
Design and Implementation of a Sickle Cell Disease Electronic Registry in Resource Limited Setting in Nigeria—A Pilot Study

 , , and
, , and
Abstract
1. Introduction
- The assessment of baseline logistics and infrastructure for SCD ER in Ahmadu Bello University Teaching Hospital Zaria (ABUTH);
- The determination of basic information required for a comprehensive SCD ER based on existing registries elsewhere;
- The design and implementation of two ERs for SCD, one for children and another for adults;
- The training of users (physicians, nurses, laboratory staff, health information/ICT staff) on the use of the ERs;
- The use of the data collected to conduct baseline analysis of the epidemiology and clinical characteristics of SCD patients.
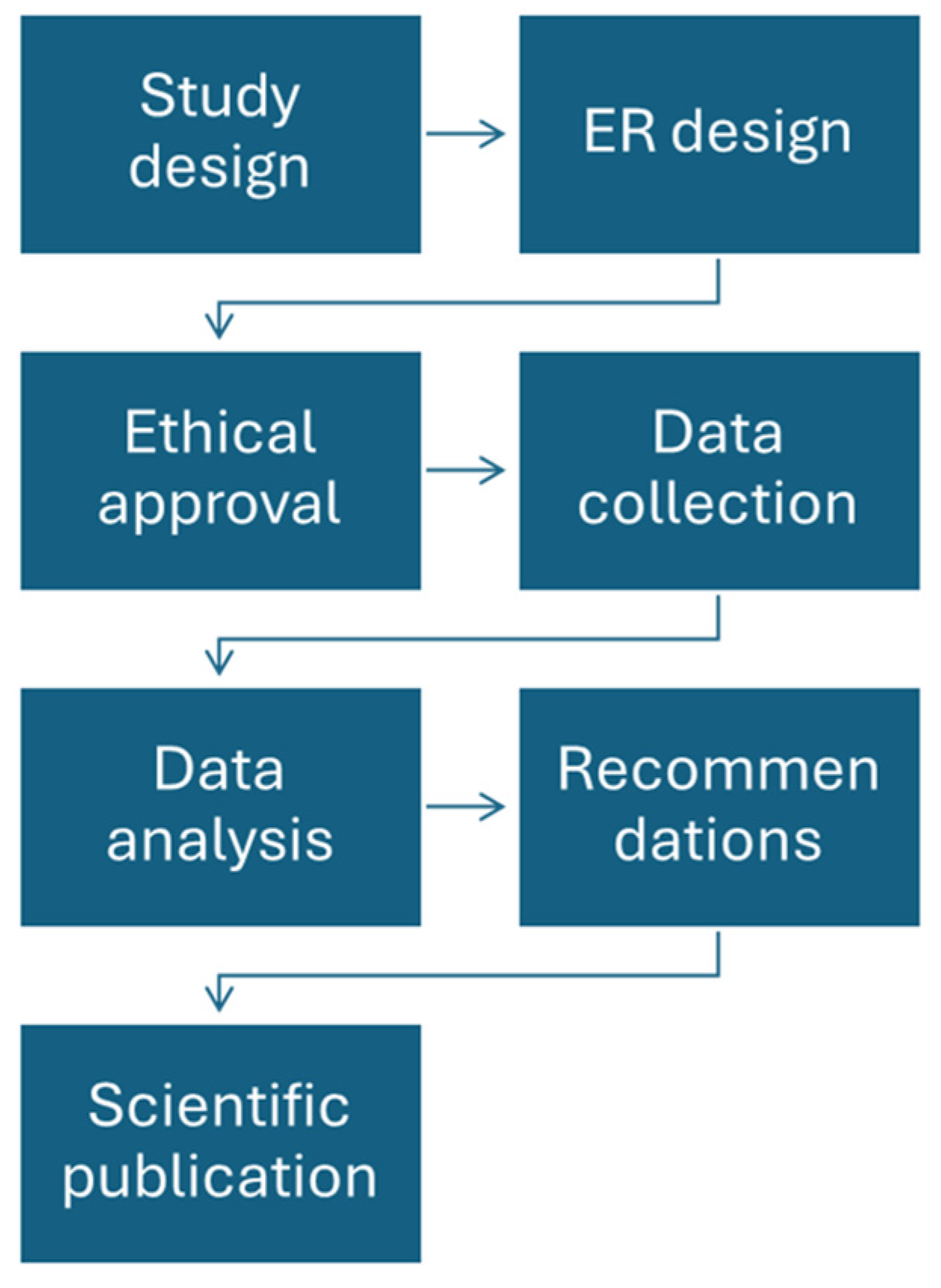
2. Materials and Methods
2.1. Study Site
2.2. Study Period
2.3. Ethical Approval
2.4. Database Design
2.5. Data Collection
2.6. Data Management
2.7. Data Sharing
3. Results
3.1. Assessment of Logistics and Infrastructure
3.2. Determination of Basic Information Required for a Comprehensive SCD ER
3.3. Design and Implementation of the ERs and Training of Users
- Paucity of resources: the study team agreed to maximise the use of the available resources to have the highest number of PRs transferred; this implied the need to minimise the budget for technical instruments, thus no new hardware nor software licenses were bought;
- Finance: the study PI and the ABUTH site lead voluntarily agreed to contribute with their own money;
- Staff: some of the needed manpower was temporarily employed and some members of the study team volunteered to be involved for free in study-related activities;
- Time: extra hours aside our daily schedules were added in order to meet up;
- Logistics: this has been taken care of by the study PI, ABUTH site lead and some members of the study team;
- Lack of embedded functionalities in MS Access: pictorial data presentations like dashboards used in advance registries were considered not essential at this stage of development.
3.4. Demographic and Clinical Information of SCD Patient Population at ABUTH
3.5. Complications
4. Discussion
Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thomson, A.M.; A McHugh, T.; Oron, A.P.; Teply, C.; Lonberg, N.; Tella, V.V.; Wilner, L.B.; Fuller, K.; Hagins, H.; Aboagye, R.G.; et al. Global, regional, and national prevalence and mortality burden of sickle cell disease, 2020–2021: A systematic analysis from the Global Burden of Disease study 2021. Lancet 2023, 10, e585–e599. [Google Scholar] [CrossRef]
- Kanter, J.; Meier, E.R.; Hankins, J.S. Improving outcomes for patients with sickle cell disease in the United States. Making the case for more resources, surveillance, and longitudinal data. JAMA Health Forum 2021, 2, e213467. [Google Scholar] [CrossRef] [PubMed]
- Sickle Cell Foundation Nigeria. Sickle Cell Disorder Registry Nigeria (SCDRN), an Initiative of Sickle Cell Foundation Nigeria and Point Care Health Initiative; Rhieos–Ventures: London, UK, 2022. [Google Scholar]
- Shahraki, A.D.; Safdari, R.; Shahmoradi, L.; Malak, J.S.; Pourghaz, B.; Ghabaee, M. Acute stroke registry planning experiences. J. Regist. Manag. 2018, 45, 37–42. [Google Scholar]
- Antoniou, Z.C.; Schiza, E.C.; Neokleous, K.C.; Angastiniotis, M.; Pattichis, C.S.; Schizas, C.N. E-health services for the European Reference Network on Rare Anaemias (eENERCA). Stud. Health Technol. Inform. 2015, 213, 153–156. [Google Scholar] [PubMed]
- Lundström, M.; Barry, P.; Brocato, L.; Fitzpatrick, C.; Henry, Y.; Rosen, P.; Stenevi, U. European registry for quality improvement in cataract surgery. Int. J. Health Care Qual. Assur. 2014, 27, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Ahern, S.; Evans, S.M.; Hopper, I.; Earnest, A. Clinical quality registries for clinician-level reporting: Strengths and limitations. Med. J. Aust. 2018, 208, 323. [Google Scholar] [CrossRef]
- Inusa, B.P.D.; Colombatti, R. European migration crises: The role of national hemoglobinopathy registries in improving patient access to care. Pediatr. Blood Cancer 2017, 64, 21–32. [Google Scholar] [CrossRef]
- Kosaryan, M.; Karami, H.; Abbas Alipour, M.D.; Akbarzadeh, R. Designing an electronic registry for patients with beta-thalassemia major for Mazandaran province. Int. J. Caring Sci. 2017, 10, 575–582. [Google Scholar]
- Davoodi, S.; Haghighi, K.; Rostam, S.; Kalhori, N.; Hosseini, N.; Mohammadzadeh, Z.; Saftdari, R. Occupational disease registries characteristics and experiences. Acta Inform. Med. 2017, 25, 136–140. [Google Scholar] [CrossRef]
- Modell, B.; Khan, M.; Darlison, M. National register for surveillance of inherited disorders: Beta-thalassaemia in the United Kingdom. Bull. World Health Organ. 2001, 79, 1006–1013. [Google Scholar]
- National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division; Board on Population Health and Public Health Practice; Committee on Addressing Sickle Cell Disease: A Strategic Plan and Blueprint for Action; Martinez, R.M.; Osei-Anto, H.A.; McCormick, M. (Eds.) Screening, Registries, and Surveillance. In Addressing Sickle Cell Disease: A Strategic Plan and Blueprint for Action; National Academies Press (US): Washington, DC, USA, 2020; Volume 3. Available online: https://www.ncbi.nlm.nih.gov/books/NBK566461/ (accessed on 21 December 2023).
- Hassan, A. and Ahmad, H.R. Data from the African Research and Innovative Initiative for Sickle Cell Education Task 2.2; Ahmadu Bello University Teaching Hospital SPARCO: Shika-Zaria, Nigeria, 2020. [Google Scholar]
- Kountouris, P.; Stephanou, C.; Archer, N.; Bonifazi, F.; Giannuzzi, V.; Kuo, K.H.M.; Maggio, A.; Makani, J.; Mañú-Pereira, M.d.M.; Michailidou, K.; et al. The International Hemoglobinopathy Research Network (INHERENT): An international initiative to study the role of genetic modifiers in hemoglobinopathies. Am. J. Hematol. 2021, 96, E416–E420. [Google Scholar] [CrossRef] [PubMed]
- Paintsil, V.; Amuzu, E.X.; Nyanor, I.; Asafo-Adjei, E.; Mohammed, A.R.; Yawnumah, S.A.; Oppong-Mensah, Y.G.; Nguah, S.B.; Obeng, P.; Dogbe, E.E.; et al. Establishing a Sickle Cell Disease Registry in Africa: Experience from the Sickle Pan-African Research Consortium, Kumasi-Ghana. Front. Genet. 2022, 13, 802355. [Google Scholar] [CrossRef]
- Telfer, P.; Coen, P.; Chakravorty, S.; Wilkey, O.; Evans, J.; Newell, H.; Smalling, B.; Amos, R.; Stephens, A.; Rogers, D.; et al. Clinical Outcomes in Children with Sickle Cell Disease Living in England: A Neonatal Cohort in East London. Haematologica 2007, 92, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, T.A.A.d.M.; de Medeiros, T.M.D.; Alves, J.J.P.; Bezerra, C.M.; Fernandes, J.V.; Serafim, S.S.; Fernandes, M.Z.; Sonati, M.d.F. Socioeconomic and Demographic Characteristics of Sickle Cell Disease Patients from a Low-Income Region of Northeastern Brazil. Rev. A Bras. Hematol. 2015, 37, 172–177. [Google Scholar] [CrossRef]
- Isa, H.; Adegoke, S.; Madu, A.; Hassan, A.-A.; Ohiaeri, C.; Chianumba, R.; Brown, B.; Okocha, E.; Ugwu, N.; Diaku-Akinwumi, I.; et al. Sickle Cell Disease Clinical Phenotypes in Nigeria: A Preliminary Analysis of the Sickle Pan Africa Research Consortium Nigeria Database. Blood Cell Mol. Dis. 2020, 84, 102438. [Google Scholar] [CrossRef]
- Asare, E.V.; Wilson, I.; Benneh-Akwasi Kuma, A.A. Burden of Sickle Cell Disease in Ghana: The Korle-Bu Experience. Adv. Hematol. 2018, 2018, 6161270. [Google Scholar] [CrossRef] [PubMed]
- Bardakdjian-Michau, J.; Bahuau, M.; Hurtrel, D.; Godart, C.; Riou, J.; Mathis, M.; Goossens, M. Neonatal Screening for Sickle Cell Disease in France. J. Clin. Pathol. 2009, 62, 31–33. [Google Scholar] [CrossRef] [PubMed]
- De Castro Lobo, C.L.; Ballas, S.K.; Domingos, A.C.B.; Moura, P.G.; Nascimento, E.M.D.; Cardoso, G.P.; de Carvalho, S.M.F. Newborn screening program for hemoglobinopathies in Rio de Janeiro, Brazil. Pediatr. Blood Cancer 2014, 61, 34–39. [Google Scholar] [CrossRef]
- Grosse, S.D.; Odame, I.; Atrash, H.K.; Amendah, D.D.; Piel, F.B.; Williams, T.N. Sickle Cell Disease in Africa: A Neglected Cause of Early Childhood Mortality. Am. J. Prev. Med. 2011, 41 (Suppl. S4), S398–S405. [Google Scholar] [CrossRef]
- Piel, F.B.; Howes, R.E.; Patil, A.P.; Nyangiri, O.A.; Gething, P.W.; Bhatt, S.; Williams, T.N.; Weatherall, D.J.; Hay, S.I. The Distribution of Haemoglobin C and its Prevalence in Newborns in Africa. Sci. Rep. 2013, 3, 1671. [Google Scholar] [CrossRef]
- Jimoh, A.O.; Adebisi, I.M.; Ndakotsu, M.A. Drug use pattern in sickle cell disease in a hematology unit of a teaching hospital in North-Western Nigeria. Ann. Niger. Med. 2014, 8, 32–36. [Google Scholar]
- Okocha, E.C.; Gyamfi, J.; Ryan, N. Barriers to Therapeutic Use of Hydroxyurea for Sickle Cell Disease in Nigeria: A Cross-Sectional Survey. Front. Genet. 2022, 12, 765958. [Google Scholar] [CrossRef]
- Galadanci, N.; Wudil, B.J.; Balogun, T.M.; Ogunrinde, G.O.; Akinsulie, A.; Hasan-Hanga, F.; Mohammed, A.S.; Kehinde, M.O.; Olaniyi, J.A.; Diaku-Akinwumi, I.N.; et al. Current Sickle Cell Disease Management Practices in Nigeria. Int. Health 2014, 6, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Adegoke, S.A.; Adeodu, O.O.; Adekile, A.D. Sickle Cell Disease Clinical Phenotypes in Children from South-Western, Nigeria. Niger. J. Clin. Pract. 2015, 18, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Adewoyin, A.S.; Oghuvwu, O.S.; Awodu, O.A. Hydroxyurea Therapy in Adult Nigerian Sickle Cell Disease: A Monocentric Survey on Pattern of Use, Clinical Effects and Patient’s Compliance. Afr. Health Sci. 2017, 17, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Adeyemo, T.A.; Diaku-Akinwunmi, I.N.; Ojewunmi, O.O. Barriers to the Use of Hydroxyurea in the Management of Sickle Cell Disease in Nigeria. Hemoglobin 2019, 43, 188–192. [Google Scholar] [CrossRef]
- Ofakunrin, A.O.; Okpe, E.S.; O Afolaranmi, T.; Olaosebikan, R.R.; Kanhu, P.U.; Adekola, K.; Dami, N.; Sagay, A.S. Level of Utilization and Provider-Related Barriers to the Use of Hydroxyurea in the Treatment of Sickle Cell Disease Patients in Jos, North-Central Nigeria. Afr. Health Sci. 2021, 21, 765–774. [Google Scholar] [CrossRef]
- Powars, D.R.; Chan, L.S.; Hiti, A.; Ramicone, E.M.; Johnson, C. Outcome of Sickle Cell Anemia. Medicine 2005, 84, 363–376. [Google Scholar] [CrossRef]
- Kanter, J.; Kruse-Jarres, R. Management of Sickle Cell Disease from Childhood through Adulthood. Blood Rev. 2013, 27, 279–287. [Google Scholar] [CrossRef]
- Shah, N.; Bhor, M.; Xie, L.; Paulose, J.; Yuce, H. Sickle Cell Disease Complications: Prevalence and Resource Utilization. PLoS ONE 2019, 14, e0214355. [Google Scholar] [CrossRef]
- Stuart, M.J.; Nagel, R.L. Sickle-cell disease. Lancet 2004, 364, 1343–1360. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lanzkron, S.; Carroll, C.P.; Haywood, C. The burden of emergency department use for sickle-cell disease: An analysis of the national emergency department sampled at a base. Am. J. Hematol. 2010, 85, 797–799. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division; Board on Population Health and Public Health Practice; Committee on Addressing Sickle Cell Disease: A Strategic Plan and Blueprint for Action; Martinez, R.M.; Osei-Anto, H.A.; McCormick, M. (Eds.) Complications of Sickle Cell Disease and Current Management Approaches. In Addressing Sickle Cell Disease: A Strategic Plan and Blueprint for Action; National Academies Press (US): Washington, DC, USA; Agency for Health Care Research and Quality: Rockville, MD, USA, 2020; Volume 4. Available online: https://www.ncbi.nlm.nih.gov/books/NBK566466/ (accessed on 21 December 2023).

{kind=link}
| Patients’ Characteristics | ||
|---|---|---|
| Gender | Total cases (2659) | Valid cases (2638) |
| Male | 1328 (49.9%) | 1328 (50.3%) |
| Female | 1310 (49.3%) | 1310 (49.7%) |
| N.a. | 21 (0.8%) | |
| Age of patients | Total cases (2659) | Valid cases (2659) |
| Paediatric subjects (<18 yrs) | 1961 (73.7%) | 1961 (73.7%) |
| Adult subjects (≥18 yrs) | 698 (26.3%) | |
| Phenotype | Total cases (2659) | Valid cases (2385) |
| SS | 2082 (78.3%) | |
| SS+F | 221 (8.3%) | |
| SC | 80 (3.0%) | |
| CC | 2 (0.1%) | |
| N.a. | 274 (10.3%) | |
| Transfusion | Total cases (2659) | |
| At least one transfusion | 861 (32.4%) | |
| Never being transfused | 1265 (47.6%) | |
| N.a. | 533 (20.0%) | |
| Pharmacological treatment | Total cases (2659) | |
| Hydroxyurea | 351 (13.2%) | |
| Not using hydroxyurea | 1657 (62.3%) | |
| N.a. | 651 (24.5%) |
| Classification | SCD-Related Complication | Frequency in Adult Subjects | Frequency in Paediatric Subjects | ||
|---|---|---|---|---|---|
| Total Cases (n = 698) | Valid Cases (n = 482) | Total Cases (n = 1961) | Valid Cases (n = 1212) | ||
| Bone/Musculoskeletal | Lower back pain | 69 (9.9%) | 69 (14.3%) | 0 (0.0%) | 0 (0.0%) |
| Osteomyelitis | 0 (0.0%) | 0 (0.0%) | 132 (6.7%) | 132 (10.9%) | |
| Difficulty in walking | 41 (5.9%) | 41 (8.5%) | 4 (0.2%) | 4 (0.3%) | |
| Dactylitis | 9 (1.3%) | 9 (1.9%) | 70 (3.6%) | 70 (5.8%) | |
| Stroke | 18 (2.6%) | 18 (3.7%) | 29 (1.5%) | 29 (2.4%) | |
| Hip pain | 24 (3.4%) | 24 (5.0%) | 0 (0.0%) | 0 (0.0%) | |
| Osteoarthritis | 13 (1.9%) | 13 (2.7%) | 8 (0.4%) | 8 (0.7%) | |
| Exertional dyspnoea | 5 (0.7%) | 5 (1.0%) | 0 (0.0%) | 0 (0.0%) | |
| Swellings | 5 (0.7%) | 5 (1.0%) | 1 (0.1%) | 1 (0.1%) | |
| Leg ulcer | 16 (2.3%) | 16 (3.3%) | 4 (0.2%) | 4 (0.3%) | |
| Abscess | 0 (0.0%) | 0 (0.0%) | 2 (0.1%) | 2 (0.2%) | |
| Respiratory | Acute chest syndrome | 27 (3.9%) | 27 (5.6%) | 44 (2.2%) | 44 (3.6%) |
| Acute respiratory infection | 0 (0.0%) | 0 (0.0%) | 14 (0.7%) | 14 (1.2%) | |
| Difficulty in breathing | 21 (3.0%) | 21 (4.4%) | 0 (0.0%) | 0 (0.0%) | |
| Chest pain | 17 (2.4%) | 17 (3.5%) | 0 (0.0%) | 0 (0.0%) | |
| Pulmonary hypertension | 5 (0.7%) | 5 (1.0%) | 0 (0.0%) | 0 (0.0%) | |
| Blood | Vaso-occlusive crises | 128 (18.3%) | 128 (26.6%) | 833 (42.5%) | 833 (68.7%) |
| Anaemia | 30 (4.3%) | 30 (6.2%) | 47 (2.4%) | 47 (3.9%) | |
| Jaundice | 25 (3.6%) | 25 (5.2%) | 0 (0.0%) | 0 (0.0%) | |
| Epistaxis | 0 (0.0%) | 0 (0.0%) | 3 (0.2%) | 3 (0.2%) | |
| Others | Priapism | 3 (0.4%) | 3 (0.6%) | 8 (0.4%) | 8 (0.7%) |
| Retinopathy | 0 (0.0%) | 0 (0.0%) | 1 (0.1%) | 1 (0.1%) | |
| Seizures | 12 (1.7%) | 12 (2.5%) | 5 (0.3%) | 5 (0.4%) | |
| Renal complications | 3 (0.4%) | 3 (0.6%) | 3 (0.2%) | 3 (0.2%) | |
| Data on complications not available | 216 (30.9%) | - | 749 (38.2%) | - | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Idris, M.A.; Ruggieri, L.; Ahmad, H.R.; Hassan, A.; Ibrahim, I.N.; Adullahi, F.J.; Awwalu, S.; Nasiru, U.; Bonifazi, F.; Inusa, B.P.D. Design and Implementation of a Sickle Cell Disease Electronic Registry in Resource Limited Setting in Nigeria—A Pilot Study. Hemato 2024, 5, 340-349. https://doi.org/10.3390/hemato5030025
Idris MA, Ruggieri L, Ahmad HR, Hassan A, Ibrahim IN, Adullahi FJ, Awwalu S, Nasiru U, Bonifazi F, Inusa BPD. Design and Implementation of a Sickle Cell Disease Electronic Registry in Resource Limited Setting in Nigeria—A Pilot Study. Hemato. 2024; 5(3):340-349. https://doi.org/10.3390/hemato5030025
Chicago/Turabian StyleIdris, Muhammad Aminu, Lucia Ruggieri, Hafsat Rufai Ahmad, Abdulaziz Hassan, Ismaila Nda Ibrahim, Faruk Jamil Adullahi, Sani Awwalu, Usman Nasiru, Fedele Bonifazi, and Baba P. D. Inusa. 2024. "Design and Implementation of a Sickle Cell Disease Electronic Registry in Resource Limited Setting in Nigeria—A Pilot Study" Hemato 5, no. 3: 340-349. https://doi.org/10.3390/hemato5030025
APA StyleIdris, M. A., Ruggieri, L., Ahmad, H. R., Hassan, A., Ibrahim, I. N., Adullahi, F. J., Awwalu, S., Nasiru, U., Bonifazi, F., & Inusa, B. P. D. (2024). Design and Implementation of a Sickle Cell Disease Electronic Registry in Resource Limited Setting in Nigeria—A Pilot Study. Hemato, 5(3), 340-349. https://doi.org/10.3390/hemato5030025