
BET Protein Inhibition Relieves MDSC-Mediated Immune Suppression in Chronic Lymphocytic Leukemia

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Inhibitors/Drugs
2.2. Murine Studies
2.3. Immunoblot Analysis
2.4. MDSC Expansion
2.5. T-Cell:MDSC Co-Culture
2.6. Flow Cytometry
2.7. Cytotoxicity Assay
2.8. Statistics
3. Results
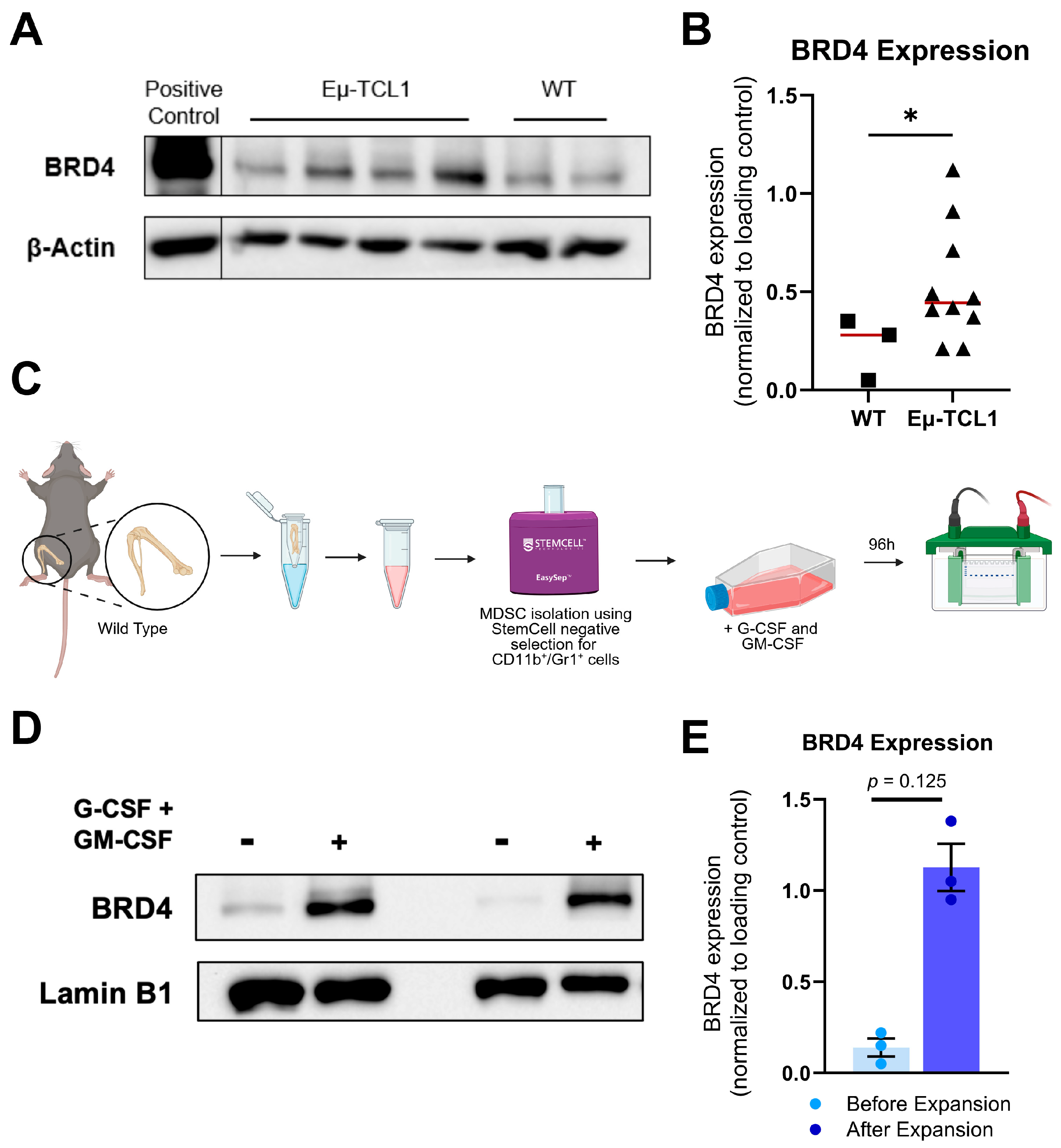
3.1. CLL-Associated MDSCs Overexpress BRD4
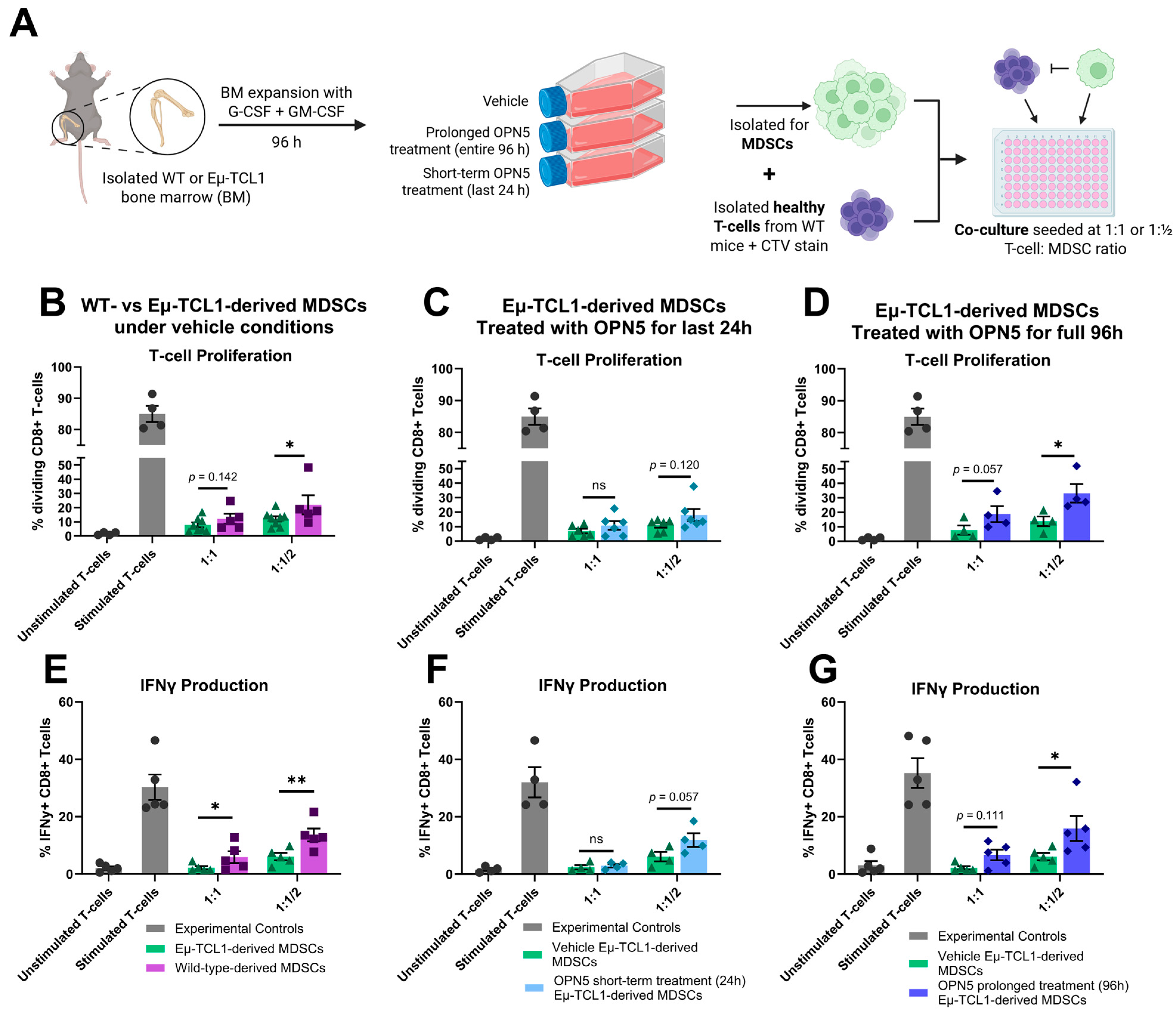
3.2. BET Protein Inhibition Alters MDSC Differentiation and Reduces Their Immune-Suppressive Activity Ex Vivo
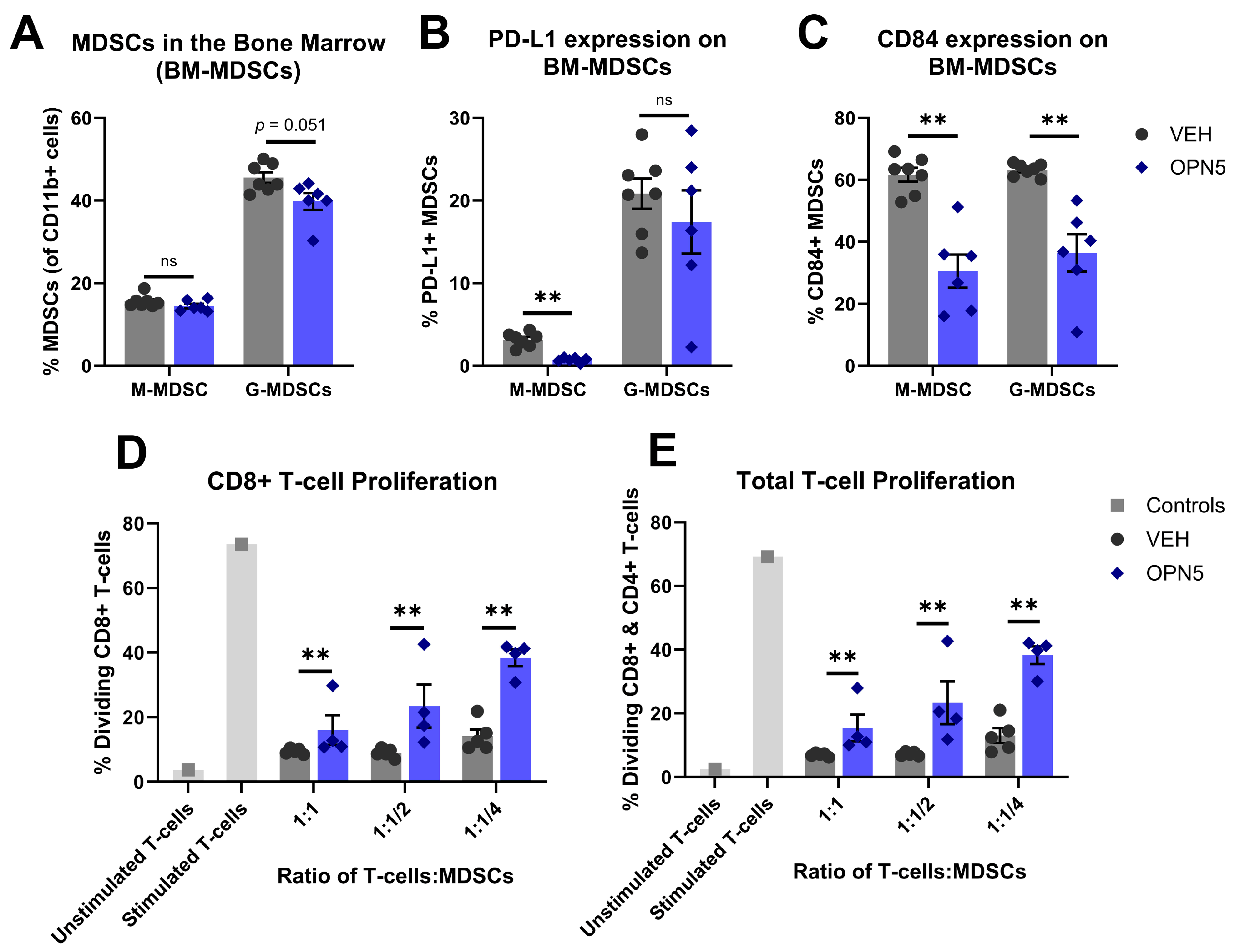
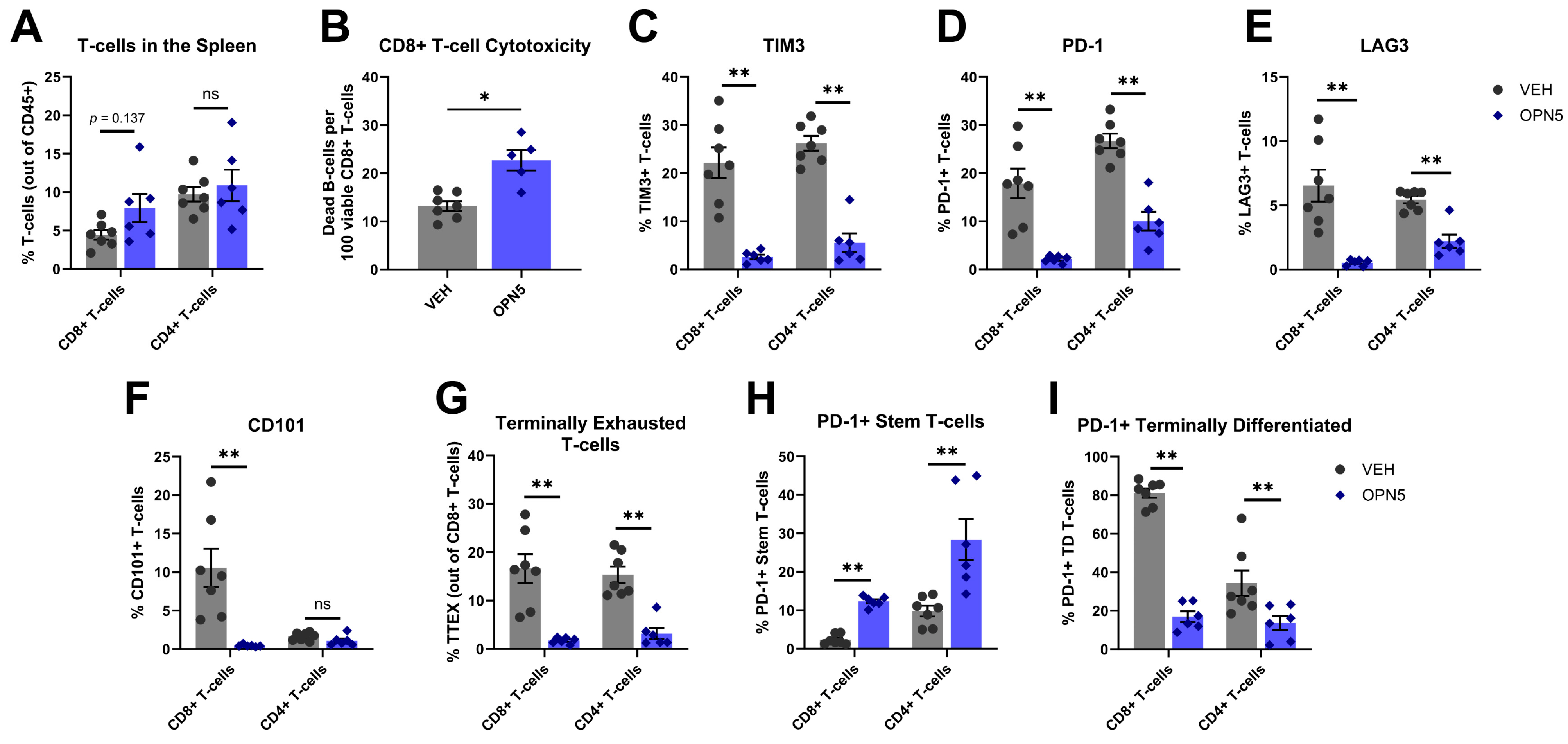
3.3. BET Protein Inhibition Reduces MDSC Immune-Suppressive Activity In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Döhner, H.; Hillmen, P.; Keating, M.; Montserrat, E.; Chiorazzi, N.; et al. iwCLL Guidelines for Diagnosis, Indications for Treatment, Response Assessment, and Supportive Management of CLL. Blood 2018, 131, 2745–2760. [Google Scholar] [CrossRef] [PubMed]
- Hallek, M.; Al-Sawaf, O. Chronic Lymphocytic Leukemia: 2022 Update on Diagnostic and Therapeutic Procedures. Am. J. Hematol. 2021, 96, 1679–1705. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Clements, V.K.; Bunt, S.K.; Albelda, S.M.; Ostrand-Rosenberg, S. Cross-Talk between Myeloid-Derived Suppressor Cells and Macrophages Subverts Tumor Immunity toward a Type 2 Response1. J. Immunol. 2007, 179, 977–983. [Google Scholar] [CrossRef]
- van Attekum, M.H.; Eldering, E.; Kater, A.P. Chronic Lymphocytic Leukemia Cells Are Active Participants in Microenvironmental Cross-Talk. Haematologica 2017, 102, 1469–1476. [Google Scholar] [CrossRef] [PubMed]
- Arruga, F.; Gyau, B.B.; Iannello, A.; Vitale, N.; Vaisitti, T.; Deaglio, S. Immune Response Dysfunction in Chronic Lymphocytic Leukemia: Dissecting Molecular Mechanisms and Microenvironmental Conditions. Int. J. Mol. Sci. 2020, 21, 1825. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Mastio, J.; Gabrilovich, D.I. Transcriptional Regulation of Myeloid-Derived Suppressor Cells. J. Leukoc. Biol. 2015, 98, 913–922. [Google Scholar] [CrossRef]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-Derived Suppressor Cells in the Era of Increasing Myeloid Cell Diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Bronte, V.; Chen, S.-H.; Colombo, M.P.; Ochoa, A.; Ostrand-Rosenberg, S.; Schreiber, H. The Terminology Issue for Myeloid-Derived Suppressor Cells. Cancer Res. 2007, 67, 425–426. [Google Scholar] [CrossRef]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cells Inhibit T Cell Activation by Depleting Cystine and Cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef]
- Jitschin, R.; Braun, M.; Büttner, M.; Dettmer-Wilde, K.; Bricks, J.; Berger, J.; Eckart, M.J.; Krause, S.W.; Oefner, P.J.; Le Blanc, K.; et al. CLL-Cells Induce IDOhi CD14+HLA-DRlo Myeloid-Derived Suppressor Cells That Inhibit T-Cell Responses and Promote TRegs. Blood 2014, 124, 750–760. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, Y.; Huang, Q.; Qiu, L. CD14+HLA-DRlow/- Expression: A Novel Prognostic Factor in Chronic Lymphocytic Leukemia. Oncol. Lett. 2015, 9, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Zarobkiewicz, M.; Kowalska, W.; Chocholska, S.; Tomczak, W.; Szymańska, A.; Morawska, I.; Wojciechowska, A.; Bojarska-Junak, A. High M-MDSC Percentage as a Negative Prognostic Factor in Chronic Lymphocytic Leukaemia. Cancers 2020, 12, 2614. [Google Scholar] [CrossRef] [PubMed]
- Zahran, A.M.; Moeen, S.M.; Thabet, A.F.; Rayan, A.; Abdel-Rahim, M.H.; Mohamed, W.M.Y.; Hetta, H.F. Monocytic Myeloid-Derived Suppressor Cells in Chronic Lymphocytic Leukemia Patients: A Single Center Experience. Leuk. Lymphoma 2020, 61, 1645–1652. [Google Scholar] [CrossRef] [PubMed]
- Fleming, V.; Hu, X.; Weber, R.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front. Immunol. 2018, 9, 398. [Google Scholar] [CrossRef]
- Hegde, S.; Leader, A.M.; Merad, M. MDSC: Markers, Development, States, and Unaddressed Complexity. Immunity 2021, 54, 875–884. [Google Scholar] [CrossRef]
- Kusmartsev, S.; Cheng, F.; Yu, B.; Nefedova, Y.; Sotomayor, E.; Lush, R.; Gabrilovich, D. All-Trans-Retinoic Acid Eliminates Immature Myeloid Cells from Tumor-Bearing Mice and Improves the Effect of Vaccination. Cancer Res. 2003, 63, 4441–4449. [Google Scholar]
- Mirza, N.; Fishman, M.; Fricke, I.; Dunn, M.; Neuger, A.M.; Frost, T.J.; Lush, R.M.; Antonia, S.; Gabrilovich, D.I. All-Trans-Retinoic Acid Improves Differentiation of Myeloid Cells and Immune Response in Cancer Patients. Cancer Res. 2006, 66, 9299–9307. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Gabrilovich, D.I. History of Myeloid Derived Suppressor Cells (MDSCs) in the Macro- and Micro-Environment of Tumour-Bearing Hosts. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef]
- Orillion, A.; Hashimoto, A.; Damayanti, N.; Shen, L.; Adelaiye-Ogala, R.; Arisa, S.; Chintala, S.; Ordentlich, P.; Kao, C.; Elzey, B.; et al. Entinostat Neutralizes Myeloid-Derived Suppressor Cells and Enhances the Antitumor Effect of PD-1 Inhibition in Murine Models of Lung and Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 5187–5201. [Google Scholar] [CrossRef]
- Rosborough, B.R.; Castellaneta, A.; Natarajan, S.; Thomson, A.W.; Turnquist, H.R. Histone Deacetylase Inhibition Facilitates GM-CSF-Mediated Expansion of Myeloid-Derived Suppressor Cells in Vitro and in Vivo. J. Leukoc. Biol. 2012, 91, 701–709. [Google Scholar] [CrossRef]
- Muller, S.; Filippakopoulos, P.; Knapp, S. Bromodomains as Therapeutic Targets. Expert. Rev. Mol. Med. 2011, 13, e29. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Bertoni, F. BET Proteins as Targets for Anticancer Treatment. Cancer Discov. 2018, 8, 24–36. [Google Scholar] [CrossRef]
- Ozer, H.G.; El-Gamal, D.; Powell, B.; Hing, Z.A.; Blachly, J.S.; Harrington, B.; Mitchell, S.; Grieselhuber, N.R.; Williams, K.; Lai, T.-H.; et al. BRD4 Profiling Identifies Critical Chronic Lymphocytic Leukemia Oncogenic Circuits and Reveals Sensitivity to PLX51107, a Novel Structurally Distinct BET Inhibitor. Cancer Discov. 2018, 8, 458–477. [Google Scholar] [CrossRef]
- Smith, A.L.; Skupa, S.A.; Eiken, A.P.; Reznicek, T.E.; Schmitz, E.; Williams, N.; Moore, D.Y.; D’Angelo, C.R.; Kallam, A.; Lunning, M.A.; et al. BET Inhibition Reforms the Immune Microenvironment and Alleviates T Cell Dysfunction in Chronic Lymphocytic Leukemia. Available online: https://insight.jci.org/articles/view/177054/pdf (accessed on 10 January 2025).
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.; Young, R.A. Super-Enhancers in the Control of Cell Identity and Disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master Transcription Factors and Mediator Establish Super-Enhancers at Key Cell Identity Genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective Inhibition of BET Bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Ferri, E.; Petosa, C.; McKenna, C.E. Bromodomains: Structure, Function and Pharmacology of Inhibition. Biochem. Pharmacol. 2016, 106, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Bichi, R.; Shinton, S.A.; Martin, E.S.; Koval, A.; Calin, G.A.; Cesari, R.; Russo, G.; Hardy, R.R.; Croce, C.M. Human Chronic Lymphocytic Leukemia Modeled in Mouse by Targeted TCL1 Expression. Proc. Natl. Acad. Sci. USA 2002, 99, 6955–6960. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Bertilaccio, M.T.S.; Chen, S.-S. Mouse Models of Chronic Lymphocytic Leukemia and Richter Transformation: What We Have Learnt and What We Are Missing. Front. Immunol. 2024, 15, 1376660. [Google Scholar] [CrossRef]
- Bresin, A.; D’Abundo, L.; Narducci, M.G.; Fiorenza, M.T.; Croce, C.M.; Negrini, M.; Russo, G. TCL1 Transgenic Mouse Model as a Tool for the Study of Therapeutic Targets and Microenvironment in Human B-Cell Chronic Lymphocytic Leukemia. Cell Death Dis. 2016, 7, e2071. [Google Scholar] [CrossRef] [PubMed]
- Goral, A.; Firczuk, M.; Fidyt, K.; Sledz, M.; Simoncello, F.; Siudakowska, K.; Pagano, G.; Moussay, E.; Paggetti, J.; Nowakowska, P.; et al. A Specific CD44lo CD25lo Subpopulation of Regulatory T Cells Inhibits Anti-Leukemic Immune Response and Promotes the Progression in a Mouse Model of Chronic Lymphocytic Leukemia. Front. Immunol. 2022, 13, 781364. [Google Scholar] [CrossRef]
- Goral, A.; Sledz, M.; Manda-Handzlik, A.; Cieloch, A.; Wojciechowska, A.; Lachota, M.; Mroczek, A.; Demkow, U.; Zagozdzon, R.; Matusik, K.; et al. Regulatory T Cells Contribute to the Immunosuppressive Phenotype of Neutrophils in a Mouse Model of Chronic Lymphocytic Leukemia. Exp. Hematol. Oncol. 2023, 12, 89. [Google Scholar] [CrossRef]
- Hanna, B.S.; McClanahan, F.; Yazdanparast, H.; Zaborsky, N.; Kalter, V.; Rößner, P.M.; Benner, A.; Dürr, C.; Egle, A.; Gribben, J.G.; et al. Depletion of CLL-Associated Patrolling Monocytes and Macrophages Controls Disease Development and Repairs Immune Dysfunction in Vivo. Leukemia 2016, 30, 570–579. [Google Scholar] [CrossRef]
- Kotaskova, J.; Tichy, B.; Trbusek, M.; Francova, H.S.; Kabathova, J.; Malcikova, J.; Doubek, M.; Brychtova, Y.; Mayer, J.; Pospisilova, S. High Expression of Lymphocyte-Activation Gene 3 (LAG3) in Chronic Lymphocytic Leukemia Cells Is Associated with Unmutated Immunoglobulin Variable Heavy Chain Region (IGHV) Gene and Reduced Treatment-Free Survival. J. Mol. Diagn. 2010, 12, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 Comes of Age as an Inhibitory Receptor. Nat. Rev. Immunol. 2020, 20, 173–185. [Google Scholar] [CrossRef]
- Zhou, Q.; Munger, M.E.; Veenstra, R.G.; Weigel, B.J.; Hirashima, M.; Munn, D.H.; Murphy, W.J.; Azuma, M.; Anderson, A.C.; Kuchroo, V.K.; et al. Coexpression of Tim-3 and PD-1 Identifies a CD8+ T-Cell Exhaustion Phenotype in Mice with Disseminated Acute Myelogenous Leukemia. Blood 2011, 117, 4501–4510. [Google Scholar] [CrossRef] [PubMed]
- Hudson, W.H.; Gensheimer, J.; Hashimoto, M.; Wieland, A.; Valanparambil, R.M.; Li, P.; Lin, J.-X.; Konieczny, B.T.; Im, S.J.; Freeman, G.J.; et al. Proliferating Transitory T Cells with an Effector-like Transcriptional Signature Emerge from PD-1+ Stem-like CD8+ T Cells during Chronic Infection. Immunity 2019, 51, 1043–1058.e4. [Google Scholar] [CrossRef]
- Niborski, L.L.; Gueguen, P.; Ye, M.; Thiolat, A.; Ramos, R.N.; Caudana, P.; Denizeau, J.; Colombeau, L.; Rodriguez, R.; Goudot, C.; et al. CD8+T Cell Responsiveness to Anti-PD-1 Is Epigenetically Regulated by Suv39h1 in Melanomas. Nat. Commun. 2022, 13, 3739. [Google Scholar] [CrossRef]
- Abdel-Hakeem, M.S.; Manne, S.; Beltra, J.-C.; Stelekati, E.; Chen, Z.; Nzingha, K.; Ali, M.-A.; Johnson, J.L.; Giles, J.R.; Mathew, D.; et al. Epigenetic Scarring of Exhausted T Cells Hinders Memory Differentiation upon Eliminating Chronic Antigenic Stimulation. Nat. Immunol. 2021, 22, 1008–1019. [Google Scholar] [CrossRef]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of Exhausted CD8+ T Cells Differentially Mediate Tumor Control and Respond to Checkpoint Blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Beltra, J.-C.; Manne, S.; Abdel-Hakeem, M.S.; Kurachi, M.; Giles, J.R.; Chen, Z.; Casella, V.; Ngiow, S.F.; Khan, O.; Huang, Y.J.; et al. Developmental Relationships of Four Exhausted CD8+ T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 2020, 52, 825–841.e8. [Google Scholar] [CrossRef]
- Wang, Z.-Q.; Zhang, Z.-C.; Wu, Y.-Y.; Pi, Y.-N.; Lou, S.-H.; Liu, T.-B.; Lou, G.; Yang, C. Bromodomain and Extraterminal (BET) Proteins: Biological Functions, Diseases and Targeted Therapy. Sig Transduct. Target. Ther. 2023, 8, 420. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Hashimoto, A.; Dweep, H.; Sanseviero, E.; De Leo, A.; Tcyganov, E.; Kossenkov, A.; Mulligan, C.; Nam, B.; Masters, G.; et al. Analysis of Classical Neutrophils and Polymorphonuclear Myeloid-Derived Suppressor Cells in Cancer Patients and Tumor-Bearing Mice. J. Exp. Med. 2021, 218, e20201803. [Google Scholar] [CrossRef]
- Lewinsky, H.; Barak, A.F.; Huber, V.; Kramer, M.P.; Radomir, L.; Sever, L.; Orr, I.; Mirkin, V.; Dezorella, N.; Shapiro, M.; et al. CD84 Regulates PD-1/PD-L1 Expression and Function in Chronic Lymphocytic Leukemia. J. Clin. Investig. 2018, 128, 5465–5478. [Google Scholar] [CrossRef] [PubMed]
- McClanahan, F.; Riches, J.C.; Miller, S.; Day, W.P.; Kotsiou, E.; Neuberg, D.; Croce, C.M.; Capasso, M.; Gribben, J.G. Mechanisms of PD-L1/PD-1–Mediated CD8 T-Cell Dysfunction in the Context of Aging-Related Immune Defects in the Eµ-TCL1 CLL Mouse Model. Blood 2015, 126, 212–221. [Google Scholar] [CrossRef]
- Brusa, D.; Serra, S.; Coscia, M.; Rossi, D.; D’Arena, G.; Laurenti, L.; Jaksic, O.; Fedele, G.; Inghirami, G.; Gaidano, G.; et al. The PD-1/PD-L1 Axis Contributes to T-Cell Dysfunction in Chronic Lymphocytic Leukemia. Haematologica 2013, 98, 953–963. [Google Scholar] [CrossRef]
- Grzywnowicz, M.; Karczmarczyk, A.; Skorka, K.; Zajac, M.; Zaleska, J.; Chocholska, S.; Tomczak, W.; Giannopoulos, K. Expression of Programmed Death 1 Ligand in Different Compartments of Chronic Lymphocytic Leukemia. Acta Haematol. 2015, 134, 255–262. [Google Scholar] [CrossRef]
- Lewinsky, H.; Gunes, E.G.; David, K.; Radomir, L.; Kramer, M.P.; Pellegrino, B.; Perpinial, M.; Chen, J.; He, T.; Mansour, A.G.; et al. CD84 Is a Regulator of the Immunosuppressive Microenvironment in Multiple Myeloma. JCI Insight 2021, 6, e141683. [Google Scholar] [CrossRef]
- Watson, R.J.; Bamborough, P.; Barnett, H.; Chung, C.-W.; Davis, R.; Gordon, L.; Grandi, P.; Petretich, M.; Phillipou, A.; Prinjha, R.K.; et al. GSK789: A Selective Inhibitor of the First Bromodomains (BD1) of the Bromo and Extra Terminal Domain (BET) Proteins. J. Med. Chem. 2020, 63, 9045–9069. [Google Scholar] [CrossRef] [PubMed]
- Zablonski, K.G.; Skupa, S.A.; Eiken, A.P.; Sundaram, S.; Mavis, C.; Gu, J.; Torka, P.; Ghione, P.; El-Gamal, D.; Hernandez-Ilizaliturri, F.J. Targeted BET Inhibition with OPN-51107 Synergizes with Venetoclax in Chronic Lymphocytic Leukemia. Leuk. Lymphoma 2024, 65, 2129–2137. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drengler, E.M.; Smith, A.L.; Skupa, S.A.; Schmitz, E.; Mohamed, E.; El-Gamal, D. BET Protein Inhibition Relieves MDSC-Mediated Immune Suppression in Chronic Lymphocytic Leukemia. Hemato 2025, 6, 14. https://doi.org/10.3390/hemato6020014
Drengler EM, Smith AL, Skupa SA, Schmitz E, Mohamed E, El-Gamal D. BET Protein Inhibition Relieves MDSC-Mediated Immune Suppression in Chronic Lymphocytic Leukemia. Hemato. 2025; 6(2):14. https://doi.org/10.3390/hemato6020014
Chicago/Turabian StyleDrengler, Erin M., Audrey L. Smith, Sydney A. Skupa, Elizabeth Schmitz, Eslam Mohamed, and Dalia El-Gamal. 2025. "BET Protein Inhibition Relieves MDSC-Mediated Immune Suppression in Chronic Lymphocytic Leukemia" Hemato 6, no. 2: 14. https://doi.org/10.3390/hemato6020014
APA StyleDrengler, E. M., Smith, A. L., Skupa, S. A., Schmitz, E., Mohamed, E., & El-Gamal, D. (2025). BET Protein Inhibition Relieves MDSC-Mediated Immune Suppression in Chronic Lymphocytic Leukemia. Hemato, 6(2), 14. https://doi.org/10.3390/hemato6020014