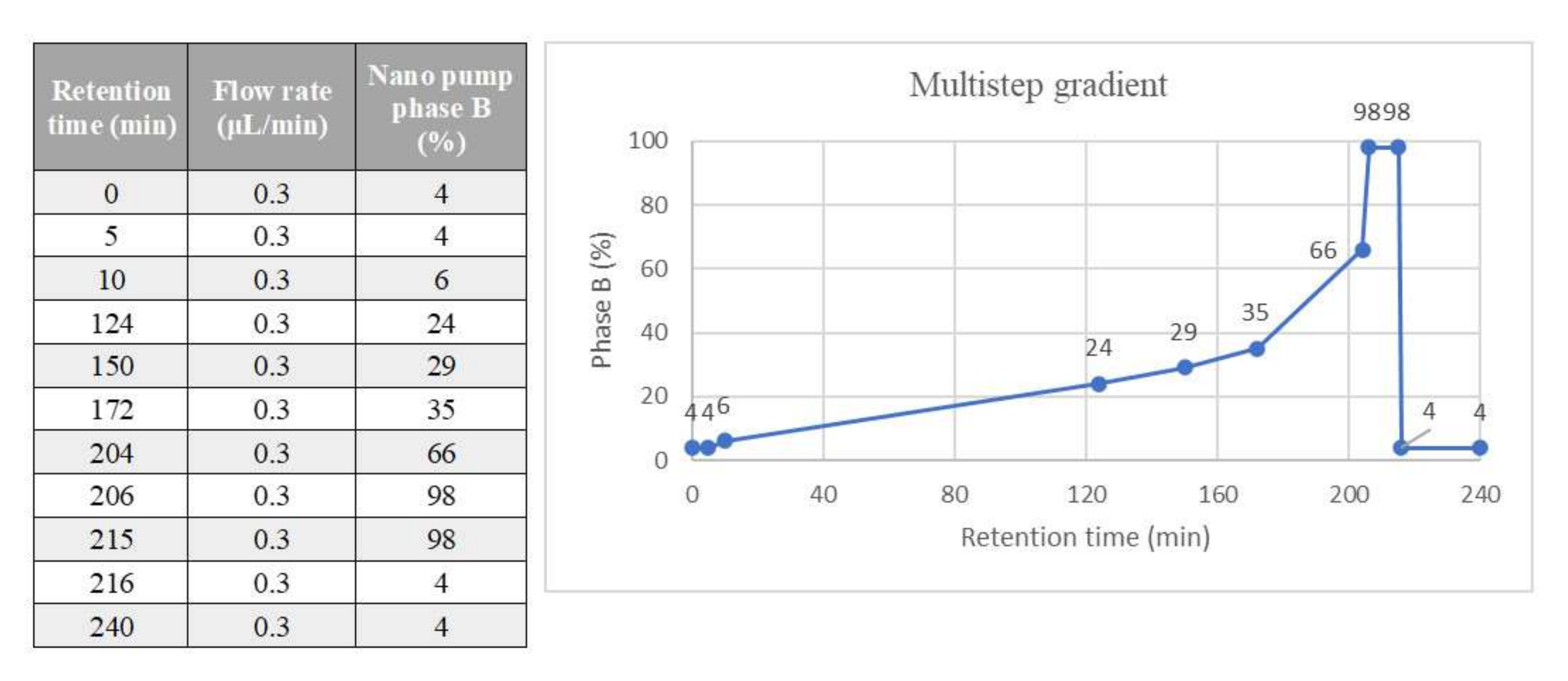
Tryptic peptides were injected into a Dionex UltiMate 3000 rapid separation (RS) LC nanosystem (Thermo Scientific, Sunnyvale, CA, USA) coupled with an Impact HDTM UHR-qToF system (Bruker Daltonics, Bremen, DE). The samples were loaded into a µ-precolumn (Thermo Scientific, Acclaim PepMap 100, 100 µm × 2 cm, nanoViper, C18, 3 µm) for a further desalting and concentration step; then, the peptides were separated in an analytical 50 cm nanocolumn (Thermo Scientific, Acclaim PepMap RSLC, 75 µm × 50 cm, nanoViper, C18, 2 µm) with a multistep 240 min gradient ranging from 4% to 98% of nanopump phase B (H
2O:ACN:FA 20:80:0.08) at a flow rate of 300 nL/min and temperature of the column in the oven of 40 ℃ (
Figure 10). The eluted peptides were ionised using a nanoBoosterCaptiveSpray™ (Bruker Daltonics) source using heated nitrogen dry gas (T = 150 ℃; 3 L/min) enriched with acetonitrile (ACN). The mass spectrometer was operated in DDA (Data Dependent Acquisition Mode), with automatic switching between full-scan MS and MS/MS acquisition. N
2 was used as a gas for CID (collision-induced dissociation) fragmentation. The software automatically selected the number of precursor ions in order to fit into a fixed cycle time: the time between two subsequent MS acquisitions was 5 s. The charge of precursor ions ranged between +2 and +5, and precursor peaks above 1575 counts, in the 300–1221 and 1225–2200
m/z windows, were selected. IDAS (Intensity Dependent Acquisition Speed) and RT2 (RealTime Re-Think) functionalities were applied. In order to achieve an improvement of mass accuracy, the mass spectrometer was calibrated using a mix of ten standards with a known mass (MMI-L Low Concentration Tuning Mix, Agilent Technologies, USA) before the sample run sequence. In addition, a specific lock mass (1221.9906
m/z) and a calibration segment (at the first 15 min of the gradient) of 10 mM sodium formate (1% NaOH 1 M and 0.1% FA) cluster solution was used.
4.7. Data Processing
Raw data were elaborated by means of Compass DataAnalysis
TM, v.4.1 (Bruker Daltonics, Hamburg, Germany). The resulting mass lists were processed using an in-house Mascot search engine (v.2.4.0) through Mascot Daemon; a SARS-CoV and SARS-CoV-2 protein database (UniProt, pre-release dataset, was downloaded from UniProt (
ftp://ftp.uniprot.org/pub/databases/uniprot/pre_release/, accessed on 6 October 2021), combined with a human database (SwissProt, released March 2020; 562,755 sequences; 202,599,198 residues) and integrated to the search engine in order to simultaneously detect specific viral proteins and the human proteome. The parameters were set as follows: trypsin as the enzyme, carbamidomethyl as the fixed modifications, deamidated-only N and oxidation (M) as the variable modifications, 20 ppm as the precursor mass tolerances and 0.05 Da for the product ions. Automatic decoy database search was applied for false discovery rate (FDR) calculation and a built-in Percolator algorithm was used for rescoring peptide spectrum matches. Only the proteins with at least one unique and significant (
p-value < 0.05) peptide were considered identified. Peaks Studio X-Plus (Bioinformatics Solutions Inc., Waterloo, ON, USA) was used for protein identification including the above described protein databases; the parameters were set as described above for the Mascot search, and FDR ≤ 1% was applied to all the analyses, and the proteins were considered only if identified with at least one unique significant peptide (
p-value ≤ 0.05). Functional annotations were performed using STRING (
https://string-db.org/, version 11.5) [
30] and the PANTHER Classification System (
http://www.pantherdb.org/, version 16.0).
4.8. Limit of Detection (LOD)
Four standard proteins purified from SARS-CoV-2 were processed, deglycosylated and enzymatically digested as previously described. Then, different quantities of the resulting peptides (10, 1, 0.1, 0.02 and 0.01 ng) were analysed by nLC-ESI-MS/MS in order to determine our limit of detection.
4.9. Limits of Viral Protein Characterisation (LOC)
A pool of saliva and plasma was prepared from healthy subjects. For each sample, 100 µL of saliva and of plasma were taken from the pool and treated with a virus inactivation protocol to simulate a real specimen from COVID-19 patients, N-deglycosylated and trypsin-digested as detailed before. Peptide mixtures were spiked with different amounts of each purified viral standard obtained by serial dilutions from a stock solution containing 25 ng/µL of each standard protein in order to inject 10 pg, 100 pg and 1000 pg of each standard in 10 µL of saliva and 33 pg, 330 pg and 3300 pg in 10 µL of plasma. Then, they were analysed by means of nLC-ESI-MS/MS.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}