What Is Still Unclear or Unresolved in Classic Hodgkin Lymphoma Pathobiology, Diagnosis, and Treatment




1. Introduction
2. Diagnostic Challenges and Uncertainties
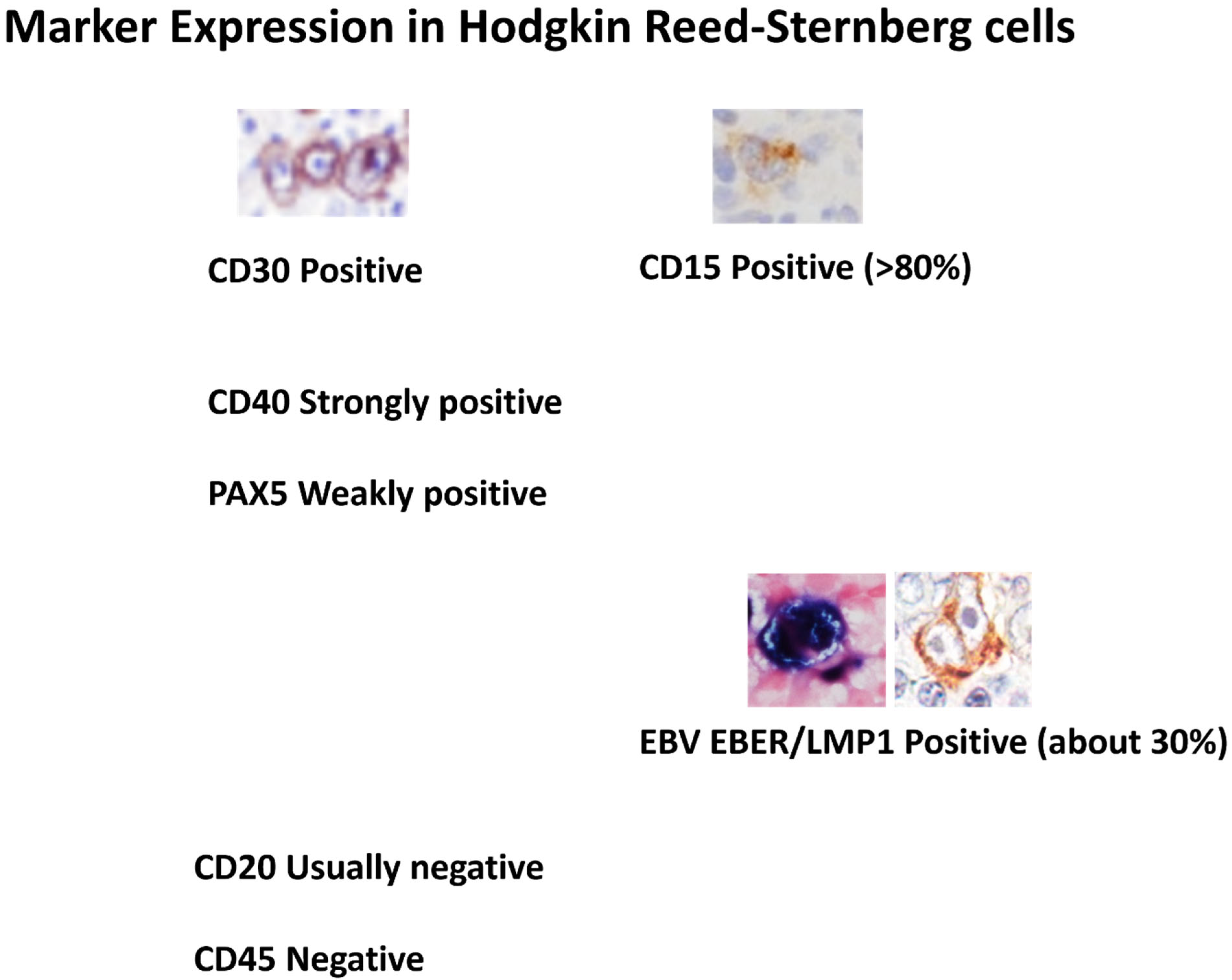
2.1. Lack of a Specific Marker for Diagnosis
2.2. Morphologic Variability of Diagnostic Tumor Cells
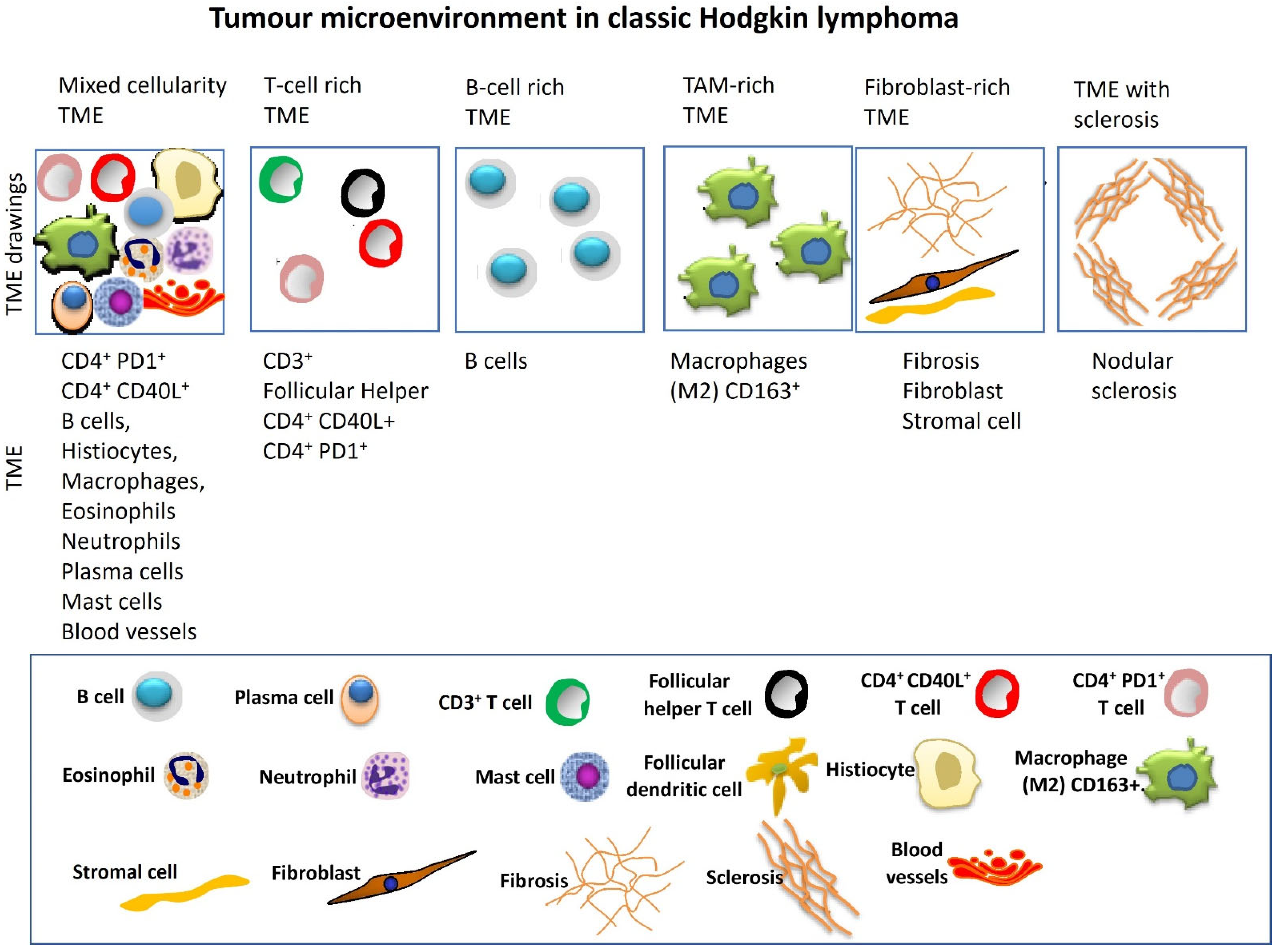
2.3. Tumor Microenvironment Variability
2.4. Extent of Disease at Presentation Variability
2.5. Overlap with Other Lymphoma Entities
3. Treatment Challenges and Unmet Needs
3.1. Refractoriness and Relapse Rates
3.2. Brentuximab and Checkpoint Inhibitor Resistance
4. Future Directions and Open Questions
4.1. Diagnostic Biomarkers
4.2. Microenvironment Targeting
4.3. Immunotherapy Optimization
4.4. Innovative Technologies
5. Conclusions
6. Summary
- Diagnostic ambiguity: cHL lacks a pathognomonic marker; CD30/CD15 expression and HRS morphology overlap with grey-zone lymphomas and EBV-driven proliferations, leading to inter-observer variability.
- Microenvironmental complexity: Spatial and single-cell studies reveal prognostic immune niches (e.g., CXCR5+ HRS–CXCL13+ macrophage axis), yet these insights have not been translated into routine risk stratification or therapy.
- Resistance mechanisms: Predictors of failure to standard ABVD, brentuximab vedotin, or PD-1 blockade remain undefined. Data implicate MDR1 upregulation, HLA-II loss, and alternative checkpoints, but no biomarkers guide clinical decisions.
- Translational opportunities: Circulating tumor DNA, refined imaging, and next-generation patient-derived models promise better monitoring and drug testing but need validation.
Author Contributions
Conflicts of Interest
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, K.; Sundquist, K.; Sundquist, J.; Försti, A.; Hemminki, A.; Li, X. Familial risks and proportions describing population landscape of familial cancer. Cancers 2021, 13, 4385. [Google Scholar] [CrossRef] [PubMed]
- Glaser, S.L.; Jarrett, R.F. 1 The epidemiology of Hodgkin’s disease. Baillière’s Clin. Haematol. 1996, 9, 401–416. [Google Scholar] [CrossRef]
- Campo, E.; Jaffe, E.S.; Cook, J.R.; Quintanilla-Martinez, L.; Swerdlow, S.H.; Anderson, K.C.; Brousset, P.; Cerroni, L.; de Leval, L.; Dirnhofer, S.; et al. The international consensus classification of mature lymphoid neoplasms: A report from the clinical advisory committee. Blood J. Am. Soc. Hematol. 2022, 140, 1229–1253. [Google Scholar] [CrossRef]
- Küppers, R.; Engert, A.; Hansmann, M.-L. Hodgkin lymphoma. J. Clin. Investig. 2012, 122, 3439–3447. [Google Scholar] [CrossRef]
- National Cancer Institute, Surveillance, Epidemiology, and End Results Program. Cancer Stat Facts: Hodgkin Lymphoma; SEER: Bethesda, MD, USA, 2019.
- Marafioti, T.; Hummel, M.; Foss, H.-D.; Laumen, H.; Korbjuhn, P.; Anagnostopoulos, I.; Lammert, H.; Demel, G.; Theil, J.; Wirth, T.; et al. Hodgkin and Reed-Sternberg cells represent an expansion of a single clone originating from a germinal center B-cell with functional immunoglobulin gene rearrangements but defective immunoglobulin transcription. Blood J. Am. Soc. Hematol. 2000, 95, 1443–1450. [Google Scholar] [CrossRef]
- Kanzler, H.; Küppers, R.; Hansmann, M.-L.; Rajewsky, K. Hodgkin and Reed-Sternberg cells in Hodgkin’s disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J. Exp. Med. 1996, 184, 1495–1505. [Google Scholar] [CrossRef]
- Randall, M.P.; Spinner, M.A. Optimizing treatment for relapsed/refractory classic Hodgkin lymphoma in the era of immunotherapy. Cancers 2023, 15, 4509. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; International Agency for Research on Cancer: Lyon, France, 2008. [Google Scholar]
- Rassidakis, G.Z.; Medeiros, L.J.; Viviani, S.; Bonfante, V.; Nadali, G.-P.; Vassilakopoulos, T.P.; Mesina, O.; Herling, M.; Angelopoulou, M.K.; Giardini, R.; et al. CD20 expression in Hodgkin and Reed-Sternberg cells of classical Hodgkin’s disease: Associations with presenting features and clinical outcome. J. Clin. Oncol. 2002, 20, 1278–1287. [Google Scholar]
- Haluska, F.G.; Brufsky, A.M.; Canellos, G.P. The cellular biology of the Reed-Sternberg cell. Blood 1994, 84, 1005–1019. [Google Scholar] [CrossRef]
- Segal, G.H.; Kjeldsberg, C.R.; Smith, G.P.; Perkins, S.L. CD 30 antigen expression in florid immunoblastic proliferations: A clinicopathologic study of 14 cases. Am. J. Clin. Pathol. 1994, 102, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Pallesen, G.; Hamilton-Dutoit, S.J. Ki-1 (CD30) antigen is regularly expressed by tumor cells of embryonal carcinoma. Am. J. Pathol. 1988, 133, 446–450. [Google Scholar]
- Stein, H.; Foss, H.-D.; DürKop, H.; Marafioti, T.; Delsol, G.; Pulford, K.; Pileri, S.; Falini, B. CD30+ anaplastic large cell lymphoma: A review of its histopathologic, genetic, and clinical features. Blood J. Am. Soc. Hematol. 2000, 96, 3681–3695. [Google Scholar] [CrossRef]
- Stein, H.; Mason, D.; Gerdes, J.; O’connor, N.; Wainscoat, J.; Pallesen, G.; Gatter, K.; Falini, B.; Delsol, G.; Lemke, H.; et al. The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: Evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood 1985, 66, 848–858. [Google Scholar] [CrossRef]
- Horie, R.; Watanabe, T. CD30: Expression and function in health and disease. Semin. Immunol. 1998, 10, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Pileri, S.; Pizzolo, G.; Dürkop, H.; Flenghi, L.; Stirpe, F.; Martelli, M.; Stein, H. CD30 (Ki-1) molecule: A new cytokine receptor of the tumor necrosis factor receptor superfamily as a tool for diagnosis and immunotherapy. Blood 1995, 85, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sarwar, S.; Tome, M.E.; Billheimer, D.; Spier, C.; Smith, C.L.; Persky, D.; Schmelz, M. Optimizing assessment of CD30 expression in Hodgkin lymphoma by controlling for low expression. Histol. Histopathol. 2024, 39, 319–331. [Google Scholar]
- Menéndez, V.; Solórzano, J.L.; Fernández, S.; Montalbán, C.; García, J.F. The Hodgkin lymphoma immune microenvironment: Turning bad news into good. Cancers 2022, 14, 1360. [Google Scholar] [CrossRef]
- Aoki, T.; Wierzbicki, K.; Sun, S.; Steidl, C.; Giulino-Roth, L. Tumor-microenvironment and molecular biology of classic Hodgkin lymphoma in children, adolescents, and young adults. Front. Oncol. 2025, 15, 1515250. [Google Scholar] [CrossRef]
- Alonso-Álvarez, S.; Vidriales, M.B.; Caballero, M.D.; Blanco, O.; Puig, N.; Martin, A.; Peñarrubia, M.J.; Zato, E.; Galende, J.; Bárez, A.; et al. The number of tumor infiltrating T-cell subsets in lymph nodes from patients with Hodgkin lymphoma is associated with the outcome after first line ABVD therapy. Leuk. Lymphoma 2017, 58, 1144–1152. [Google Scholar] [CrossRef]
- Barros, M.H.M.; Vera-Lozada, G.; Soares, F.A.; Niedobitek, G.; Hassan, R. Tumor microenvironment composition in pediatric classical Hodgkin lymphoma is modulated by age and Epstein-Barr virus infection. Int. J. Cancer 2012, 131, 1142–1152. [Google Scholar] [CrossRef]
- Chetaille, B.; Bertucci, F.; Finetti, P.; Esterni, B.; Stamatoullas, A.; Picquenot, J.M.; Copin, M.C.; Morschhauser, F.; Casasnovas, O.; Petrella, T.; et al. Molecular profiling of classical Hodgkin lymphoma tissues uncovers variations in the tumor microenvironment and correlations with EBV infection and outcome. Blood J. Am. Soc. Hematol. 2009, 113, 2765–3775. [Google Scholar] [CrossRef]
- Pavlovic, A.; Durdov, M.G.; Capkun, V.; Pitesa, J.J.; Sakic, M.B. Classical hodgkin lymphoma with positive Epstein-Barr virus status is associated with more FOXP3 regulatory T cells. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2016, 22, 2340–2346. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Steidl, C. Novel insights into Hodgkin lymphoma biology by single-cell analysis. Blood 2023, 141, 1791–1801. [Google Scholar] [CrossRef] [PubMed]
- Carey, C.D.; Gusenleitner, D.; Lipschitz, M.; Roemer, M.G.; Stack, E.C.; Gjini, E.; Hu, X.; Redd, R.; Freeman, G.J.; Neuberg, D.; et al. Topological analysis reveals a PD-L1-associated microenvironmental niche for Reed-Sternberg cells in Hodgkin lymphoma. Blood J. Am. Soc. Hematol. 2017, 130, 2420–2430. [Google Scholar] [CrossRef]
- Cader, F.Z.; Schackmann, R.C.; Hu, X.; Wienand, K.; Redd, R.; Chapuy, B.; Ouyang, J.; Paul, N.; Gjini, E.; Lipschitz, M.; et al. Mass cytometry of Hodgkin lymphoma reveals a CD4+ regulatory T-cell–rich and exhausted T-effector microenvironment. Blood J. Am. Soc. Hematol. 2018, 132, 825–836. [Google Scholar] [CrossRef]
- Aoki, T.; Chong, L.C.; Takata, K.; Milne, K.; Hav, M.; Colombo, A.; Chavez, E.A.; Nissen, M.; Wang, X.; Miyata-Takata, T.; et al. Single-cell transcriptome analysis reveals disease-defining T-cell subsets in the tumor microenvironment of classic Hodgkin lymphoma. Cancer Discov. 2020, 10, 406–421. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Jiang, A.; Xu, A.; Yin, Y.; Gamboa, A.; Milne, K.; Takata, K.; Miyata-Takata, T.; Chung, S.; Rai, S.; et al. Spatially resolved tumor microenvironment predicts treatment outcomes in relapsed/refractory Hodgkin lymphoma. J. Clin. Oncol. 2024, 42, 1077–1087. [Google Scholar] [CrossRef]
- Xu, A.; Jiang, A.; Aoki, T.; Gamboa, A.; Chong, L.; Colombo, A.; Yin, Y.; Lownik, J.; Takata, K.; Hav, M.; et al. 125 Single cell spatial analysis and biomarker discovery in hodgkin lymphoma. J. Immunother. Cancer 2023, 11, 141. [Google Scholar]
- Cader, F.Z.; Hu, X.; Goh, W.L.; Wienand, K.; Ouyang, J.; Mandato, E.; Redd, R.; Lawton, L.N.; Chen, P.-H.; Weirather, J.L.; et al. A peripheral immune signature of responsiveness to PD-1 blockade in patients with classical Hodgkin lymphoma. Nat. Med. 2020, 26, 1468–1479. [Google Scholar] [CrossRef]
- Calabretta, E.; d’Amore, F.; Carlo-Stella, C. Immune and inflammatory cells of the tumor microenvironment represent novel therapeutic targets in classical hodgkin lymphoma. Int. J. Mol. Sci. 2019, 20, 5503. [Google Scholar] [CrossRef]
- Ferrarini, I.; Rigo, A.; Visco, C.; Krampera, M.; Vinante, F. The evolving knowledge on T and NK cells in classic Hodgkin lymphoma: Insights into novel subsets populating the immune microenvironment. Cancers 2020, 12, 3757. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Elliott, L.A.; Doherty, G.A.; Sheahan, K.; Ryan, E.J. Human tumor-infiltrating myeloid cells: Phenotypic and functional diversity. Front. Immunol. 2017, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Parisi, L.; Gini, E.; Baci, D.; Tremolati, M.; Fanuli, M.; Bassani, B.; Farronato, G.; Bruno, A.; Mortara, L. Macrophage polarization in chronic inflammatory diseases: Killers or builders? J. Immunol. Res. 2018, 2018, 8917804. [Google Scholar] [CrossRef]
- Oshi, M.; Tokumaru, Y.; Asaoka, M.; Yan, L.; Satyananda, V.; Matsuyama, R.; Matsuhashi, N.; Futamura, M.; Ishikawa, T.; Yoshida, K.; et al. M1 Macrophage and M1/M2 ratio defined by transcriptomic signatures resemble only part of their conventional clinical characteristics in breast cancer. Sci. Rep. 2020, 10, 16554. [Google Scholar] [CrossRef]
- Henry, M.; Buck, S.; Savaşan, S. Flow cytometry for assessment of the tumor microenvironment in pediatric Hodgkin lymphoma. Pediatr. Blood Cancer 2018, 65, e27307. [Google Scholar] [CrossRef]
- Scott, D.W.; Chan, F.C.; Hong, F.; Rogic, S.; Tan, K.L.; Meissner, B.; Ben-Neriah, S.; Boyle, M.; Kridel, R.; Telenius, A.; et al. Gene expression–based model using formalin-fixed paraffin-embedded biopsies predicts overall survival in advanced-stage classical Hodgkin lymphoma. J. Clin. Oncol. 2013, 31, 692–700. [Google Scholar] [CrossRef]
- Johnston, R.L.; Mottok, A.; Chan, F.C.; Jiang, A.; Diepstra, A.; Visser, L.; Telenius, A.; Gascoyne, R.D.; Friedman, D.L.; Schwartz, C.L.; et al. A gene expression–based model predicts outcome in children with intermediate-risk classical Hodgkin lymphoma. Blood J. Am. Soc. Hematol. 2022, 139, 889–893. [Google Scholar] [CrossRef]
- Castellino, S.M.; Pei, Q.; Parsons, S.K.; Hodgson, D.; McCarten, K.; Horton, T.; Cho, S.; Wu, Y.; Punnett, A.; Dave, H.; et al. Brentuximab vedotin with chemotherapy in pediatric high-risk Hodgkin’s lymphoma. N. Engl. J. Med. 2022, 387, 1649–1660. [Google Scholar] [CrossRef]
- Clarke, C.A.; Glaser, S.L.; Keegan, T.H.; Stroup, A. Neighborhood socioeconomic status and Hodgkin’s lymphoma incidence in California. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1441–1447. [Google Scholar] [CrossRef] [PubMed]
- Gulley, M.L.; Eagan, P.A.; Quintanilla-Martinez, L.; Picado, A.L.; Smir, B.N.; Childs, C.; Dunn, C.D.; Craig, F.E.; Williams, J.W., Jr.; Banks, P.M. Epstein-Barr virus DNA is abundant and monoclonal in the Reed-Sternberg cells of Hodgkin’s disease: Association with mixed cellularity subtype and Hispanic American ethnicity. Blood 1994, 83, 1595–1602. [Google Scholar] [CrossRef] [PubMed]
- Karube, K.; Niino, D.; Kimura, Y.; Ohshima, K. Classical Hodgkin lymphoma, lymphocyte depleted type: Clinicopathological analysis and prognostic comparison with other types of classical Hodgkin lymphoma. Pathol.-Res. Pract. 2013, 209, 201–207. [Google Scholar] [CrossRef]
- Klimm, B.; Franklin, J.; Stein, H.; Eichenauer, D.A.; Haverkamp, H.; Diehl, V.; Fuchs, M.; Borchmann, P.; Engert, A. Lymphocyte-depleted classical Hodgkin’s lymphoma: A comprehensive analysis from the German Hodgkin study group. J. Clin. Oncol. 2011, 29, 3914–3920. [Google Scholar] [CrossRef]
- Clavel, J.; Steliarova-Foucher, E.; Berger, C.; Danon, S.; Valerianova, Z. Hodgkin’s disease incidence and survival in European children and adolescents (1978–1997): Report from the Automated Cancer Information System project. Eur. J. Cancer 2006, 42, 2037–2049. [Google Scholar] [CrossRef] [PubMed]
- Diehl, V.; Sextro, M.; Franklin, J.; Hansmann, M.-L.; Harris, N.; Jaffe, E.; Poppema, S.; Harris, M.; Franssila, K.; van Krieken, J.; et al. Clinical presentation, course, and prognostic factors in lymphocyte-predominant Hodgkin’s disease and lymphocyte-rich classical Hodgkin’s disease: Report from the European Task Force on Lymphoma Project on Lymphocyte-Predominant Hodgkin’s Disease. J. Clin. Oncol. 1999, 17, 776. [Google Scholar] [CrossRef]
- Musshoff, K. Prognostic and therapeutic implications of staging in extranodal Hodgkin’s disease. Cancer Res. 1971, 31, 1814–1827. [Google Scholar]
- Bosch-Schips, J.; Granai, M.; Quintanilla-Martinez, L.; Fend, F. The grey zones of classic Hodgkin lymphoma. Cancers 2022, 14, 742. [Google Scholar] [CrossRef]
- Zhang, M.L.; Sohani, A.R. Lymphomas of the mediastinum and their differential diagnosis. Semin. Diagn. Pathol. 2020, 37, 156–165. [Google Scholar] [CrossRef]
- Egan, C.; Pittaluga, S. Into the Gray-Zone: Update on the Diagnosis and Classification of a Rare Lymphoma; Taylor & Francis: Abingdon, UK, 2020; pp. 1–3. [Google Scholar]
- Sarkozy, C.; Hung, S.S.; Chavez, E.A.; Duns, G.; Takata, K.; Chong, L.C.; Aoki, T.; Jiang, A.; Miyata-Takata, T.; Telenius, A.; et al. Mutational landscape of gray zone lymphoma. Blood J. Am. Soc. Hematol. 2021, 137, 1765–1776. [Google Scholar] [CrossRef]
- Elsayed, A.A.; Satou, A.; Eladl, A.E.; Kato, S.; Nakamura, S.; Asano, N. Grey zone lymphoma with features intermediate between diffuse large B-cell lymphoma and classical Hodgkin lymphoma: A clinicopathological study of 14 Epstein–Barr virus-positive cases. Histopathology 2017, 70, 579–594. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla-Martinez, L.; de Jong, D.; de Mascarel, A.; Hsi, E.D.; Kluin, P.; Natkunam, Y.; Parrens, M.; Pileri, S.; Ott, G. Gray zones around diffuse large B cell lymphoma. Conclusions based on the workshop of the XIV meeting of the European Association for Hematopathology and the Society of Hematopathology in Bordeaux, France. J. Hematop. 2009, 2, 211–236. [Google Scholar] [CrossRef]
- Eberle, F.C.; Rodriguez-Canales, J.; Wei, L.; Hanson, J.C.; Killian, J.K.; Sun, H.-W.; Adams, L.G.; Hewitt, S.M.; Wilson, W.H.; Pittaluga, S.; et al. Methylation profiling of mediastinal gray zone lymphoma reveals a distinctive signature with elements shared by classical Hodgkin’s lymphoma and primary mediastinal large B-cell lymphoma. Haematologica 2011, 96, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Eberle, F.C.; Salaverria, I.; Steidl, C.; Summers Jr, T.A.; Pittaluga, S.; Neriah, S.B.; Rodriguez-Canales, J.; Xi, L.; Ylaya, K.; Liewehr, D.; et al. Gray zone lymphoma: Chromosomal aberrations with immunophenotypic and clinical correlations. Mod. Pathol. 2011, 24, 1586–1597. [Google Scholar] [CrossRef]
- Seliem, R.M.; Ferry, J.A.; Hasserjian, R.P.; Harris, N.L.; Zukerberg, L.R. Nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL) with CD30-positive lymphocyte-predominant (LP) cells. J. Hematop. 2011, 4, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Montes-Moreno, S.; Odqvist, L.; Diaz-Perez, J.A.; Lopez, A.B.; De Villambrosía, S.G.; Mazorra, F.; E Castillo, M.; Lopez, M.; Pajares, R.; García, J.F.; et al. EBV-positive diffuse large B-cell lymphoma of the elderly is an aggressive post-germinal center B-cell neoplasm characterized by prominent nuclear factor-kB activation. Mod. Pathol. 2012, 25, 968–982. [Google Scholar] [CrossRef]
- Ok, C.Y.; Papathomas, T.G.; Medeiros, L.J.; Young, K.H. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood J. Am. Soc. Hematol. 2013, 122, 328–340. [Google Scholar] [CrossRef]
- Ikeda, T.; Gion, Y.; Sakamoto, M.; Tachibana, T.; Nishikori, A.; Nishimura, M.F.; Yoshino, T.; Sato, Y. Clinicopathological analysis of 34 Japanese patients with EBV-positive mucocutaneous ulcer. Mod. Pathol. 2020, 33, 2437–2448. [Google Scholar] [CrossRef]
- Oyama, T.; Ichimura, K.; Suzuki, R.; Suzumiya, J.; Ohshima, K.; Yatabe, Y.; Takio, Y.; Masaru, K.; Yoshikazu, K.; Hirofumi, T.; et al. Senile EBV+ B-cell lymphoproliferative disorders: A clinicopathologic study of 22 patients. Am. J. Surg. Pathol. 2003, 27, 16–26. [Google Scholar] [CrossRef]
- Pitman, S.D.; Huang, Q.; Zuppan, C.W.; Rowsell, E.H.; Cao, J.D.; Berdeja, J.G.; Weiss, L.M.; Wang, J. Hodgkin lymphoma-like posttransplant lymphoproliferative disorder (HL-like PTLD) simulates monomorphic B-cell PTLD both clinically and pathologically. Am. J. Surg. Pathol. 2006, 30, 470–476. [Google Scholar] [CrossRef]
- Oyama, T.; Yamamoto, K.; Asano, N.; Oshiro, A.; Suzuki, R.; Kagami, Y.; Morishima, Y.; Takeuchi, K.; Izumo, T.; Mori, S.; et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: A study of 96 patients. Clin. Cancer Res. 2007, 13, 5124–5132. [Google Scholar] [CrossRef]
- Müschen, M.; Rajewsky, K.; Bräuninger, A.; Baur, A.S.; Oudejans, J.J.; Roers, A.; Hansmann, M.-L.; Küppers, R. Rare occurrence of classical Hodgkin’s disease as a T cell lymphoma. J. Exp. Med. 2000, 191, 387–394. [Google Scholar] [CrossRef]
- Krenacs, L.; Wellmann, A.; Sorbara, L.; Himmelmann, A.W.; Bagdi, E.; Jaffe, E.S.; Raffeld, M. Cytotoxic cell antigen expression in anaplastic large cell lymphomas of T-and null-cell type and Hodgkin’s disease: Evidence for distinct cellular origin. Blood J. Am. Soc. Hematol. 1997, 89, 980–989. [Google Scholar] [CrossRef]
- Johnson, P.; Federico, M.; Kirkwood, A.; Fosså, A.; Berkahn, L.; Carella, A.; D’aMore, F.; Enblad, G.; Franceschetto, A.; Fulham, M.; et al. Adapted treatment guided by interim PET-CT scan in advanced Hodgkin’s lymphoma. N. Engl. J. Med. 2016, 374, 2419–2429. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M. Hodgkin lymphoma: 2018 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2018, 93, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Voorhees, T.J.; Beaven, A.W. Therapeutic updates for relapsed and refractory classical Hodgkin lymphoma. Cancers 2020, 12, 2887. [Google Scholar] [CrossRef]
- Voorhees, T.J.; McLaughlin, E.M.; Torka, P.; Florindez, J.; Kim, N.H.; Moyo, T.K.; Reves, H.; Sumransub, N.; Deshpande, S.; Rose, A.; et al. Outcomes in patients with classic Hodgkin lymphoma refractory or intolerant to brentuximab vedotin and anti-PD-1 therapy: A real world analysis from 15 US academic centers. Blood Cancer J. 2025, 15, 45. [Google Scholar] [CrossRef]
- Chen, R.; Hou, J.; Newman, E.; Kim, Y.; Donohue, C.; Liu, X.; Thomas, S.H.; Forman, S.J.; Kane, S.E. CD30 downregulation, MMAE resistance, and MDR1 upregulation are all associated with resistance to brentuximab vedotin. Mol. Cancer Ther. 2015, 14, 1376–1384. [Google Scholar] [CrossRef]
- Van der Weyden, C.; Pileri, S.; Feldman, A.; Whisstock, J.; Prince, H. Understanding CD30 biology and therapeutic targeting: A historical perspective providing insight into future directions. Blood Cancer J. 2017, 7, e603. [Google Scholar] [CrossRef]
- Nathwani, N.; Krishnan, A.Y.; Huang, Q.; Kim, Y.; Karanes, C.; Smith, E.P.; Forman, S.J.; Sievers, E.; Thomas, S.H.; Chen, R.W. Persistence of CD30 expression in Hodgkin lymphoma following brentuximab vedotin (SGN-35) treatment failure. Leuk. Lymphoma 2012, 53, 2051–2053. [Google Scholar] [CrossRef]
- Bera, D.; Roy, D. Brentuximab vedotin resistance in classic Hodgkin’s lymphoma and its therapeutic strategies: A review. Future J. Pharm. Sci. 2024, 10, 15. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Seebacher, N.A.; Krchniakova, M.; Stacy, A.E.; Skoda, J.; Jansson, P.J. Tumour microenvironment stress promotes the development of drug resistance. Antioxidants 2021, 10, 1801. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.P.; Paes Leme, A.F.; Hallek, M. Role of ADAM10 as a CD30 sheddase in classical Hodgkin lymphoma. Front. Immunol. 2020, 11, 398. [Google Scholar] [CrossRef]
- Kushekhar, K.; van den Berg, A.; Nolte, I.; Hepkema, B.; Visser, L.; Diepstra, A. Genetic associations in classical hodgkin lymphoma: A systematic review and insights into susceptibility mechanisms. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2737–2747. [Google Scholar] [CrossRef] [PubMed]
- Diepstra, A.; van Imhoff, G.W.; Karim-Kos, H.E.; van den Berg, A.; te Meerman, G.J.; Niens, M.; Nolte, I.M.; Bastiaannet, E.; Schaapveld, M.; Vellenga, E.; et al. HLA class II expression by Hodgkin Reed-Sternberg cells is an independent prognostic factor in classical Hodgkin’s lymphoma. J. Clin. Oncol. 2007, 25, 3101–3108. [Google Scholar] [CrossRef]
- Liu, Y.; Abdul Razak, F.; Terpstra, M.; Chan, F.; Saber, A.; Nijland, M.; Van Imhoff, G.; Visser, L.; Gascoyne, R.; Steidl, C.; et al. The mutational landscape of Hodgkin lymphoma cell lines determined by whole-exome sequencing. Leukemia 2014, 28, 2248–2251. [Google Scholar] [CrossRef]
- Steidl, C.; Shah, S.P.; Woolcock, B.W.; Rui, L.; Kawahara, M.; Farinha, P.; Johnson, N.A.; Zhao, Y.; Telenius, A.; Neriah, S.B.; et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature 2011, 471, 377–381. [Google Scholar] [CrossRef]
- Niens, M.; Visser, L.; Nolte, I.M.; Van Der Steege, G.; Diepstra, A.; Cordano, P.; Jarrett, R.F.; Meerman, G.J.T.; Poppema, S.; Van Der Berg, A. Serum chemokine levels in Hodgkin lymphoma patients: Highly increased levels of CCL17 and CCL22. Br. J. Haematol. 2008, 140, 527–536. [Google Scholar] [CrossRef]
- Ishida, T.; Ishii, T.; Inagaki, A.; Yano, H.; Komatsu, H.; Iida, S.; Inagaki, H.; Ueda, R. Specific recruitment of CC chemokine receptor 4–positive regulatory T cells in Hodgkin lymphoma fosters immune privilege. Cancer Res. 2006, 66, 5716–5722. [Google Scholar] [CrossRef] [PubMed]
- Ricciuti, B.; Leonardi, G.C.; Puccetti, P.; Fallarino, F.; Bianconi, V.; Sahebkar, A.; Baglivo, S.; Chiari, R.; Pirro, M. Targeting indoleamine-2, 3-dioxygenase in cancer: Scientific rationale and clinical evidence. Pharmacol. Ther. 2019, 196, 105–116. [Google Scholar] [CrossRef]
- Liu, X.-Q.; Lu, K.; Feng, L.-L.; Ding, M.; Gao, J.-M.; Ge, X.-L.; Wang, X. Up-regulated expression of indoleamine 2, 3-dioxygenase 1 in non-Hodgkin lymphoma correlates with increased regulatory T-cell infiltration. Leuk. Lymphoma 2014, 55, 405–414. [Google Scholar] [CrossRef]
- Young, A.; Ngiow, S.F.; Gao, Y.; Patch, A.-M.; Barkauskas, D.S.; Messaoudene, M.; Lin, G.; Coudert, J.D.; Stannard, K.A.; Zitvogel, L.; et al. A2AR adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Cancer Res. 2018, 78, 1003–1016. [Google Scholar] [CrossRef]
- Vari, F.; Arpon, D.; Keane, C.; Hertzberg, M.S.; Talaulikar, D.; Jain, S.; Cui, Q.; Han, E.; Tobin, J.; Bird, R.; et al. Immune evasion via PD-1/PD-L1 on NK cells and monocyte/macrophages is more prominent in Hodgkin lymphoma than DLBCL. Blood J. Am. Soc. Hematol. 2018, 131, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Veldman, J.; Visser, L.; van den Berg, A.; Diepstra, A. Primary and acquired resistance mechanisms to immune checkpoint inhibition in Hodgkin lymphoma. Cancer Treat. Rev. 2020, 82, 101931. [Google Scholar] [CrossRef] [PubMed]
- Camus, V.; Viennot, M.; Lequesne, J.; Viailly, P.-J.; Bohers, E.; Bessi, L.; Marcq, B.; Etancelin, P.; Dubois, S.; Picquenot, J.-M.; et al. Targeted genotyping of circulating tumor DNA for classical Hodgkin lymphoma monitoring: A prospective study. Haematologica 2020, 106, 154–162. [Google Scholar] [CrossRef]
- Sobesky, S.; Mammadova, L.; Cirillo, M.; Drees, E.E.; Mattlener, J.; Dörr, H.; Altmüller, J.; Shi, Z.; Bröckelmann, P.J.; Weiss, J.; et al. In-depth cell-free DNA sequencing reveals genomic landscape of Hodgkin’s lymphoma and facilitates ultrasensitive residual disease detection. Med 2021, 2, 1171–1193.e11. [Google Scholar] [CrossRef]
- Alig, S.K.; Shahrokh Esfahani, M.; Garofalo, A.; Li, M.Y.; Rossi, C.; Flerlage, T.; Flerlage, J.E.; Adams, R.; Binkley, M.S.; Shukla, N.; et al. Distinct Hodgkin lymphoma subtypes defined by noninvasive genomic profiling. Nature 2024, 625, 778–787. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Diagnostic Challenge | Details |
|---|---|
| Lack of disease-specific biomarkers | CD30 expression alone or CD30 and CD15 co-expression are non-specific, since they overlap with atypical lymphoproliferations and other lymphomas. |
| Morphologic variability of HRS cells | Diverse appearances (mononuclear, multinucleated, or lacunar); sometimes subtype-specific; variations cause diagnostic ambiguity. |
| Grey-zone lymphomas | Overlap with PMBL, NLPHL, DLBCL, PTCL, and EBV-driven proliferations, complicating definitive classification. |
| TME heterogeneity | Significant variation influenced by age, EBV status, and spatial organization complicates a standard diagnostic approach. |
| Inter-observer variability | Due to sparse malignant cells, variable morphology, fixation artifacts, or small biopsies. |
| Treatment Challenge | Details |
|---|---|
| Refractoriness and relapse rates | ~15–20% of cHL patients fail initial therapy; ~50% cure rate with salvage therapy (high-dose chemo + transplant) |
| Resistance to brentuximab vedotin | Driven by MDR1-mediated payload efflux, linker–payload cleavage issues, TME-mediated immune evasion, ectodomain shedding (ADAM10/17) |
| Resistance to PD-1 inhibitors | Involves impaired antigen presentation (CIITA/HLA-DM alterations), chemokine-driven T-cell suppression (Tregs), IDO and adenosine accumulation, alternative checkpoints (LAG-3, TIM-3) |
| Lack of predictive biomarkers | No validated biomarkers to predict therapeutic resistance or guide immunotherapy vs. chemotherapy decisions |
| Limited therapeutic options for double-refractory patients | Patients resistant to both BV and PD-1 inhibitors have poor outcomes; few effective alternatives (e.g., CAR-T or allo-transplantation) |
| Open Question | Potential Approaches and Opportunities |
|---|---|
| Diagnostic biomarkers | Development of molecular diagnostics, novel IHC markers, genomic signatures, and ctDNA assays |
| Targeting the tumor microenvironment | Exploiting TME biology, trials with LAG-3, CSF1R inhibitors, and biomarker-driven stratification |
| Optimization of immunotherapy | Clarifying the benefit of frontline vs. salvage checkpoint inhibitors; personalized therapy using biomarkers |
| Integrative technologies (spatial transcriptomics, single-cell profiling, liquid biopsy) | Integration into clinical trials; prognostic models like RHL4S, routine monitoring of minimal residual disease |
| Resolving grey-zone cases | Defining molecular criteria to distinguish true biological hybrids from phenotypic mimics |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carbone, A.; Alibrahim, M.N.; Gloghini, A. What Is Still Unclear or Unresolved in Classic Hodgkin Lymphoma Pathobiology, Diagnosis, and Treatment. Hemato 2025, 6, 20. https://doi.org/10.3390/hemato6030020
Carbone A, Alibrahim MN, Gloghini A. What Is Still Unclear or Unresolved in Classic Hodgkin Lymphoma Pathobiology, Diagnosis, and Treatment. Hemato. 2025; 6(3):20. https://doi.org/10.3390/hemato6030020
Chicago/Turabian StyleCarbone, Antonino, Mohamed Nazem Alibrahim, and Annunziata Gloghini. 2025. "What Is Still Unclear or Unresolved in Classic Hodgkin Lymphoma Pathobiology, Diagnosis, and Treatment" Hemato 6, no. 3: 20. https://doi.org/10.3390/hemato6030020
APA StyleCarbone, A., Alibrahim, M. N., & Gloghini, A. (2025). What Is Still Unclear or Unresolved in Classic Hodgkin Lymphoma Pathobiology, Diagnosis, and Treatment. Hemato, 6(3), 20. https://doi.org/10.3390/hemato6030020