Abstract
Runt-related transcription factor 1 (RUNX1) is a key transcription factor in hematopoiesis, producing multiple major isoforms, RUNX1A, B, and C, via alternative promoter usage and splicing. These isoforms have distinct roles in hematopoiesis and leukemogenesis. Imbalances in isoform expression, such as RUNX1A overexpression or RUNX1C loss, contribute to leukemogenesis in disorders. RUNX1 isoform expression is regulated by transcriptional, epigenetic, and splicing mechanisms and is further influenced by genome architecture. Pathogenic variants, including truncations and fusion proteins, disrupt isoform homeostasis and transcriptional control for the target genes in hematopoiesis. Recent therapeutic strategies aim to restore isoform balance rather than inhibit RUNX1 globally. Approaches include splice-switching oligonucleotides, CRISPR-based promoter modulation, and enhancer-targeted therapies. Understanding isoform-specific RUNX1 biology offers new opportunities for precision treatment of hematologic malignancies.
1. Introduction
RUNX1 is a pivotal transcriptional factor that regulates hematopoietic development and differentiation [1,2,3]. It is essential for definitive hematopoiesis during embryonic development and continues to play a vital role in balancing stem cell maintenance and lineage commitment in adult hematopoiesis. Additionally, it is a frequent target of somatic mutations in hematologic malignancies, such as acute myeloid leukemia (AML), myelodysplastic syndrome (MDS), and acute lymphoblastic leukemia (ALL) [1,2,3,4,5,6,7,8,9].
RUNX1 is unique among hematopoietic transcription factors in that it exists as multiple isoforms, generated through alternative usage of two promoters, P1 (distal promoter) and P2 (proximal promoter), and alternative splicing [10,11,12,13]. The major isoforms, including RUNX1A, RUNX1B, and RUNX1C, differ in their N- and C-terminal structures and possess distinct (Figure 1), sometimes antagonistic functions in hematopoietic development and leukemogenesis [11,14,15,16,17,18]. Their structural diversity broadens RUNX1’s functional capacity, allowing it to fine-tune hematopoietic development and maintain cellular homeostasis in both normal and disease states.
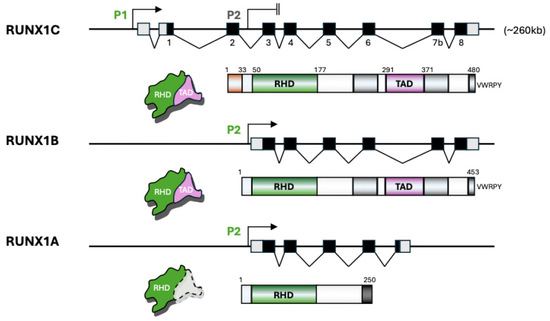
Figure 1.
Structure and domain composition of major RUNX1 isoforms. This schematic illustrates the gene structure, promoter usage, and protein domain organization of the three major RUNX1 isoforms: RUNX1C, RUNX1B, and RUNX1A. The RUNX1 gene spans approximately 260 kb and contains two promoters, a distal promoter (P1) that drives the expression of RUNX1C and a proximal promoter (P2) that generates RUNX1B and RUNX1A transcripts. Black boxes indicate exons and light gray boxes indicate untranslated regions for the genomic maps. RUNX1C includes all eight exons, initiating from promoter P1, and encodes a full-length protein comprising the Runt Homology Domain (RHD, green) responsible for DNA binding, followed by the Transactivation Domain (TAD, purple) and other regulatory regions including the VWRPY motif. RUNX1B, transcribed from promoter P2, shares the RHD and TAD domains but has a distinct N-terminus and lacks the first exon used in RUNX1C. RUNX1A, also transcribed from P2, lacks several exons encoding the TAD, resulting in a truncated protein that contains the RHD but lacks functional transactivation capacity (indicated by dashed lines). The differences in exon composition and domain structure among these isoforms underlie their distinct functional roles in hematopoiesis and disease.
Recent applications of high-resolution genomic and transcriptomic methods, including targeted CRISPR-based genome editing, single-cell RNA sequencing, and long-read RNA sequencing, have substantially elucidated the regulatory mechanisms and functional diversity of RUNX1 isoforms. CRISPR-based genome editing has enabled precise dissection of RUNX1 cis-regulatory elements and isoform-specific promoters, revealing their distinct roles in hematopoietic specification [17,19,20]. Meanwhile, single-cell and long-read transcriptomic analyses have uncovered pronounced heterogeneity in RUNX1 isoform expression and delineated complex patterns of alternative splicing across hematopoietic developmental trajectories [21,22]. These approaches are reframing our understanding of how isoform-specific RUNX1 regulation underpins lineage commitment and disease susceptibility.
It has been well recognized that dysregulation of RUNX1 isoform expression is mechanistically implicated in the pathogenesis of several hematologic malignancies. In Down syndrome-associated myeloid leukemia (ML-DS), a disproportionate increase in the RUNX1A isoform relative to RUNX1C drives leukemogenesis [22]. Germline RUNX1 mutations causing familial platelet disorder with predisposition to myeloid malignancy (FPDMM) and somatic alterations in RUNX1 in acute myeloid leukemia (AML) similarly disrupt isoform function or balance, contributing to disease development [23,24]. Collectively, these observations underscore the critical importance of understanding the specific roles and regulatory mechanisms governing individual RUNX1 isoforms in both normal hematopoiesis and malignant transformation.
In this review, we summarize the current understanding of RUNX1 isoform biology, explore regulatory mechanisms at multiple levels, and discuss how isoform imbalance contributes to hematologic diseases. Finally, we examine potential therapeutic strategies that target RUNX1 isoform dynamics in pathogenic conditions.
2. Distinctive Features of RUNX1 Isoforms and Variants
The RUNX1 gene spans over 260 kb located on chromosome 21q22 and contains dual promoters, a distal promoter (P1) and a proximal promoter (P2), which produce different isoforms with distinct 5′ and 3′ untranslated regions (UTRs) and exons (Figure 1). P1 is located ~160 kb upstream of the main coding region and is primarily active during early development and in specific hematopoietic contexts, while P2 is used more broadly in adult hematopoiesis [2,11,12,13,25]. This dual promoter architecture enables precise temporal and tissue-specific expression of RUNX1 isoforms. Promoter choice not only affects transcript abundance but also influences isoform-specific translation initiation and post-translational regulation. Alternative splicing further increases isoform complexity [26,27,28,29]. RUNX1A is derived from P2 and lacks exons encoding the transactivation domain (TAD), resulting in a truncated protein with dominant negative activity [7,26,27,28,29] (Figure 1 and Figure 2). RUNX1B and RUNX1C are longer isoforms generated from P2 and P1, respectively.
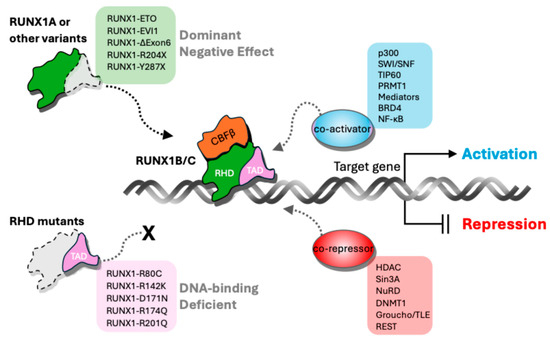
Figure 2.
Mechanism of RUNX1 isoform-mediated gene regulation. RUNX1 regulates gene expression through isoform-/domain-dependent interactions with co-activators and co-repressors. The full-length RUNX1B/C isoforms contain both the DNA-binding Runt Homology Domain (RHD) and the Transactivation Domain (TAD), enabling them to recruit co-activators, such as p300, SWI/SNF, TIP60, PRMT1, Mediators, BRD4, and NF-κB, which promote transcriptional activation of target genes. In contrast, RUNX1 variants lacking a functional TAD—including exon 6-skipped forms (RUNX1A, RUNX1- ΔExon6), nonsense mutants (R204X, Y287X), and fusion proteins (RUNX1-ETO, RUNX1-EVI1)—exert dominant-negative effects by occupying DNA binding sites without activating transcription. Mutants with defective or lost RHD fail to bind DNA, rendering them unable to recruit co-regulators. RUNX1 also interacts with co-repressors, such as HDAC, Sin3A, NuRD, DNMT1, Groucho/TLE, and REST to repress gene expression. This balance between activation and repression mediated by RUNX1 isoforms and mutants shapes hematopoietic gene regulation and influences leukemogenic potential.
All RUNX1 isoforms share the Runt homology domain (RHD), which mediates DNA binding and heterodimerization with CBFβ, a non-DNA-binding partner that enhances DNA binding affinity and protein stability [7,17,27] (Figure 1 and Figure 2). The full-length RUNX1B/C isoforms contain both the RHD and the TAD, enabling them to recruit co-activators, such as p300, SWI/SNF, TIP60, PRMT1, Mediators, BRD4, and NF-κB, which promote transcriptional activation of target genes [7,13,30,31]. In contrast, RUNX1 variants lacking a functional TAD, including RUNX1A, RUNX1-ΔExon6, nonsense mutants (e.g., R204X, Y287X), and fusion proteins (RUNX1-ETO, RUNX1-EVI1), exert dominant negative effects by occupying DNA binding sites without activating transcription [15,21,32] (Figure 2). Mutants with defective or lost RHD fail to bind DNA, rendering them unable to recruit co-regulators [23]. RUNX1 also interacts with co-repressors, such as HDAC, Sin3A, NuRD, DNMT1, Groucho/TLE, and REST, to repress gene expression [25,33]. Thus, the dynamic equilibrium of co-activator and co-repressor recruitment by different RUNX1 isoforms and mutants modulates hematopoietic gene expression and impacts leukemic transformation (Figure 2).
3. Isoform-Specific Roles of RUNX1 in Hematopoiesis and Leukemogenesis
RUNX1 is dispensable for the initial wave of primitive hematopoiesis in the yolk sac but modulates early gene expression programs critical for hematopoietic specification. The RUNX1A isoform has been shown to support progenitor expansion predominantly in leukemia models; however, its physiological role during normal development remains unclear.
RUNX1C is expressed at the developmental stages and anatomical sites of definitive hematopoietic stem cell (HSC) emergence, such as the aorta-gonad-mesonephros (AGM) region, in both murine and human systems. RUNX1 expression in hemogenic endothelial cells is essential for their endothelial-to-hematopoietic transition (EHT) and subsequent HSC formation. Conditional knockout studies deleting RUNX1 in endothelial cells abolish HSC emergence, underscoring its indispensable role at this critical event [5,34,35]. Furthermore, rescue experiments in Runx1-null models demonstrate that restoration of RUNX1C expression alone is sufficient to reinstate definitive HSC production and sustain long-term hematopoiesis, confirming RUNX1C’s functional indispensability [5]. In contrast, RUNX1A does not naturally appear at these sites or time points critical for HSC emergence in the mouse and fails to rescue HSC formation or sustain long-term hematopoiesis in knockout contexts. The physiological role of RUNX1A during normal hematopoiesis remains elusive, with most evidence suggesting it performs distinct functions insufficient for HSC rescue. The complete loss of all RUNX1 isoforms results in embryonic lethality due to failed definitive hematopoiesis, highlighting the necessity of isoforms containing the full transactivation domain for normal blood development.
The RUNX1B isoform is less characterized compared to RUNX1C. Recent research indicates that RUNX1B is expressed early in development within a subset of endothelial cells marked by hemogenic potential. RUNX1B expression levels regulate hemogenic competency in a dose-dependent manner, effectively acting as a gatekeeper of the EHT during hematopoietic initiation [5,12]. Accordingly, RUNX1B serves as an early marker and key determinant of hemogenic transition rather than primarily functioning during intermediate progenitor stages. Its role is not to broadly regulate progenitor differentiation or maintain lineage fidelity but rather to mark cells transitioning to hematopoietic fate. This stage-specific and dosage-dependent regulation highlights RUNX1B’s unique position in orchestrating the initial commitment from endothelium to blood, distinct from the functions of other RUNX1 isoforms that act later in hematopoiesis. In adult hematopoiesis, RUNX1B expression is heterogeneous and has been associated specifically with fate decisions between megakaryocytic and erythroid lineages at the progenitor level, where promoter-specific isoform expression influences lineage outcomes [36]. Understanding how RUNX1 isoform expression patterns translate into functional consequences at different developmental stages also requires appropriate disease models that accurately reflect human biology.
In small animal models of familial platelet disorder with predisposition to myeloid malignancy (FPDMM), the full spectrum of the human disease phenotype caused by RUNX1 haploinsufficiency is not completely recapitulated, reflecting important species-specific differences in hematopoietic regulation and disease manifestation [3]. For example, murine Runx1+/− models commonly exhibit partial hematopoietic defects, such as altered megakaryocytic differentiation and modest myeloid abnormalities but fail to develop persistent thrombocytopenia or leukemic progression characteristic of human FPDMM [3,37]. In contrast, recent non-human primate models (e.g., rhesus macaques) engineered via CRISPR/Cas9 to harbor heterozygous RUNX1 loss-of-function mutations more faithfully recapitulate key FPDMM features, including thrombocytopenia, megakaryocyte dysplasia, impaired polyploidization, and hematopoietic stem and progenitor cell (HSPC) defects, emphasizing the species context as a critical factor in disease modeling [38].
Notably, the RUNX1A isoform, which is expressed uniquely in humans and absent in mouse models, is enriched in early hematopoietic progenitors and leukemia contexts, including Down syndrome-associated myeloid leukemia (ML-DS). Dysregulated expression of RUNX1 isoforms plays a central role in leukemogenesis. Overexpression of RUNX1A in murine models, despite its absence in normal mouse hematopoiesis, induces myeloproliferation and cooperates with cooperating oncogenic events, such as GATA1s mutations, to initiate leukemia. The dominant-negative nature of RUNX1A disrupts normal transcriptional programs by displacing RUNX1C from its target binding sites, consequently impairing differentiation [1,13,22,39].
Additionally, RUNX1 isoform imbalance frequently cooperates with isoform-specific partner proteins to exacerbate disease phenotypes. For example, RUNX1A not only synergizes with GATA1s mutations in ML-DS but also interferes with the normal cooperative binding of RUNX1C and GATA1 at megakaryocytic enhancers [22]. Similarly, PU.1 redistribution in leukemogenesis has been shown to depend on altered RUNX1 isoform binding, leading to survival advantages in malignant progenitors [40]. Isoform-selective interactions with Menin further contribute to the stabilization of leukemic transcriptional networks [41]. These findings emphasize that disruption of RUNX1 isoform–partner interactions is a critical pathogenic mechanism and represents a promising avenue for therapeutic intervention.
In trisomy 21, RUNX1 expression is globally upregulated but disproportionately favors RUNX1A due to altered promoter usage and splicing regulation. This increased RUNX1A:RUNX1C ratio is associated with impaired megakaryocytic differentiation and heightened leukemic susceptibility within the fetal liver. RUNX1A cooperates with GATA1s, a common co-mutation in ML-DS, to drive pre-leukemic expansion and transformation [22]. Conversely, haploinsufficient RUNX1 mutations reduce the total functional RUNX1 isoform pool, particularly RUNX1C, leading to defective megakaryocyte maturation, thrombocytopenia, and increased myeloid malignancy risk [13,21,23,37]. Some FPDMM mutations may selectively affect promoter or splicing regulatory elements, further disrupting RUNX1 isoform distribution and function [13,15,21,24]. This suggests that aberrant RUNX1A expression may also contribute functionally to leukemogenesis in FPDMM, although direct mechanistic evidence linking RUNX1A to FPDMM pathogenesis remains limited and warrants further study.
Induced pluripotent stem cell (iPSC)-based models derived from FPDMM patients and human CD34+-derived megakaryocyte differentiation model in RUNX1 depletion more accurately recapitulate megakaryopoietic defects than murine models and reveal aberrant activation of stress-associated signaling pathways such as TGF-β1 and JNK. These pathways impair megakaryocyte development and represent potential therapeutic targets, as pharmacological inhibition partially restores megakaryopoiesis [37,42]. Similar stress response patterns have been observed in ML-DS and acute myeloid leukemia, where RUNX1 dysregulation disrupts normal hematopoiesis.
Beyond its central role in hematopoietic differentiation, emerging evidence demonstrates that RUNX1 modulates cell cycle progression, in part by regulating key proliferation-associated genes such as CENPE and by influencing the activity of cyclin/CDK complexes, thereby promoting leukemic cell growth and survival. Dysregulated RUNX1 activity can corrupt normal cell cycle control and facilitate malignant expansion [43].
Additionally, RUNX1 has been implicated in the regulation of inflammatory signaling pathways, including modulation of cytokine responses and chromatin accessibility at pro-inflammatory gene loci [44,45,46]. These effects impact both the leukemic niche and immune evasion, further fostering malignant progression.
Recent studies also highlight RUNX1’s role in controlling ribosomal RNA gene expression and nucleolar function, processes that are tightly linked to cell growth and metabolic adaptation. Aberrant regulation of ribosome biogenesis by RUNX1 contributes to the increased proliferative and anabolic demands of leukemia cells [21,47]. These diverse functions underscore the multifaceted impact of RUNX1 dysregulation in leukemogenesis. Inclusion of these mechanisms provides a more comprehensive understanding of RUNX1 as an integrative driver of malignant transformation and therapeutic resistance in hematologic malignancies.
Taken together, these findings highlight the intricate and isoform-specific functions of RUNX1 in both normal hematopoietic development and leukemogenesis. The complex regulation of RUNX1 expression, encompassing total RUNX1 levels, promoter usage, and isoform balance, critically influences hematopoietic cell fate decisions and disease predisposition. Notably, human-specific isoforms such as RUNX1A play distinct, often dominant negative roles that are absent in traditional animal models, underscoring the limitations of cross-species disease modeling. As illustrated in Figure 3, deviations from the tightly regulated RUNX1 isoform equilibrium, whether through haploinsufficiency or isoform overexpression, lead to abortive differentiation and expansion of leukemogenic progenitors developing clonal evolution. Within a defined “balanced zone”, characterized by physiologically normal RUNX1 levels and an optimal isoform ratio, HSPCs can efficiently differentiate into mature megakaryocytes (Mk). This emphasizes the importance of considering isoform dynamics and dosage effects when investigating RUNX1-related hematologic disorders and developing targeted therapies (Figure 3).
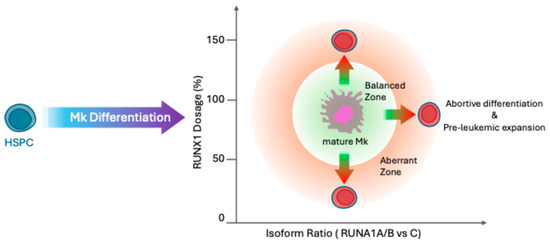
Figure 3.
Fine-tuned balance of RUNX1 dosage and isoform ratio regulates hematopoietic stem and progenitor cell differentiation. This schematic depicts how precise regulation of RUNX1 expression levels and isoform balance controls differentiation of hematopoietic stem and progenitor cells (HSPCs) into mature megakaryocytes (Mk). The central green “Balanced Zone” indicates the optimal range for RUNX1 dosage and isoform ratio that supports normal Mk differentiation. The surrounding red “Aberrant Zone” represents regions where either RUNX1 dosage or isoform ratio is imbalanced, leading to abortive differentiation and pre-leukemic cell expansion. Both overexpression and deficiency of RUNX1, whether due to altered isoform ratios or RUNX1 variants, result in abortive differentiation, preventing effective maturation of progenitors. The balance is critical: excessive RUNX1 leads to differentiation arrest, while insufficient RUNX1 impairs lineage commitment, highlighting RUNX1’s dosage-sensitive role in hematopoiesis and the importance of maintaining isoform equilibrium for normal blood cell development.
4. Multilayered Regulation of RUNX1 Isoform Expression
The expression and function of RUNX1 isoforms are regulated through a multilayered system that integrates transcriptional, epigenetic, and post-transcriptional control. These mechanisms govern not only the overall expression level of RUNX1 but also the relative abundance of individual isoforms in a cell-type and developmental stage-specific manner. Understanding these regulatory layers is essential for elucidating how physiological expression patterns are maintained and how their disruption contributes to hematologic disease.
RUNX1 transcription itself is controlled by two promoters. Promoter usage is tightly regulated by developmental signals and cell context. For example, in embryonic stem cells and early hematopoietic progenitors, P1 is more active, favoring RUNX1C expression. Cloning the RUNX1C P1 promoter and +24 enhancer to drive reporter expression specifically marks emerging hematopoietic progenitors derived from human pluripotent stem cells, indicating P1-driven RUNX1C predominance in these cells. The RUNX1C isoform is the first to be induced in hematoendothelial progenitors derived from embryonic stem cells, correlating with P1 activity [11,48,49,50,51]. During megakaryopoiesis and in fetal liver hematopoiesis, P2 activity increases, resulting in more RUNX1B transcripts [36,48]. Transcription factors, such as GATA2, PU.1, and C/EBPα, bind selectively to promoter-associated enhancers and may bias promoter usage [40,52,53,54].
Furthermore, recent studies show that distal promoter P1 is enriched in activating histone marks such as H3K4me3 and H3K27ac during early development, consistent with active transcription in embryonic and early hematopoietic progenitors [55]. In contrast, the proximal promoter P2 becomes increasingly marked by these epigenetic modifications during later lineage commitment stages, such as megakaryopoiesis, reflecting its activation dynamics [55,56,57].
RUNX1 isoforms contribute to complex feedback mechanisms. RUNX1C and RUNX1B isoforms can bind upstream regulatory elements of the P1 promoter and repress its activity, forming a negative feedback loop that limits RUNX1A expression. This auto-repression helps maintain appropriate isoform balance during hematopoietic differentiation. When RUNX1A is overexpressed, or when DNA-binding or co-repressor recruitment is impaired, this autoregulatory repression is weakened, potentially disrupting the isoform equilibrium critical for normal lineage progression. This balance between P1 and P2 promoter activity, modulated by histone modifications and RUNX1 isoform-mediated feedback, is especially important during the transition from proliferative progenitors to terminally differentiating cells. Tight repression of P1 and relative enhancement of P2 activity ensures a proper shift in RUNX1 isoform expression that supports normal hematopoietic maturation and prevents leukemogenic transformations [3,7,13,26,36,48,49,55,58]. Promoter selection can be substantially influenced by genomic architecture that constrains which promoters are physically accessible to enhancers and transcription machinery [59]. The P1 and P2 promoters are indeed located in separate chromatin domains with differential enhancer connectivity, influencing cell type–specific isoform expression [13,28]. Disruption of CTCF/cohesin sites, mutations in TAD boundary elements, or architectural protein loss can alter chromatin looping, resulting in inappropriate enhancer–promoter interactions or separations. This can shift promoter choice, alter isoform usage, or lead to misexpression seen in development and disease [60,61].
Splicing decisions governing inclusion or skipping of RUNX1 exon 6, which generate the RUNX1C/B isoforms (included exon 6) or the RUNX1A isoform (skipped exon 6), are tightly regulated by a network of RNA-binding proteins (RBPs) with distinct mechanistic roles. Among these, the serine/arginine-rich splicing factor SRSF2 plays a critical role by binding to exonic splicing enhancers within exon 6, thereby promoting its inclusion in the mature transcript. This splicing factor is essential not only for normal hematopoiesis, but its mutations are also recurrent in myeloid malignancies, implicating dysregulated exon 6 inclusion in RUNX1-associated leukemogenesis [15,21]. Conversely, heterogeneous nuclear ribonucleoprotein K (hnRNP K) acts as a splicing repressor by interacting with intronic splicing silencer elements flanking exon 6, facilitating exon skipping and increasing RUNX1A isoform production. Supporting this model, experimental perturbations including RNA interference, CRISPR-based RBP screens, and splicing reporter assays indicate that SRSF2 binding motifs correlate strongly with exon 6 inclusion efficiency, while hnRNP K enrichment around the intronic silencers correlates with exon skipping bias [17,62].
The regulatory interplay between these RBPs integrates with broader splicing factor networks and is further influenced by the chromatin state and transcriptional kinetics. For example, RUNX1 fusion proteins in leukemia, such as RUNX1/RUNX1T1, indirectly modulate the expression of various splicing regulators, dynamically reshaping exon usage patterns involved in leukemic gene expression programs [21]. This highlights the complex layering of transcriptional and post-transcriptional control in RUNX1 isoform homeostasis. Overall, this finely tuned RBP-mediated splicing regulation at exon 6 is a key determinant of RUNX1 functional isoform output and represents a mechanistically compelling target for therapeutic modulation in RUNX1-driven hematopoietic disorders [15,21].
The chromatin state also affects RNA polymerase II elongation rate, which can influence splice site recognition via co-transcriptional coupling. Thus, RUNX1 isoform expression is governed by an intricate and dynamic interplay of cis-regulatory architecture, coordinated splicing regulation, and isoform-specific autoregulatory feedback, collectively ensuring the appropriate balance of RUNX1 variants required for normal hematopoietic function and preventing malignant transformation [61].
5. Therapeutic Strategies Targeting RUNX1 Isoform Regulation
RUNX1 has long been regarded as a challenging therapeutic target due to its indispensable role in normal hematopoiesis. Nevertheless, accumulating evidence indicates that therapeutic strategies focusing on modulating isoform-specific functions, rather than global RUNX1 inhibition, may provide a more precise and safer approach. Current efforts are investigating methods to selectively target transcriptional regulation, isoform-specific transcripts or protein domains, and upstream pathways that govern isoform switching and activity, aiming to correct pathogenic RUNX1 dysregulation while preserving normal hematopoietic function.
Therapeutic correction of RUNX1 isoform imbalance is particularly relevant in disorders such as ML-DS and FPDMM, where pathological elevation of the RUNX1A:RUNX1C ratio disrupts normal hematopoiesis and promotes leukemogenesis. Splice-switching oligonucleotides (SSOs) can be engineered to enhance inclusion of exon 6 during RUNX1 pre-mRNA processing, thereby reducing RUNX1A isoform production and favoring the expression of full-length RUNX1C. Although SSOs have not yet been applied experimentally to modulate RUNX1 splicing in ML-DS or FPDMM, this strategy is mechanistically compelling and has shown success in targeting aberrant splicing in other hematologic genes. For example, SSOs have been shown to induce a pro-apoptotic BCL-X(S) isoform, decreasing cancer cell viability in preclinical leukemia models [40], and SSOs were designed to skip NOTCH1 exon 27, restoring protein function in model systems with the NOTCH1 splicing defect [3,22,63,64].
SSO-mediated restoration of RUNX1 isoform balance holds significant promise as a precision therapy for RUNX1-driven hematologic malignancies. Based on current knowledge from splice-switching therapies and delivery technologies, it would be valuable for future studies to explore and carefully access RUNX1-targeted splice-switching oligonucleotides as a potential strategy to modulate leukemogenic processes in hematopoietic malignancies.
The expression of RUNX1 isoforms is tightly linked to chromatin state and enhancer accessibility. Targeting these upstream regulators may offer indirect but effective modulation of RUNX1 function. BET inhibitors (e.g., JQ1) disrupt enhancer–promoter interactions and have been shown to downregulate aberrant RUNX1A expression in some leukemias. HDAC inhibitors and DNA methyltransferase inhibitors may reprogram promoter accessibility, particularly in leukemias with epigenetic silencing of P2. Changes in chromatin structure, such as those caused by cohesin mutations, can alter the accessibility and function of RUNX1 regulatory elements, implying that upstream regulators of chromatin state could indirectly modulate RUNX1 isoform output. These epigenetic drugs are already in clinical use or trials for AML and MDS and may be repurposed or combined with isoform-specific therapies [13,26,57].
Recent studies have identified tissue- and lineage-specific enhancers that regulate RUNX1 promoter usage and transcriptional output. For example, enhancers bound by GATA1 and TAL1 in megakaryocytes preferentially loop to P2. Deletion of specific enhancers can shift expression toward RUNX1C or RUNX1A. This opens avenues for enhancer-targeted therapies, including CRISPR-mediated promoter editing may enable selective activation or repression of P1 or P2, rebalancing isoform expression. Although these approaches remain largely preclinical, their potential for precision therapy is high, especially in pediatric leukemias with defined RUNX1 dysregulation. CRISPR-mediated promoter editing (CRISPRi/CRISPRa) can selectively activate or repress P1 or P2, rationally rebalancing RUNX1 isoform expression. The specificity of these strategies could allow modulation of RUNX1 isoforms in leukemic cells while sparing normal hematopoiesis [28,48,65,66].
Recent mechanistic studies directly support the existence of lineage-specific enhancers that regulate RUNX1 promoter usage and isoform output [28,48,65]. Disrupting these enhancers via CRISPRi, small molecules, or PROTACs is a plausible, though still emerging, strategy for modulating RUNX1 isoforms with lineage specificity [66,67,68,69]. While the therapeutic concept is robust, direct clinical application to RUNX1 enhancers awaits further preclinical validation.
6. Conclusions
RUNX1 isoforms exemplify how alternative promoter usage and splicing diversify transcription factor function, profoundly influencing hematopoietic development and leukemogenesis. The balanced interplay of RUNX1A, RUNX1B, and RUNX1C isoforms is essential for maintaining hematopoietic stem cell emergence, progenitor expansion, and lineage fidelity. Disruption of this balance—whether through germline mutations, somatic alterations, splicing defects, or epigenetic dysregulation—underlies pathogenesis in multiple hematologic malignancies, including Down syndrome-associated myeloid leukemia (ML-DS), familial platelet disorder with predisposition to myeloid malignancy (FPDMM), and RUNX1-mutated acute myeloid leukemia (AML).
Emerging mechanistic insights, facilitated by single-cell, long-read transcriptomics and CRISPR-based genomic dissection, highlight significant species-specific isoform regulation and reveal the critical roles of human-specific isoforms like RUNX1A. These discoveries underscore the limitations of traditional animal models and emphasize the need for human-relevant systems in studying RUNX1-driven diseases.
Therapeutic development is shifting toward isoform-specific strategies aimed at restoring normal RUNX1 isoform balance rather than global inhibition. Promising approaches include splice-switching oligonucleotides to correct RUNX1A overexpression, CRISPR-mediated promoter modulation to rebalance isoform expression, and enhancer-targeted therapies that selectively modulate transcriptional regulation [66,67]. Adjunctive modalities, such as epigenetic drugs, small molecule inhibitors targeting isoform-specific pathways, and immunotherapies exploiting RUNX1-associated surface markers, offer further opportunities for tailored intervention.
Future research priorities should focus on: (i) elucidating isoform-specific transcriptional networks, (ii) identifying the splicing factors and epigenetic regulators that govern isoform expression, and (iii) developing and refining isoform-selective therapeutics with high specificity and minimal off-target effects.
By embracing the complexity and nuances of RUNX1 isoform biology, we can deepen our understanding of hematopoietic regulation and leukemogenesis and pave the way for precision medicine strategies that improve outcomes in RUNX1-driven hematologic malignancies.
Author Contributions
Conceptualization, K.L.; literature research and screening, K.L. and S.K.; figure design and preparation, K.L.; writing—original draft, K.L. and S.K.; writing—review and editing, K.L. and S.K.; funding acquisition, K.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Hankuk University of Foreign Studies Research Fund; the Gyeonggi Regional Innovation System & Education Project (Gyeonggi RISE Project), funded by the Ministry of Education and Gyeonggi Province; and the National Research Foundation of Korea (NRF) grant funded by the Ministry of Science and ICT (MSIT) (grant number RS-2025-00556460).
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
We thank Mortimer Poncz and Hyun Sook Ahn for their insights and support.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ichikawa, M.; Asai, T.; Chiba, S.; Kurokawa, M.; Ogawa, S. Runx1/AML-1 ranks as a master regulator of adult hematopoiesis. Cell Cycle 2004, 3, 720–722. [Google Scholar] [CrossRef]
- Bonifer, C.; Levantini, E.; Kouskoff, V.; Lacaud, G. Runx1 Structure and Function in Blood Cell Development. Adv. Exp. Med. Biol. 2017, 962, 65–81. [Google Scholar]
- Sood, R.; Kamikubo, Y.; Liu, P. Role of RUNX1 in hematological malignancies. Blood 2017, 129, 2070–2082. [Google Scholar] [PubMed]
- Okuda, T.; van Deursen, J.; Hiebert, S.W.; Grosveld, G.; Downing, J.R. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 1996, 84, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.J.; Yokomizo, T.; Zeigler, B.M.; Dzierzak, E.; Speck, N.A. Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature 2009, 457, 887–891. [Google Scholar] [CrossRef]
- Kuo, M.C.; Liang, D.C.; Huang, C.F.; Shih, Y.S.; Wu, J.H.; Lin, T.L.; Shih, L.Y. RUNX1 mutations are frequent in chronic myelomonocytic leukemia and mutations at the C-terminal region might predict acute myeloid leukemia transformation. Leukemia 2009, 23, 1426–1431. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.; Zhang, D.E. RUNX1 and RUNX1-ETO: Roles in hematopoiesis and leukemogenesis. Front. Biosci. (Landmark Ed.) 2012, 17, 1120–1139. [Google Scholar] [CrossRef]
- Yokota, A.; Huo, L.; Lan, F.; Wu, J.; Huang, G. The Clinical, Molecular, and Mechanistic Basis of RUNX1 Mutations Identified in Hematological Malignancies. Mol. Cells 2020, 43, 145–152. [Google Scholar]
- Hayashi, Y.; Harada, Y.; Harada, H. Myeloid neoplasms and clonal hematopoiesis from the RUNX1 perspective. Leukemia 2022, 36, 1203–1214. [Google Scholar] [CrossRef]
- Sroczynska, P.; Lancrin, C.; Kouskoff, V.; Lacaud, G. The differential activities of Runx1 promoters define milestones during embryonic hematopoiesis. Blood 2009, 114, 5279–5289. [Google Scholar] [CrossRef]
- Challen, G.A.; Goodell, M.A. Runx1 isoforms show differential expression patterns during hematopoietic development but have similar functional effects in adult hematopoietic stem cells. Exp. Hematol. 2010, 38, 403–416. [Google Scholar] [CrossRef]
- Menegatti, S.; Potts, B.; Garcia-Alegria, E.; Paredes, R.; Lie-A.-Ling, M.; Lacaud, G.; Kouskoff, V. The RUNX1b Isoform Defines Hemogenic Competency in Developing Human Endothelial Cells. Front. Cell Dev. Biol. 2021, 9, 812639. [Google Scholar] [CrossRef]
- Guan, L.; Voora, D.; Myers, R.; Del Carpio-Cano, F.; Rao, A.K. RUNX1 isoforms regulate RUNX1 and target genes differentially in platelets-megakaryocytes: Association with clinical cardiovascular events. J. Thromb. Haemost. 2024, 22, 3581–3598. [Google Scholar] [CrossRef]
- Brady, G.; Elgueta Karstegl, C.; Farrell, P.J. Novel function of the unique N-terminal region of RUNX1c in B cell growth regulation. Nucleic Acids Res. 2013, 41, 1555–1568. [Google Scholar] [CrossRef]
- Komeno, Y.; Yan, M.; Matsuura, S.; Lam, K.; Lo, M.C.; Huang, Y.J.; Tenen, D.G.; Downing, J.R.; Zhang, D.E. Runx1 exon 6-related alternative splicing isoforms differentially regulate hematopoiesis in mice. Blood 2014, 123, 3760–3769. [Google Scholar] [CrossRef] [PubMed]
- Draper, J.E.; Sroczynska, P.; Tsoulaki, O.; Leong, H.S.; Fadlullah, M.Z.; Miller, C.; Kouskoff, V.; Lacaud, G. RUNX1B Expression Is Highly Heterogeneous and Distinguishes Megakaryocytic and Erythroid Lineage Fate in Adult Mouse Hematopoiesis. PLoS Genet. 2016, 12, e1005814. [Google Scholar]
- Davis, A.G.; Einstein, J.M.; Zheng, D.; Jayne, N.D.; Fu, X.D.; Tian, B.; Yeo, G.W.; Zhang, D.E. A CRISPR RNA-binding protein screen reveals regulators of RUNX1 isoform generation. Blood Adv. 2021, 5, 1310–1323. [Google Scholar] [CrossRef] [PubMed]
- Jayne, N.D.; Liang, Z.; Lim, D.H.; Chen, P.B.; Diaz, C.; Arimoto, K.I.; Xia, L.; Liu, M.; Ren, B.; Fu, X.D.; et al. RUNX1 C-terminal mutations impair blood cell differentiation by perturbing specific enhancer-promoter networks. Blood Adv. 2024, 8, 2410–2423. [Google Scholar] [CrossRef]
- Neldeborg, S.; Soerensen, J.F.; Møller, C.T.; Bill, M.; Gao, Z.; Bak, R.O.; Holm, K.; Sorensen, B.; Nyegaard, M.; Luo, Y.; et al. Dual intron-targeted CRISPR-Cas9-mediated disruption of the AML RUNX1-RUNX1T1 fusion gene effectively inhibits proliferation and decreases tumor volume in vitro and in vivo. Leukemia 2023, 37, 1792–1801. [Google Scholar] [CrossRef]
- Khan, N.M.; Wilderman, A.; Kaiser, J.M.; Kamalakar, A.; Goudy, S.L.; Cotney, J.; Drissi, H. Enhanced osteogenic potential of iPSC-derived mesenchymal progenitor cells following genome editing of GWAS variants in the RUNX1 gene. Bone Res. 2024, 12, 70. [Google Scholar] [CrossRef]
- Grinev, V.V.; Barneh, F.; Ilshonak, I.M.; Nakjang, S.; Smink, J.; van Oort, A.; Clough, R.; Seyani, M.; McNeill, H.; Reza, M.; et al. RUNX1/RUNX1T1 mediates alternative splicing and reorganises the transcriptional landscape in leukemia. Nat. Commun. 2021, 12, 520. [Google Scholar] [CrossRef]
- Gialesaki, S.; Bräuer-Hartmann, D.; Issa, H.; Bhayadia, R.; Alejo-Valle, O.; Verboon, L.; Schmell, A.L.; Laszig, S.; Regényi, E.; Schuschel, K.; et al. RUNX1 isoform disequilibrium promotes the development of trisomy 21-associated myeloid leukemia. Blood 2023, 141, 1105–1118. [Google Scholar] [CrossRef]
- Decker, M.; Lammens, T.; Ferster, A.; Erlacher, M.; Yoshimi, A.; Niemeyer, C.M.; Ernst, M.P.T.; Raaijmakers, M.H.G.P.; Duployez, N.; Flaum, A.; et al. Functional classification of RUNX1 variants in familial platelet disorder with associated myeloid malignancies. Leukemia 2021, 35, 3304–3308. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Deuitch, N.; Merguerian, M.; Cunningham, L.; Davis, J.; Bresciani, E.; Diemer, J.; Andrews, E.; Young, A.; Donovan, F.; et al. Genomic landscape of patients with germline RUNX1 variants and familial platelet disorder with myeloid malignancy. Blood Adv. 2024, 8, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Levanon, D.; Glusman, G.; Bangsow, T.; Ben-Asher, E.; Male, D.A.; Avidan, N.; Bangsow, C.; Hattori, M.; Taylor, T.D.; Taudien, S.; et al. Architecture and anatomy of the genomic locus encoding the human leukemia-associated transcription factor RUNX1/AML1. Gene 2001, 262, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Hinojosa, M.; Trombly, D.; Morin, V.; Stein, J.; Stein, G.; Javed, A.; Gutierrez, S.E. Transcriptional Auto-Regulation of RUNX1 P1 Promoter. PLoS ONE 2016, 11, e0149119. [Google Scholar] [CrossRef]
- Riddell, A.; McBride, M.; Braun, T.; Nicklin, S.A.; Cameron, E.; Loughrey, C.M.; Martin, T.P. RUNX1: An emerging therapeutic target for cardiovascular disease. Cardiovasc. Res. 2020, 116, 1410–1423. [Google Scholar] [CrossRef]
- Owens, D.D.G.; Anselmi, G.; Oudelaar, A.M.; Downes, D.J.; Cavallo, A.; Harman, J.R.; Schwessinger, R.; Bucakci, A.; Greder, L.; de Ornellas, S.; et al. Dynamic Runx1 chromatin boundaries affect gene expression in hematopoietic development. Nat. Commun. 2022, 13, 773. [Google Scholar] [CrossRef]
- Miyoshi, H.; Ohira, M.; Shimizu, K.; Mitani, K.; Hirai, H.; Imai, T.; Yokoyama, K.; Soeda, E.; Ohki, M. Alternative splicing and genomic structure of the AML1 gene involved in acute myeloid leukemia. Nucleic Acids Res. 1995, 23, 2762–2769. [Google Scholar] [CrossRef] [PubMed]
- Barutcu, A.R.; Hong, D.; Lajoie, B.R.; McCord, R.P.; van Wijnen, A.J.; Lian, J.B.; Stein, J.L.; Dekker, J.; Imbalzano, A.N.; Stein, G.S. RUNX1 contributes to higher-order chromatin organization and gene regulation in breast cancer cells. Biochim. Biophys. Acta 2016, 1859, 1389–1397. [Google Scholar] [CrossRef]
- Imai, Y.; Kurokawa, M.; Tanaka, K.; Friedman, A.D.; Ogawa, S.; Mitani, K.; Yazaki, Y.; Hirai, H. TLE, the human homolog of Groucho, interacts with AML1 and acts as a repressor of AML1-induced transactivation. Biochem. Biophys. Res. Commun. 1998, 252, 582–589. [Google Scholar] [CrossRef]
- Kellaway, S.G.; Keane, P.; Edginton-White, B.; Regha, K.; Kennett, E.; Bonifer, C. Different mutant RUNX1 oncoproteins program alternate haematopoietic differentiation trajectories. Life Sci. Alliance 2021, 4, e202000864. [Google Scholar] [CrossRef] [PubMed]
- Lutterbach, B.; Hiebert, S.W. Role of the transcription factor AML-1 in acute leukemia and hematopoietic differentiation. Gene 2000, 245, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Yzaguirre, A.D.; de Bruijn, M.F.; Speck, N.A. The Role of Runx1 in Embryonic Blood Cell Formation. Adv. Exp. Med. Biol. 2017, 962, 47–64. [Google Scholar]
- Morino-Koga, S.; Yokomizo, T. Deciphering hematopoietic stem cell development: Key signaling pathways and mechanisms. Front. Cell Dev. Biol. 2024, 12, 1510198. [Google Scholar] [CrossRef]
- Draper, J.E.; Sroczynska, P.; Leong, H.S.; Fadlullah, M.Z.H.; Miller, C.; Kouskoff, V.; Lacaud, G. Mouse RUNX1C regulates premegakaryocytic/erythroid output and maintains survival of megakaryocyte progenitors. Blood 2017, 130, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Estevez, B.; Borst, S.; Jarocha, D.; Sudunagunta, V.; Gonzalez, M.; Garifallou, J.; Hakonarson, H.; Gao, P.; Tan, K.; Liu, P.; et al. RUNX-1 haploinsufficiency causes a marked deficiency of megakaryocyte-biased hematopoietic progenitor cells. Blood 2021, 137, 2662–2675. [Google Scholar] [CrossRef]
- Lee, B.C.; Zhou, Y.; Bresciani, E.; Ozkaya, N.; Dulau-Florea, A.; Carrington, B.; Shin, T.H.; Baena, V.; Syed, Z.A.; Hong, S.G.; et al. A RUNX1-FPDMM rhesus macaque model reproduces the human phenotype and predicts challenges to curative gene therapies. Blood 2023, 141, 231–237. [Google Scholar] [CrossRef]
- Sakurai, H.; Harada, Y.; Ogata, Y.; Kagiyama, Y.; Shingai, N.; Doki, N.; Ohashi, K.; Kitamura, T.; Komatsu, N.; Harada, H. Overexpression of RUNX1 short isoform has an important role in the development of myelodysplastic/myeloproliferative neoplasms. Blood Adv. 2017, 1, 1382–1386. [Google Scholar] [CrossRef][Green Version]
- Bender, A.; Boydere, F.; Jayavelu, A.K.; Tibello, A.; König, T.; Aleth, H.; Meyer Zu Hörste, G.; Vogl, T.; Rosenbauer, F. Redistribution of PU.1 partner transcription factor RUNX1 binding secures cell survival during leukemogenesis. EMBO J. 2024, 43, 6291–6309. [Google Scholar] [CrossRef]
- Wilkinson, A.; Ballabio, E.; Geng, H.; North, P.; Tapia, M.; Kerry, J.; Biswas, D.; Roeder, R.; Allis, C.; Melnick, A.; et al. RUNX1 is a key target in t(4;11) leukemias that contributes to gene activation through an AF4-MLL complex interaction. Cell Rep. 2013, 3, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Ahn, H.S.; Estevez, B.; Poncz, M. RUNX1-deficient human megakaryocytes demonstrate thrombopoietic and platelet half-life and functional defects. Blood 2023, 141, 260–270. [Google Scholar] [CrossRef]
- Liu, S.; Yang, J.; Sun, G.; Zhang, Y.; Cheng, C.; Xu, J.; Yen, K.; Lu, T. RUNX1 Upregulates CENPE to Promote Leukemic Cell Proliferation. Front. Mol. Biosci. 2021, 8, 692880. [Google Scholar] [CrossRef] [PubMed]
- Bellissimo, D.; Chen, C.; Zhu, Q.; Bagga, S.; Lee, C.; He, B.; Wertheim, G.; Jordan, M.; Tan, K.; Worthen, G.; et al. Runx1 negatively regulates inflammatory cytokine production by neutrophils in response to Toll-like receptor signaling. Blood Adv. 2020, 4, 1145–1158. [Google Scholar] [CrossRef]
- Zezulin, A.; Yen, D.; Ye, D.; Howell, E.; Bresciani, E.; Diemer, J.; Ren, J.; Ahmad, M.; Castilla, L.; Touw, I.; et al. RUNX1 is required in granulocyte-monocyte progenitors to attenuate inflammatory cytokine production by neutrophils. Genes. Dev. 2023, 37, 605–620. [Google Scholar] [CrossRef]
- Mohammadhosseini, M.; Enright, T.; Duvall, A.; Chitsazan, A.; Lin, H.; Ors, A.; Davis, B.; Nikolova, O.; Bresciani, E.; Diemer, J.; et al. Targeting the CD74 signaling axis suppresses inflammation and rescues defective hematopoiesis in RUNX1-familial platelet disorder. Sci. Transl. Med. 2025, 17, eadn9832. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Gao, L.; Teng, L.; Ge, J.; Oo, Z.; Kumar, A.; Gilliland, D.; Mason, P.; Tan, K.; Speck, N. Runx1 Deficiency Decreases Ribosome Biogenesis and Confers Stress Resistance to Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 2015, 17, 165–177. [Google Scholar] [CrossRef]
- Bee, T.; Liddiard, K.; Swiers, G.; Bickley, S.R.; Vink, C.S.; Jarratt, A.; Hughes, J.R.; Medvinsky, A.; de Bruijn, M.F. Alternative Runx1 promoter usage in mouse developmental hematopoiesis. Blood Cells Mol. Dis. 2009, 43, 35–42. [Google Scholar] [CrossRef]
- Bee, T.; Swiers, G.; Muroi, S.; Pozner, A.; Nottingham, W.; Santos, A.C.; Li, P.S.; Taniuchi, I.; de Bruijn, M.F. Nonredundant roles for Runx1 alternative promoters reflect their activity at discrete stages of developmental hematopoiesis. Blood 2010, 115, 3042–3050. [Google Scholar] [CrossRef]
- Ferrell, P.I.; Xi, J.; Ma, C.; Adlakha, M.; Kaufman, D.S. The RUNX1 +24 enhancer and P1 promoter identify a unique subpopulation of hematopoietic progenitor cells derived from human pluripotent stem cells. Stem Cells 2015, 33, 1130–1141. [Google Scholar] [CrossRef][Green Version]
- Navarro-Montero, O.; Ayllon, V.; Lamolda, M.; López-Onieva, L.; Montes, R.; Bueno, C.; Ng, E.; Guerrero-Carreno, X.; Romero, T.; Romero-Moya, D.; et al. RUNX1c Regulates Hematopoietic Differentiation of Human Pluripotent Stem Cells Possibly in Cooperation with Proinflammatory Signaling. Stem Cells 2017, 35, 2253–2266. [Google Scholar] [CrossRef]
- Guo, H.; Ma, O.; Speck, N.A.; Friedman, A.D. Runx1 deletion or dominant inhibition reduces Cebpa transcription via conserved promoter and distal enhancer sites to favor monopoiesis over granulopoiesis. Blood 2012, 119, 4408–4418. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.; Guo, H.; Friedman, A.D. The +37 kb Cebpa Enhancer Is Critical for Cebpa Myeloid Gene Expression and Contains Functional Sites that Bind SCL, GATA2, C/EBPα, PU.1, and Additional Ets Factors. PLoS ONE 2015, 10, e0126385. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ohmori, S.; Moriguchi, T.; Noguchi, Y.; Ikeda, M.; Kobayashi, K.; Tomaru, N.; Ishijima, Y.; Ohneda, O.; Yamamoto, M.; Ohneda, K. GATA2 is critical for the maintenance of cellular identity in differentiated mast cells derived from mouse bone marrow. Blood 2015, 125, 3306–3315. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Zhou, L.; Wang, H.; Chen, N.; Jia, L.; Wang, C.; Wang, Y.; Chen, J.; Wen, X.; Niu, C.; et al. Profiling the epigenetic interplay of lncRNA RUNXOR and oncogenic RUNX1 in breast cancer cells by gene in situ cis-activation. Am. J. Cancer Res. 2019, 9, 1635–1649. [Google Scholar]
- Webber, B.R.; Iacovino, M.; Choi, S.H.; Tolar, J.; Kyba, M.; Blazar, B.R. DNA methylation of Runx1 regulatory regions correlates with transition from primitive to definitive hematopoietic potential in vitro and in vivo. Blood 2013, 122, 2978–2986. [Google Scholar] [CrossRef]
- Thomas, A.L.; Marsman, J.; Antony, J.; Schierding, W.; O’Sullivan, J.M.; Horsfield, J.A. Transcriptional Regulation of RUNX1: An Informatics Analysis. Genes 2021, 12, 1175. [Google Scholar] [CrossRef]
- Yi, H.; He, Y.; Zhu, Q.; Fang, L. RUNX Proteins as Epigenetic Modulators in Cancer. Cells 2022, 11, 3687. [Google Scholar] [CrossRef]
- Phillips-Cremins, J.E.; Sauria, M.E.; Sanyal, A.; Gerasimova, T.I.; Lajoie, B.R.; Bell, J.S.; Ong, C.T.; Hookway, T.A.; Guo, C.; Sun, Y.; et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 2013, 153, 1281–1295. [Google Scholar] [CrossRef]
- Pugacheva, E.M.; Kubo, N.; Loukinov, D.; Tajmul, M.; Kang, S.; Kovalchuk, A.L.; Strunnikov, A.V.; Zentner, G.E.; Ren, B.; Lobanenkov, V.V. CTCF mediates chromatin looping via N-terminal domain-dependent cohesin retention. Proc. Natl. Acad. Sci. USA 2020, 117, 2020–2031. [Google Scholar] [CrossRef]
- Davidson, I.F.; Barth, R.; Zaczek, M.; van der Torre, J.; Tang, W.; Nagasaka, K.; Janissen, R.; Kerssemakers, J.; Wutz, G.; Dekker, C.; et al. CTCF is a DNA-tension-dependent barrier to cohesin-mediated loop extrusion. Nature 2023, 616, 822–827. [Google Scholar] [CrossRef]
- Mercatante, D.R.; Mohler, J.L.; Kole, R. Cellular response to an antisense-mediated shift of Bcl-x pre-mRNA splicing and antineoplastic agents. J. Biol. Chem. 2002, 277, 49374–49382. [Google Scholar] [CrossRef]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Tijssen, M.R.; Cvejic, A.; Joshi, A.; Hannah, R.L.; Ferreira, R.; Forrai, A.; Bellissimo, D.C.; Oram, S.H.; Smethurst, P.A.; Wilson, N.K.; et al. Genome-wide analysis of simultaneous GATA1/2, RUNX1, FLI1, and SCL binding in megakaryocytes identifies hematopoietic regulators. Dev. Cell 2011, 20, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, F.; Barthélémy, A.; Peyrouze, P.; Fenwarth, L.; Preudhomme, C.; Duployez, N.; Cheok, M. Targeting RUNX1 in acute myeloid leukemia: Preclinical innovations and therapeutic implications. Expert. Opin. Ther. Targets 2021, 4, 299–309. [Google Scholar] [CrossRef]
- Mill, C.; Fiskus, W.; DiNardo, C.; Qian, Y.; Raina, K.; Rajapakshe, K.; Perera, D.; Coarfa, C.; Kadia, T.; Khoury, J.; et al. RUNX1-targeted therapy for AML expressing somatic or germline mutation in RUNX1. Blood 2019, 134, 59–73. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef]
- Ottis, P.; Crews, C.M. Proteolysis-Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy. ACS Chem. Biol. 2017, 12, 892–898. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).