Figure 1.
Classification of hematopoietic neoplasms. This figure shows a simplified version of the classification with the most frequent and/or characteristic lymphoma subtypes.
Figure 1.
Classification of hematopoietic neoplasms. This figure shows a simplified version of the classification with the most frequent and/or characteristic lymphoma subtypes.
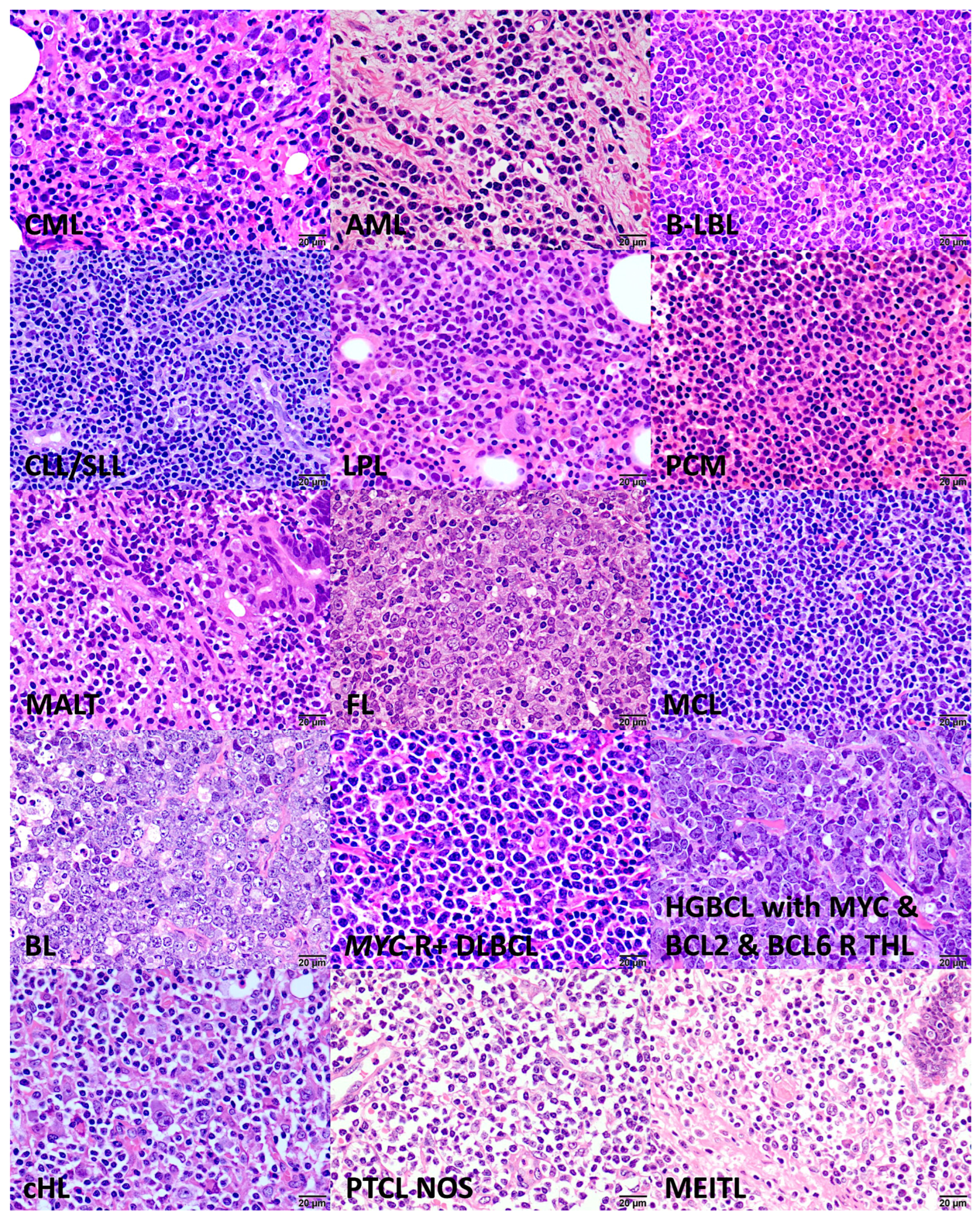
Figure 2.
Histological images of lymphoma subtypes (Hematoxylin and Eosin staining; original magnification 400×). Chronic myeloid leukemia (CML), acute myeloid leukemia (AML), B lymphoblastic lymphoma (B-LBL), chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), lymphoplasmacytic lymphoma (LPL), plasma cell myeloma (PCM), mucosa-associated lymphoid tissue (MALT) lymphoma, follicular lymphoma (FL), mantle cell lymphoma (MCL), Burkitt lymphoma (BL), diffuse large B-cell lymphoma (DLBCL) with MYC rearrangement (MYC-R+), high-grade B-cell lymphoma with MYC, BCL2, and BCL6 rearrangement (triple-hit lymphoma (THL)), classical Hodgkin lymphoma (cHL), peripheral T-cell lymphoma (PTCL), not otherwise specified (NOS), and monomorphic epitheliotropic intestinal T-cell lymphoma (MEITL).
Figure 2.
Histological images of lymphoma subtypes (Hematoxylin and Eosin staining; original magnification 400×). Chronic myeloid leukemia (CML), acute myeloid leukemia (AML), B lymphoblastic lymphoma (B-LBL), chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), lymphoplasmacytic lymphoma (LPL), plasma cell myeloma (PCM), mucosa-associated lymphoid tissue (MALT) lymphoma, follicular lymphoma (FL), mantle cell lymphoma (MCL), Burkitt lymphoma (BL), diffuse large B-cell lymphoma (DLBCL) with MYC rearrangement (MYC-R+), high-grade B-cell lymphoma with MYC, BCL2, and BCL6 rearrangement (triple-hit lymphoma (THL)), classical Hodgkin lymphoma (cHL), peripheral T-cell lymphoma (PTCL), not otherwise specified (NOS), and monomorphic epitheliotropic intestinal T-cell lymphoma (MEITL).
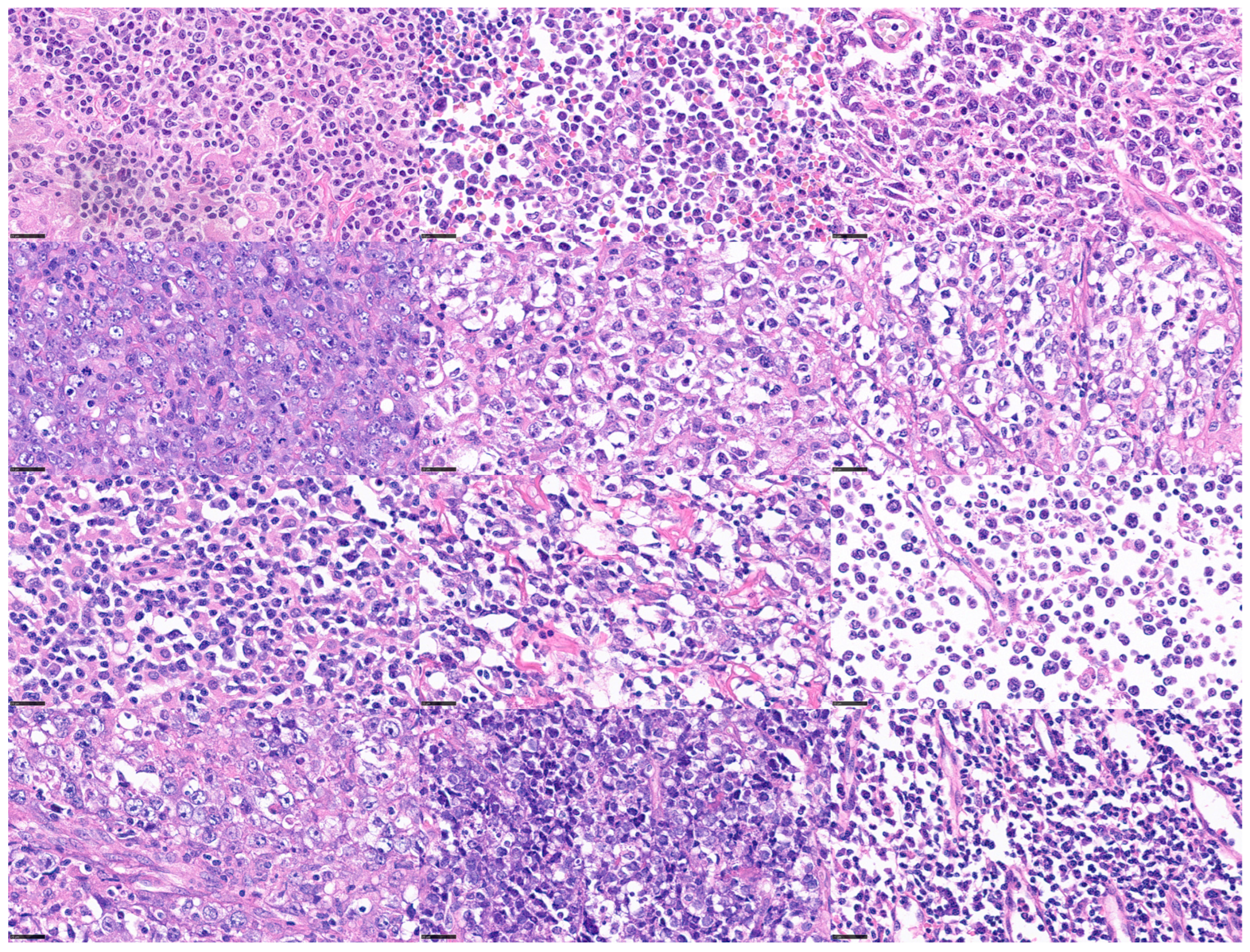
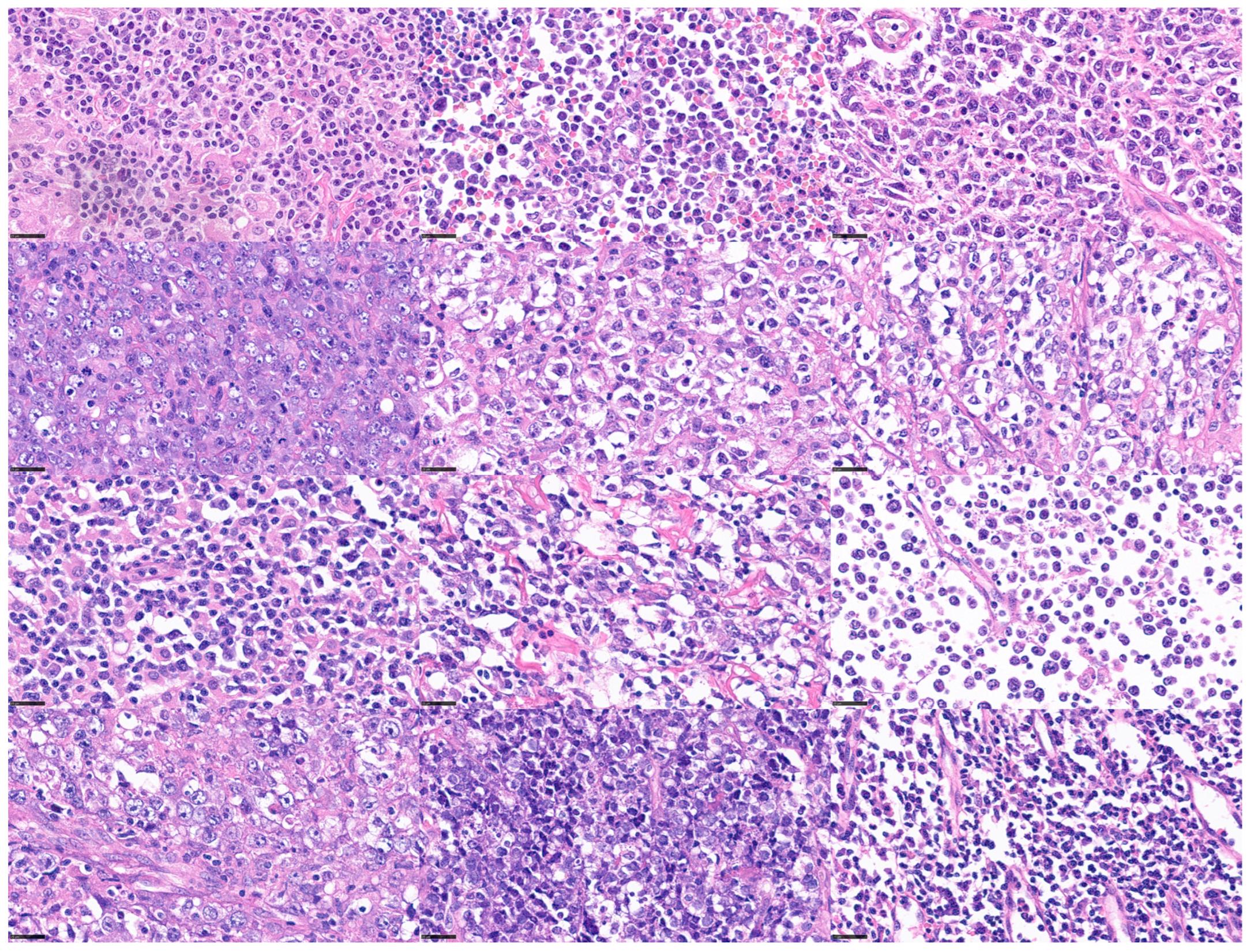
Figure 3.
Histological variability of diffuse large B-cell lymphoma (DLBCL). DLBCL is one of the most frequent mature B-cell neoplasms. DLBCL is a heterogeneous disease with differentmorphologic, genetic, and biologic characteristics. scale bar = 25 μm.
Figure 3.
Histological variability of diffuse large B-cell lymphoma (DLBCL). DLBCL is one of the most frequent mature B-cell neoplasms. DLBCL is a heterogeneous disease with differentmorphologic, genetic, and biologic characteristics. scale bar = 25 μm.
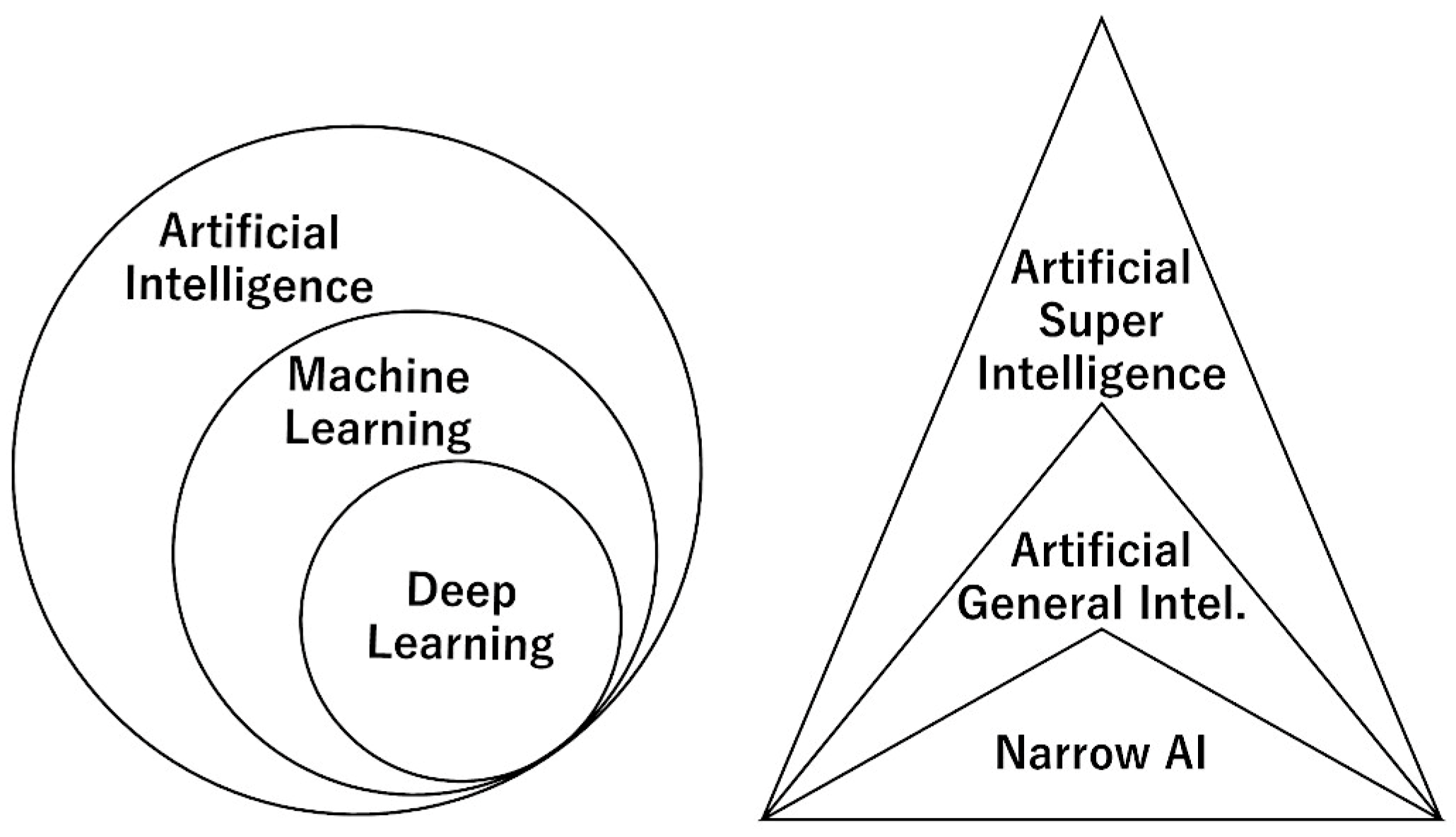
Figure 4.
Types of artificial intelligence. Artificial intelligence (AI) is a broad term that includes several analytical techniques, such machine learning and deep learning (artificial neural networks). AI can also be classified according to comparison with the human intellect, which is also based on an organic neural network, such as narrow AI, artificial general intelligence (AGI), and artificial superintelligence (ASI).
Figure 4.
Types of artificial intelligence. Artificial intelligence (AI) is a broad term that includes several analytical techniques, such machine learning and deep learning (artificial neural networks). AI can also be classified according to comparison with the human intellect, which is also based on an organic neural network, such as narrow AI, artificial general intelligence (AGI), and artificial superintelligence (ASI).
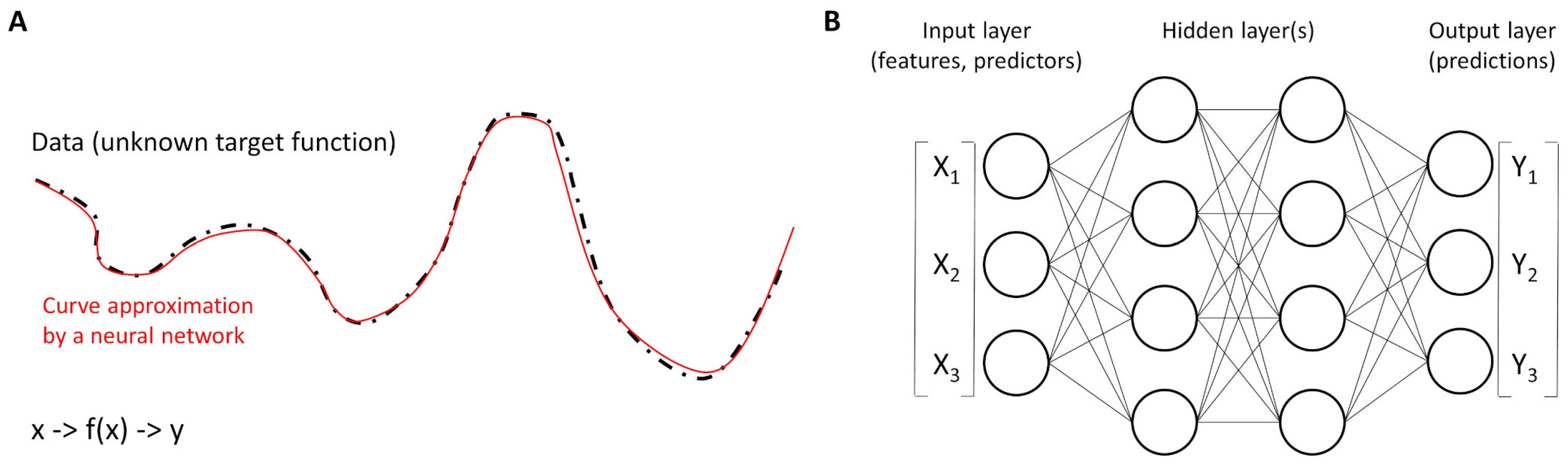
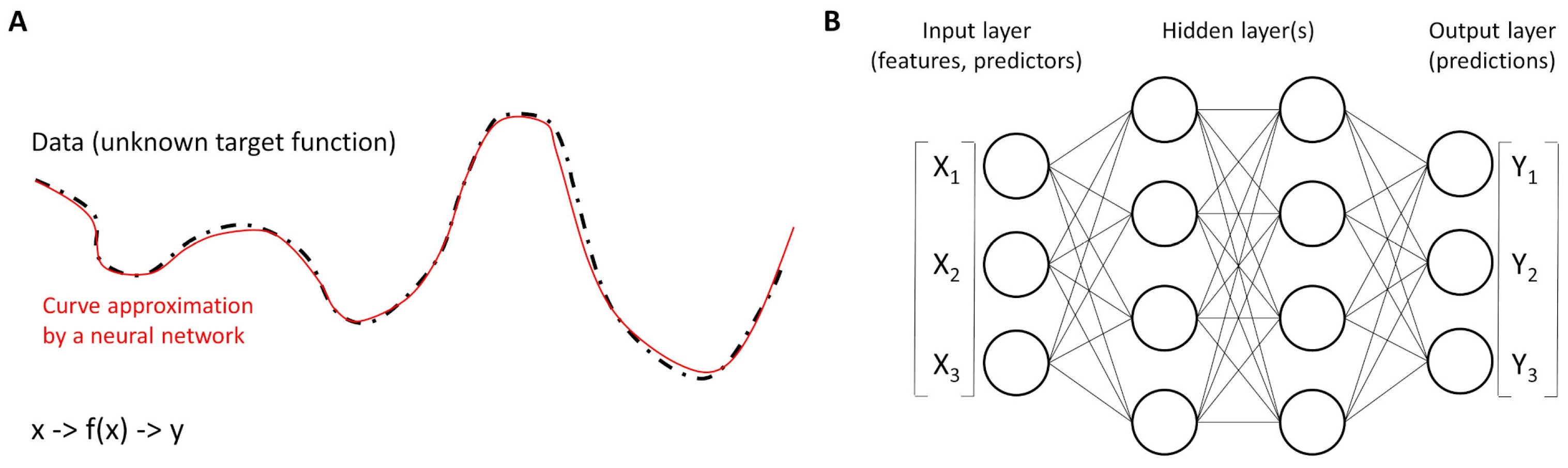
Figure 5.
Functions and neural networks. (A) Neural networks work as universal function approximators to different curves of the dataset. In other words, a neural network is a function that approximates an unknown target function. (B) The basic units are called neurons and are organized into layers. The structure of a neural network is characterized by three parts: the input layer that contains the input fields (variables), one or more hidden layers, and an output layer (with a unit or units that represent the target fields). The units are connected by different connection strengths (weights).
Figure 5.
Functions and neural networks. (A) Neural networks work as universal function approximators to different curves of the dataset. In other words, a neural network is a function that approximates an unknown target function. (B) The basic units are called neurons and are organized into layers. The structure of a neural network is characterized by three parts: the input layer that contains the input fields (variables), one or more hidden layers, and an output layer (with a unit or units that represent the target fields). The units are connected by different connection strengths (weights).
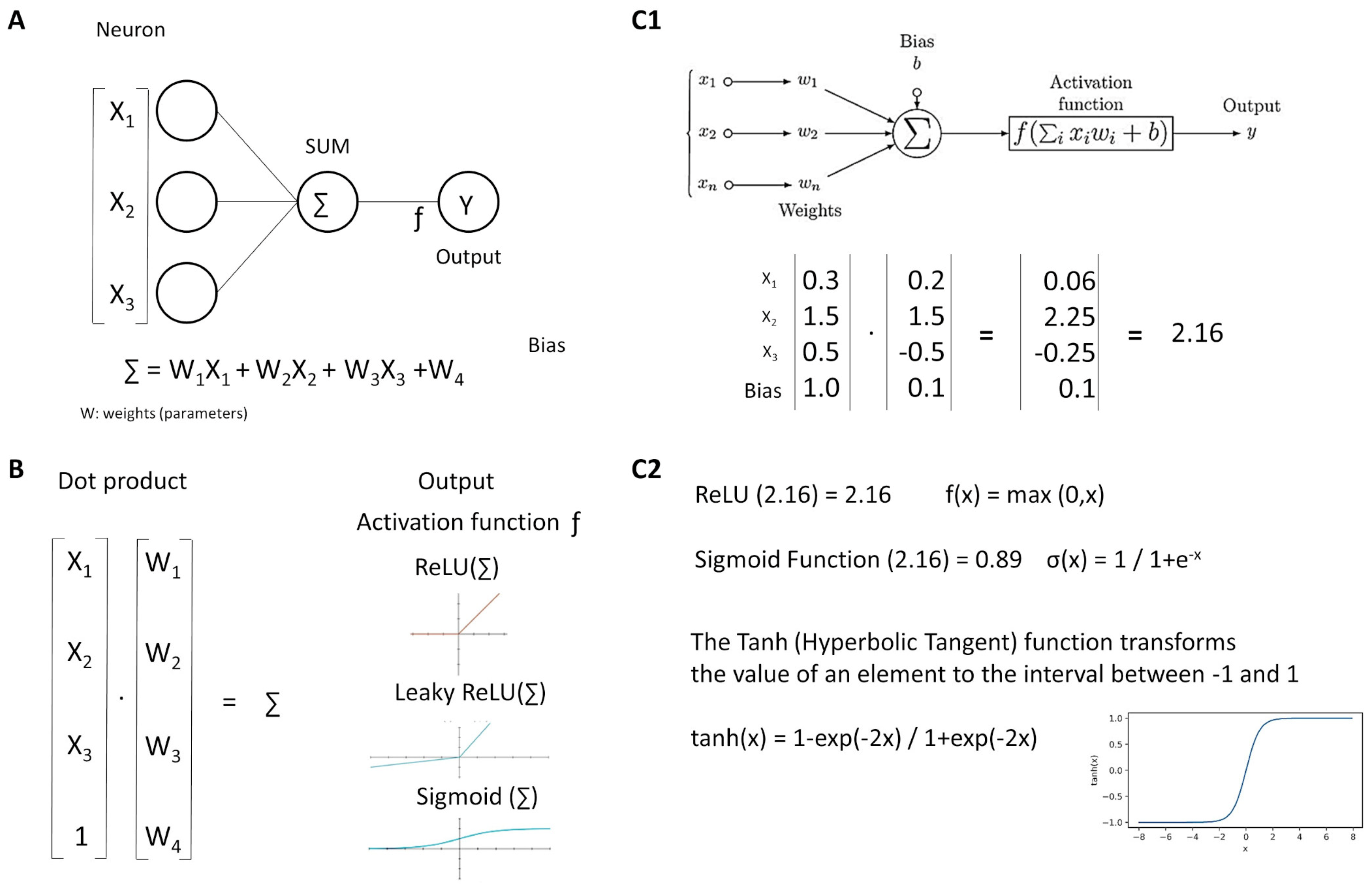
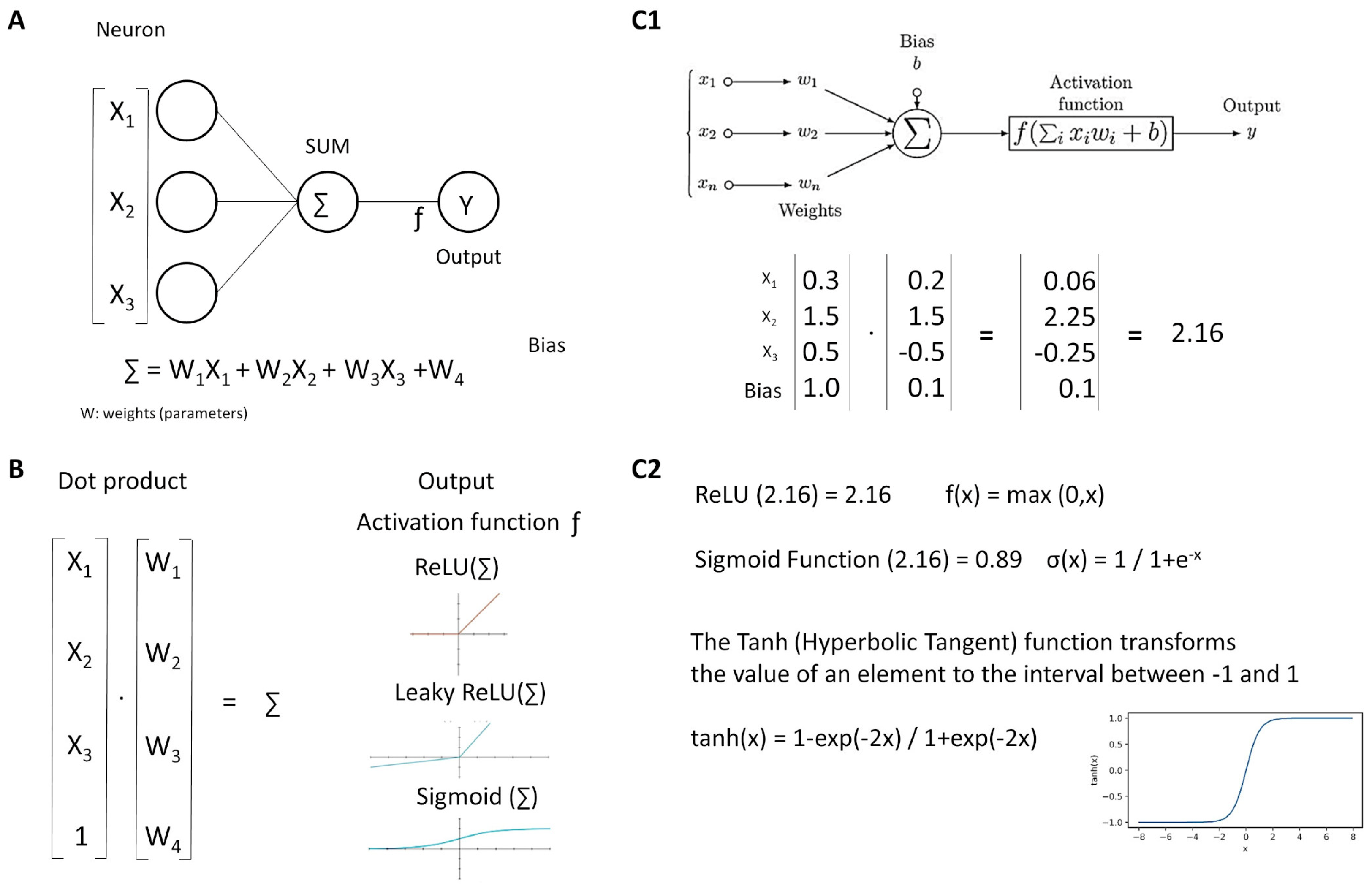
Figure 6.
Neurons. Neural networks comprise several simple functions called neurons (A). Each input is multiplied by its weight, all values (including the bias) are added, and the product is transformed by the activation function (B). An example is shown (C1,C2).
Figure 6.
Neurons. Neural networks comprise several simple functions called neurons (A). Each input is multiplied by its weight, all values (including the bias) are added, and the product is transformed by the activation function (B). An example is shown (C1,C2).
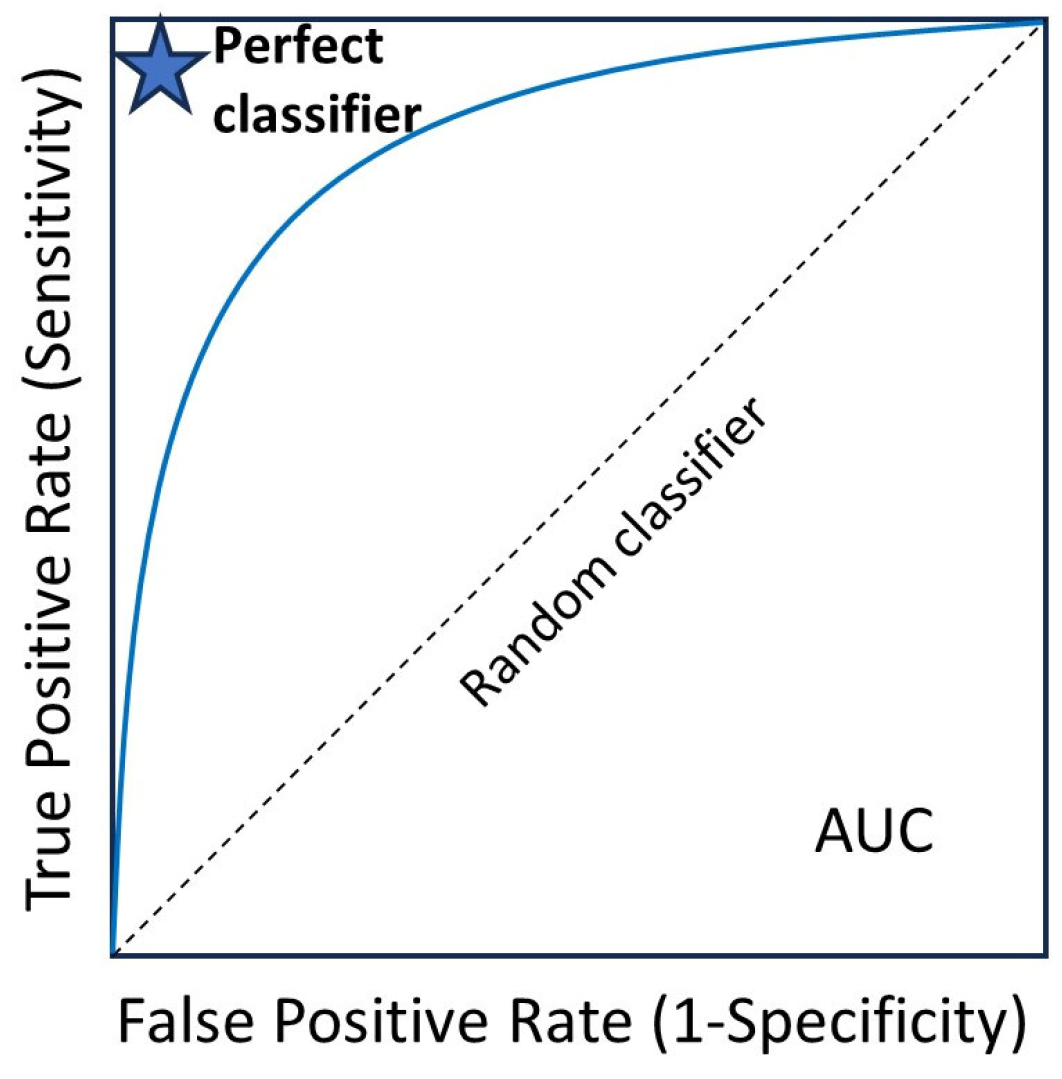
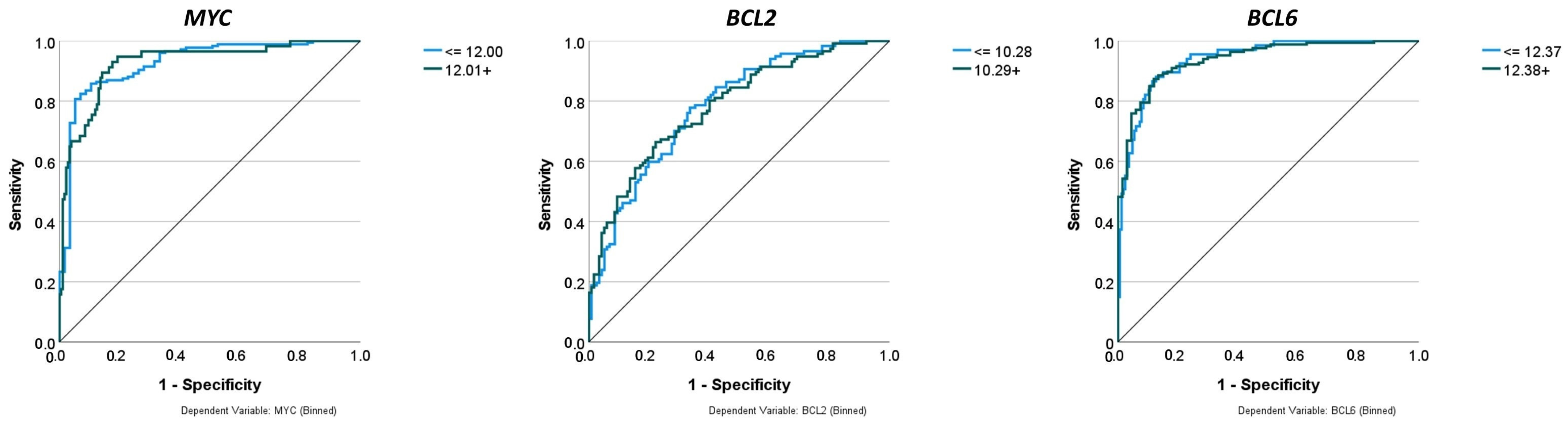
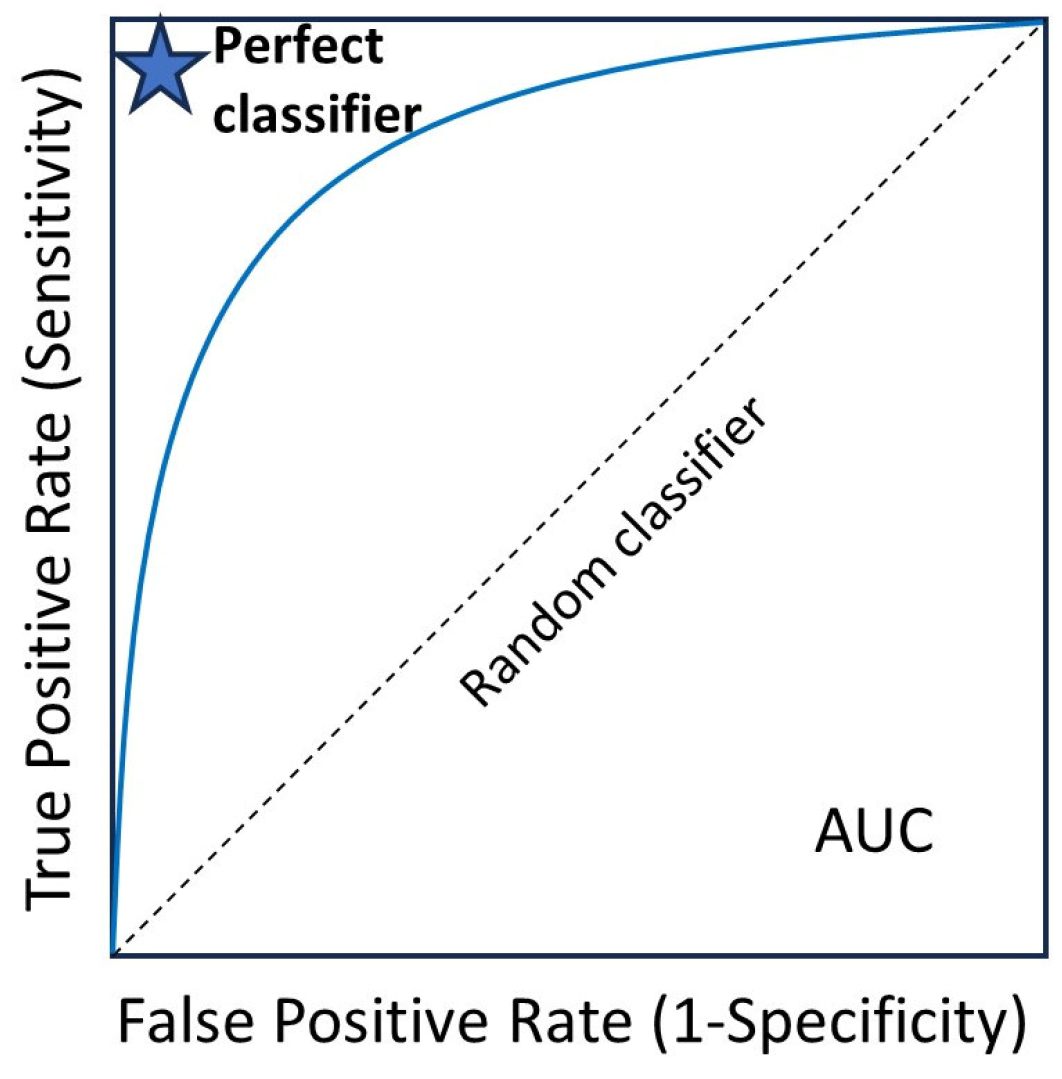
Figure 7.
Receiver operating characteristic (ROC) curve. The area under the curve (AUC) ranges from 0 to 1, and larger AUC values indicate better performance. An AUC of 0.5 indicates no discriminative power. The “blue star” indicates the position of the AUC of 1.0.
Figure 7.
Receiver operating characteristic (ROC) curve. The area under the curve (AUC) ranges from 0 to 1, and larger AUC values indicate better performance. An AUC of 0.5 indicates no discriminative power. The “blue star” indicates the position of the AUC of 1.0.
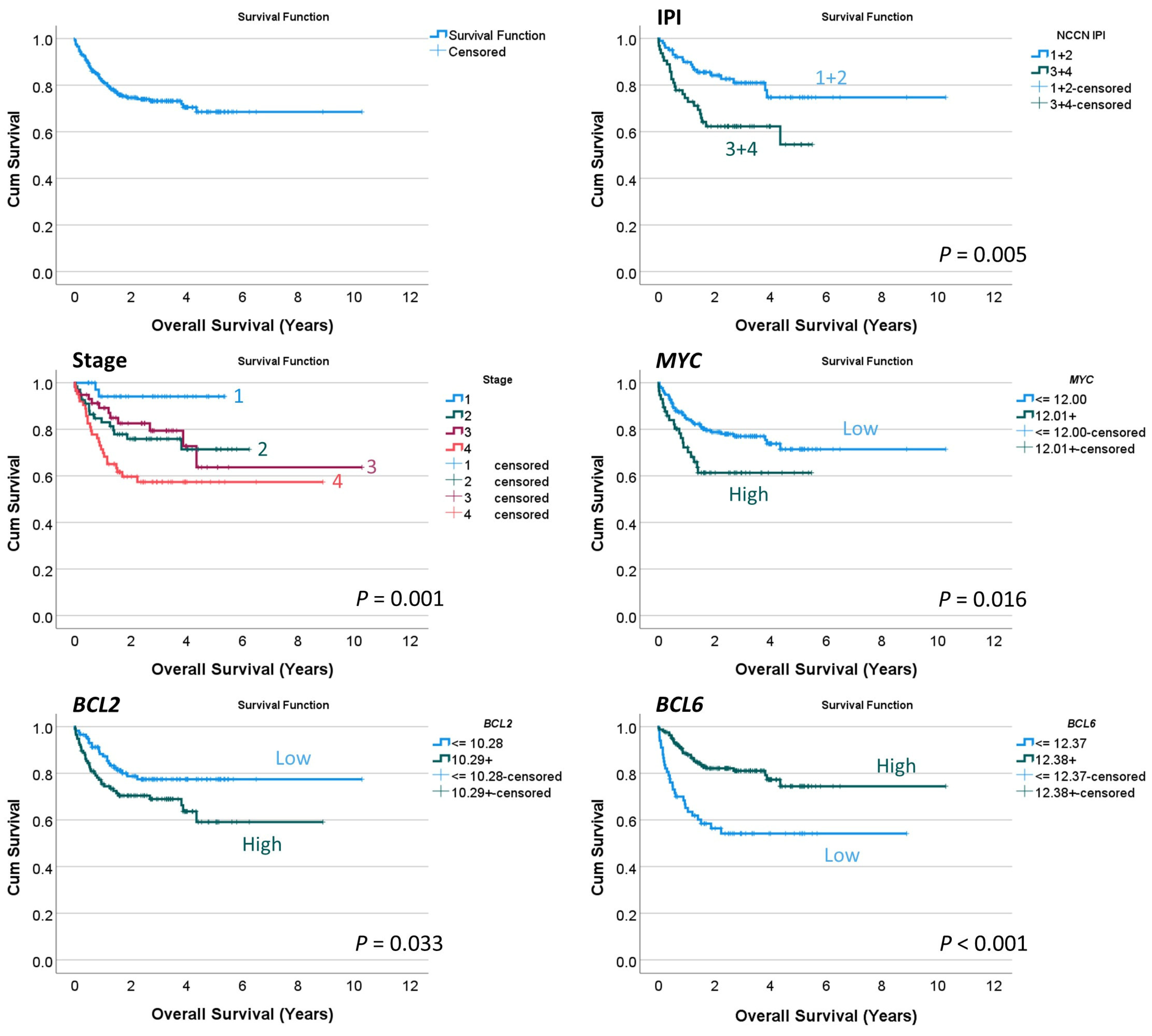
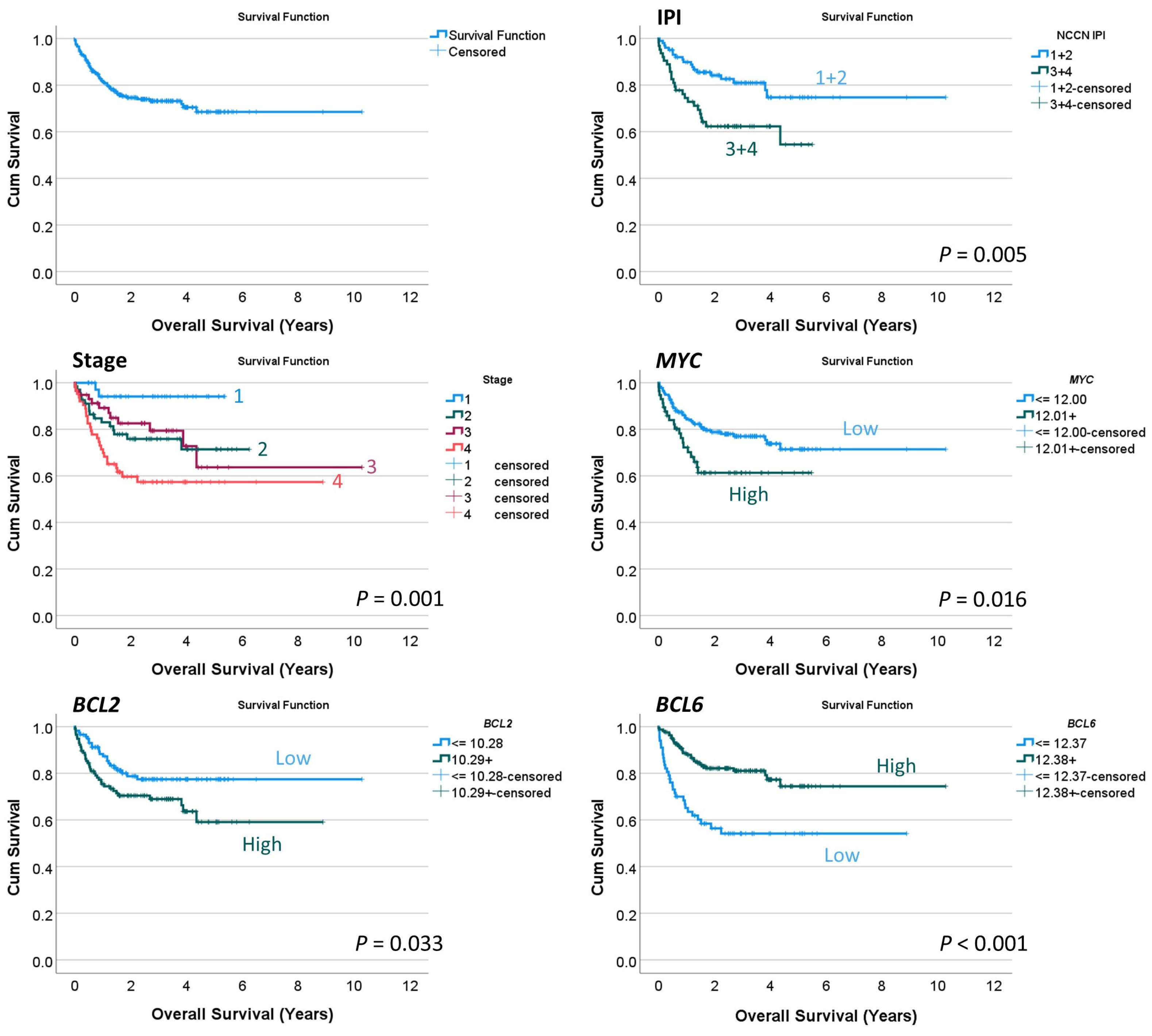
Figure 8.
Overall survival analysis. This study used a conventional series of diffuse large B-cell lymphoma (DLBCL), as shown by the International Prognostic Index (IPI) and clinical stage that stratified patients according to survival. By gene expression, high MYC and BCL2 levels were associated with poor overall survival. Conversely, high BCL6 levels were associated with a favorable outcome.
Figure 8.
Overall survival analysis. This study used a conventional series of diffuse large B-cell lymphoma (DLBCL), as shown by the International Prognostic Index (IPI) and clinical stage that stratified patients according to survival. By gene expression, high MYC and BCL2 levels were associated with poor overall survival. Conversely, high BCL6 levels were associated with a favorable outcome.
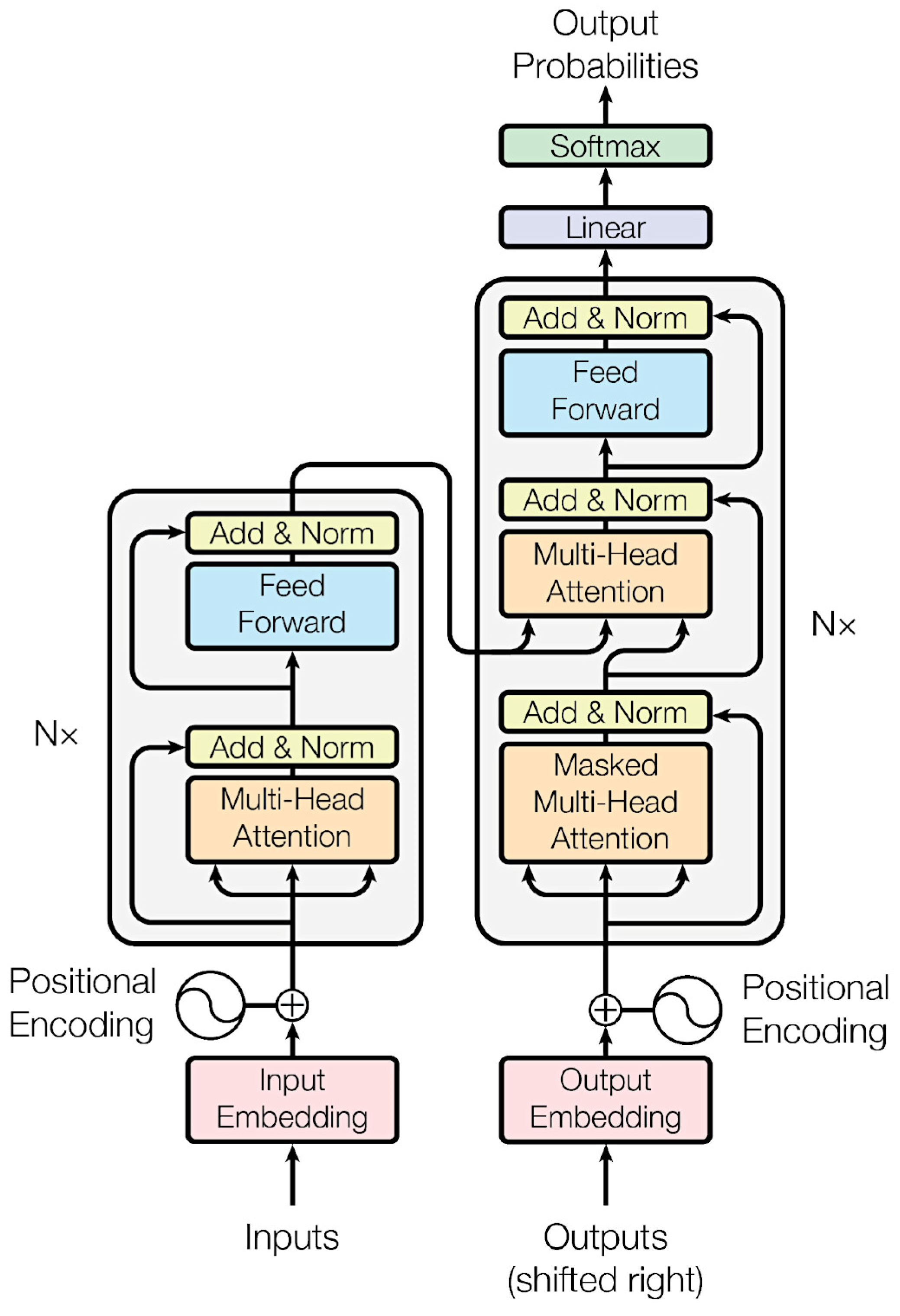
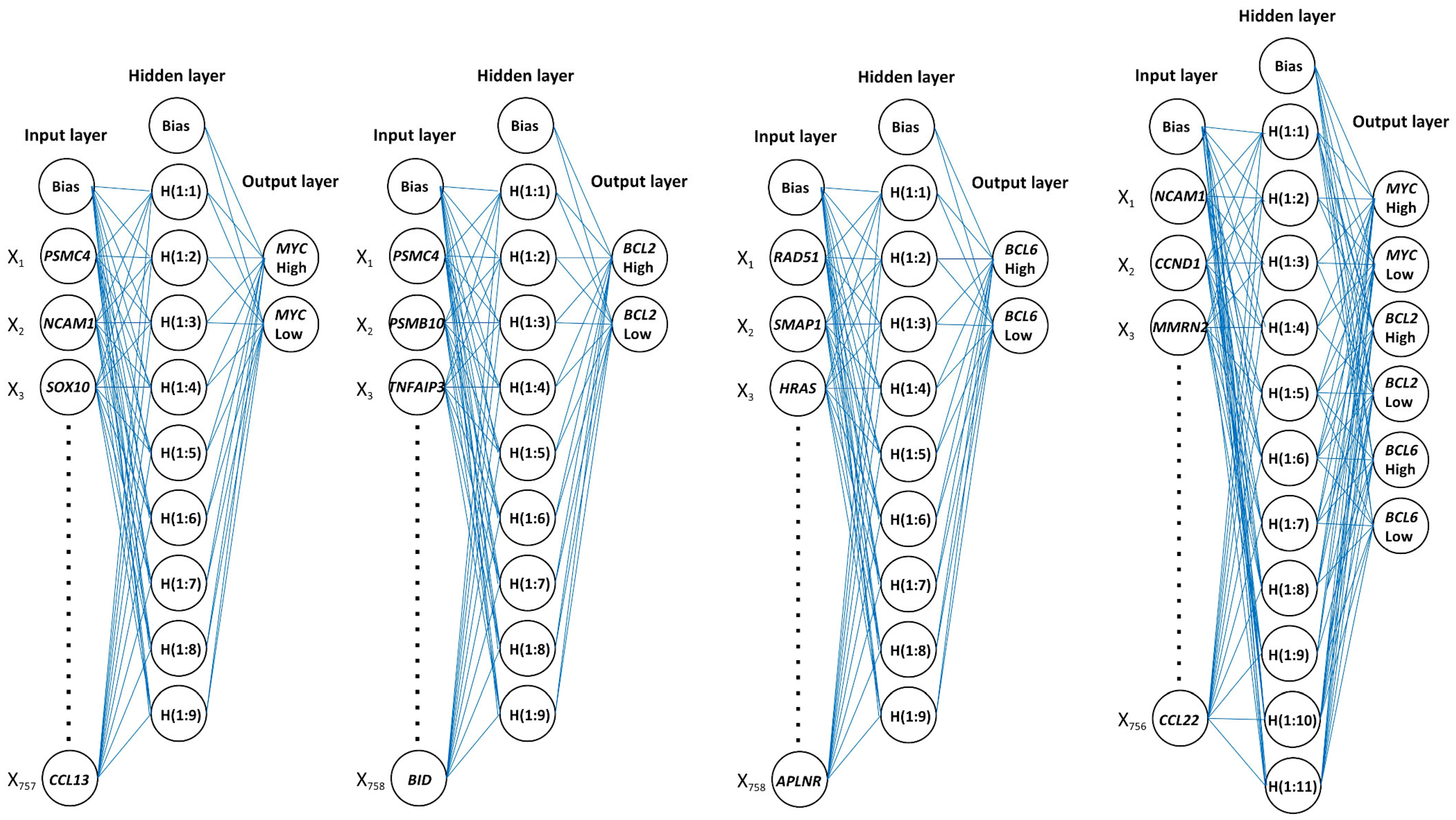
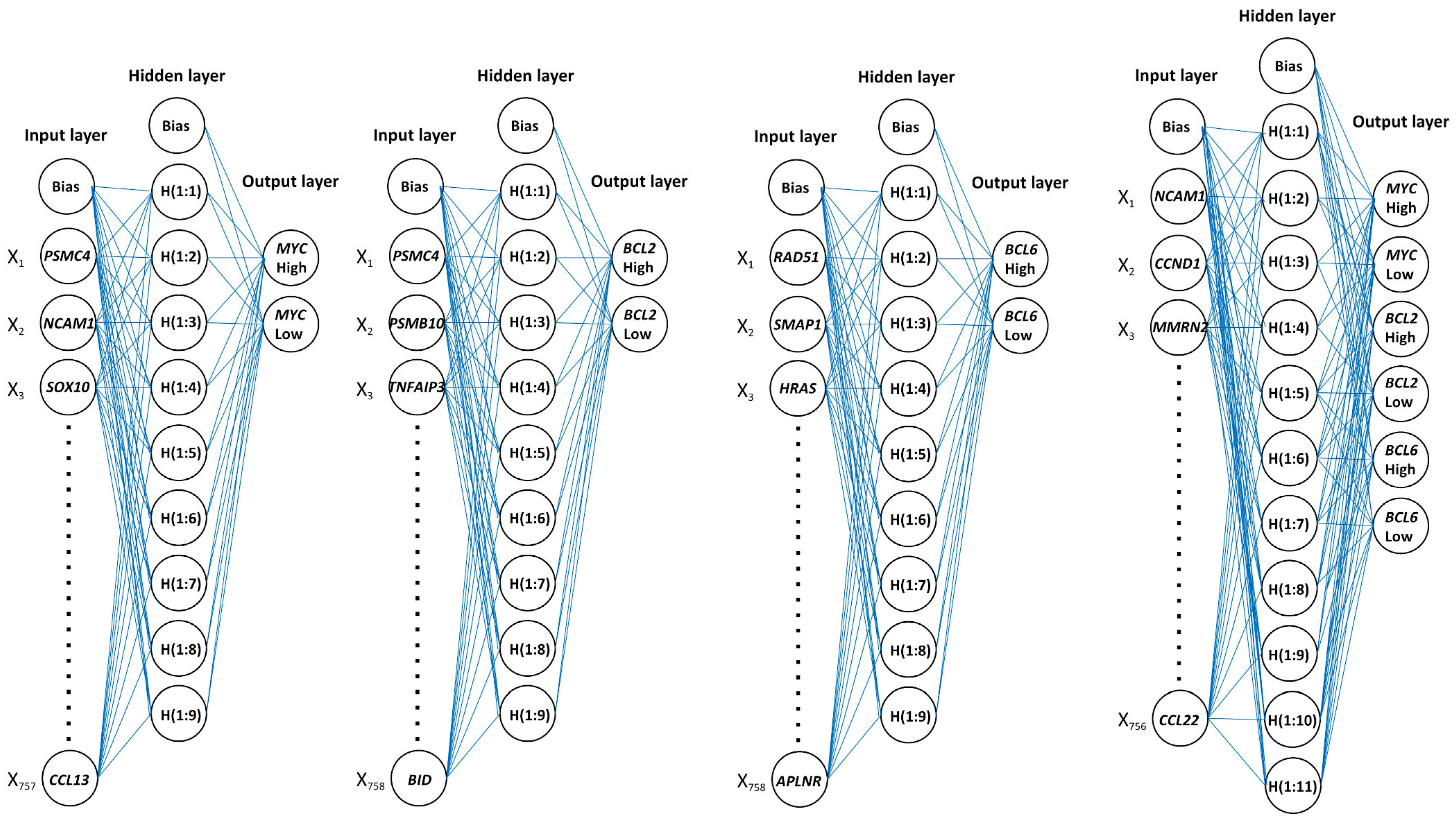
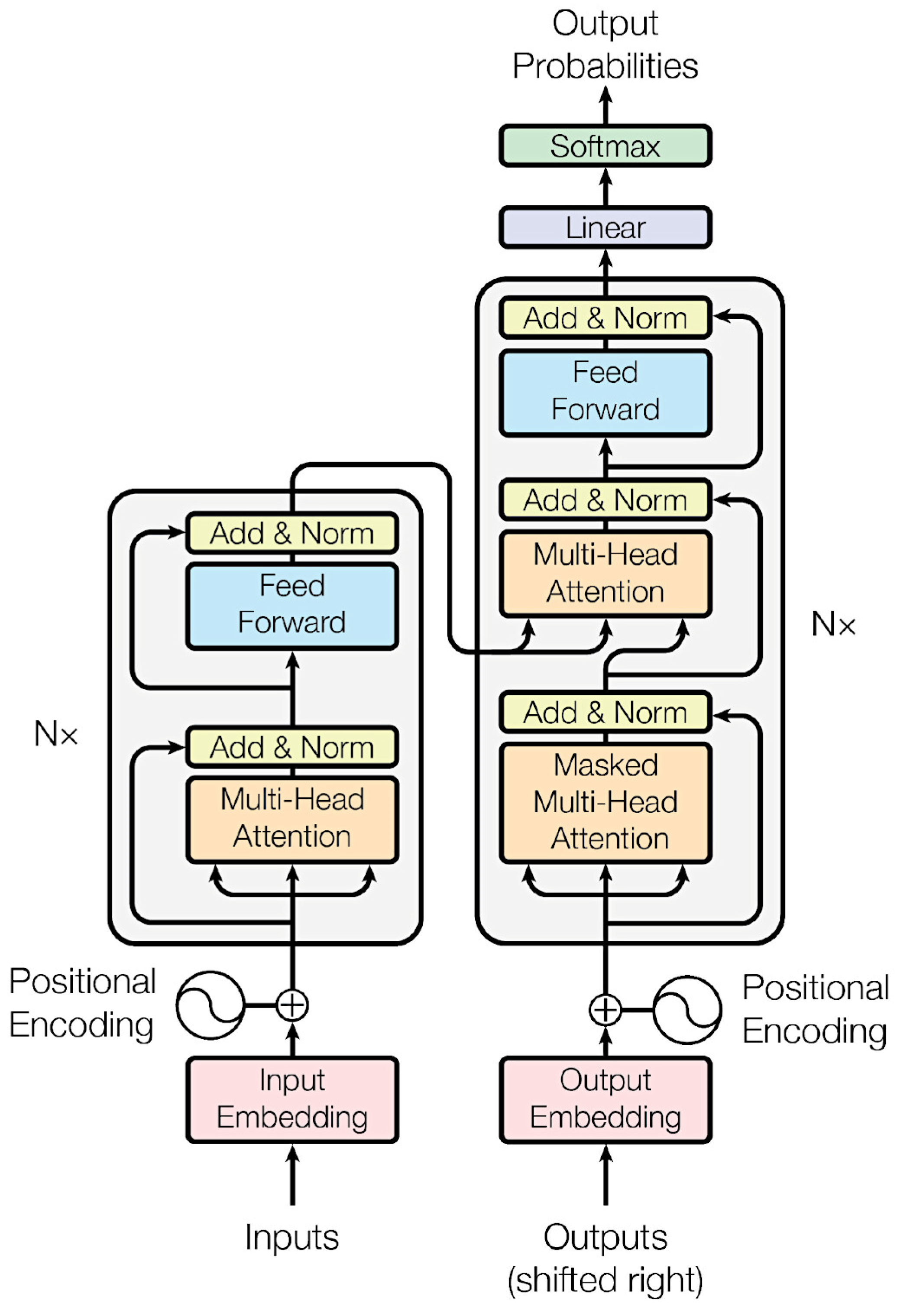
Figure 9.
Architecture of neural networks.
Figure 9.
Architecture of neural networks.
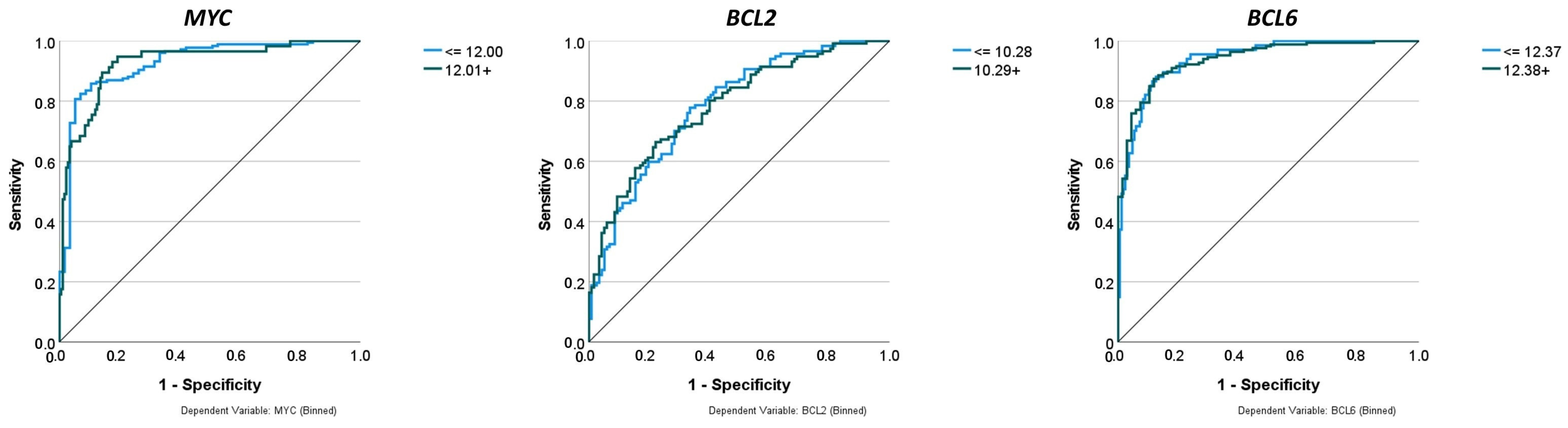
Figure 10.
Comparison of performance using receiver operating characteristic (ROC) curves. The neural networks predicted the gene expression of MYC, BCL2, and BCL6 as binary variables (high vs. low). The predictors were 758 genes of a pan-cancer panel of immuno-oncology and translational research that includes clinically relevant actionable genes and pathways. The areas under the ROC curves were 0.925, 0.783, and 0.939, respectively.
Figure 10.
Comparison of performance using receiver operating characteristic (ROC) curves. The neural networks predicted the gene expression of MYC, BCL2, and BCL6 as binary variables (high vs. low). The predictors were 758 genes of a pan-cancer panel of immuno-oncology and translational research that includes clinically relevant actionable genes and pathways. The areas under the ROC curves were 0.925, 0.783, and 0.939, respectively.
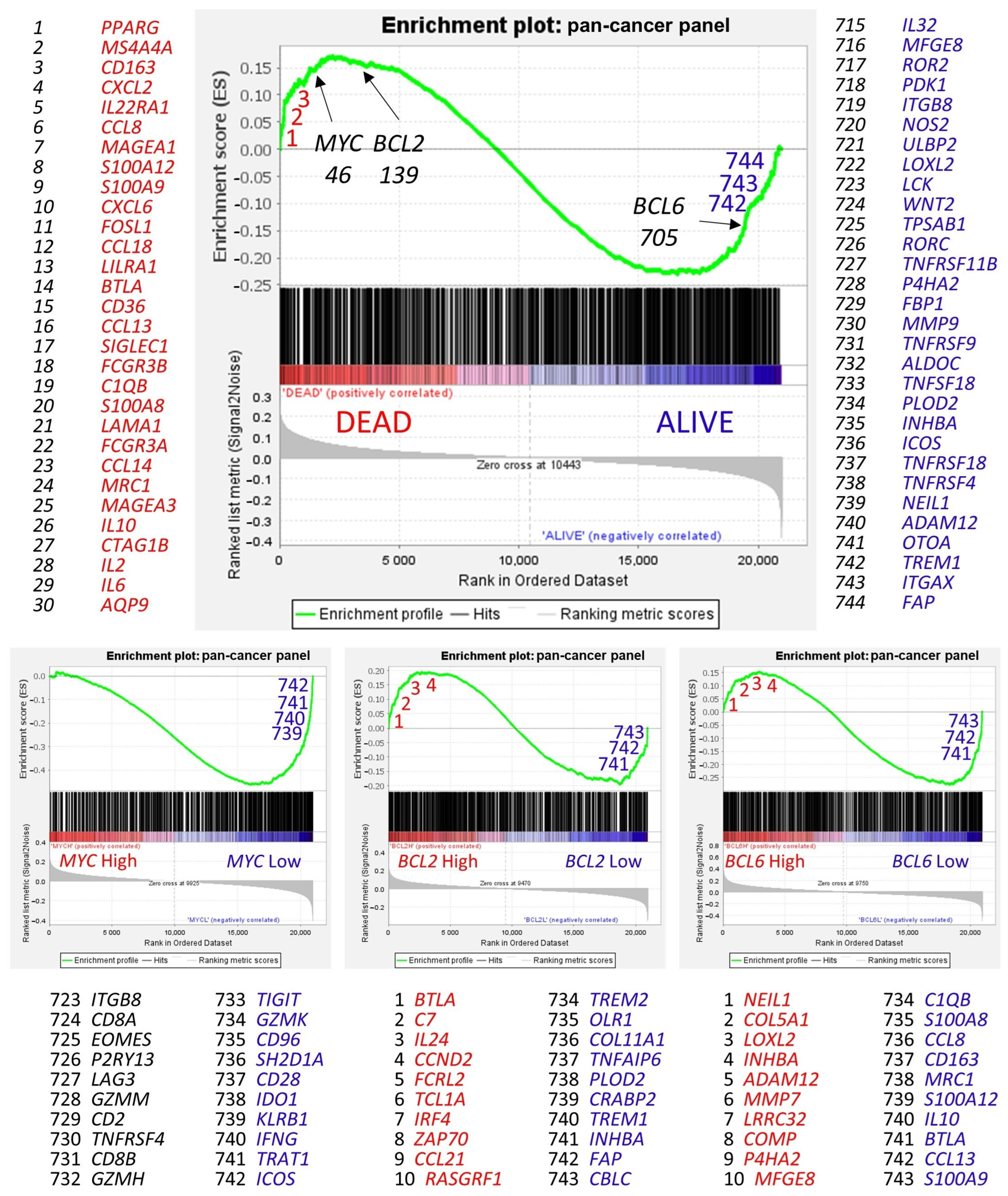
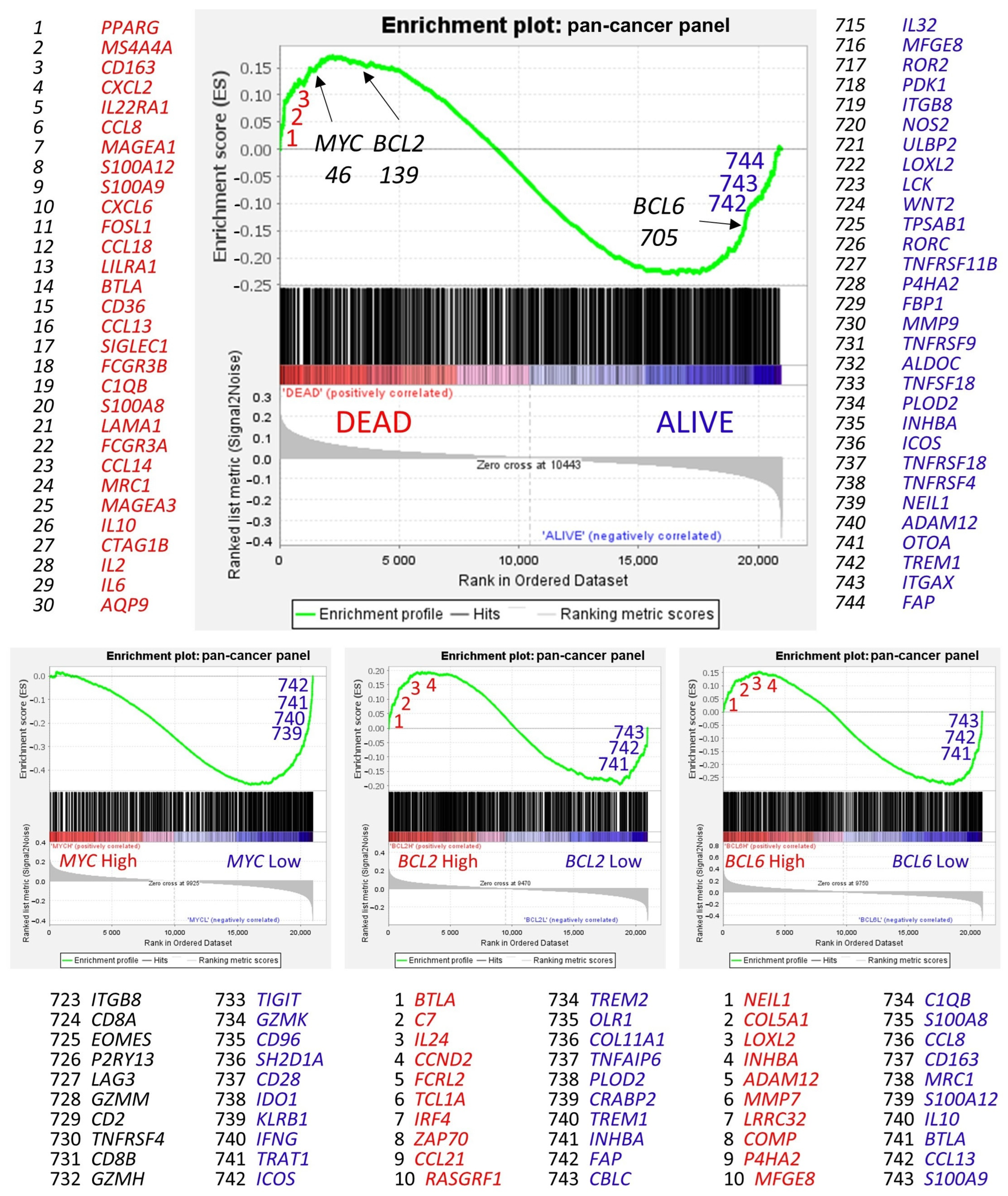
Figure 11.
Gene set enrichment analysis (GSEA). Gene set enrichment analysis (GSEA) is a computational method that determines whether an a priori defined set of genes shows statistically significant, concordant differences between two biological states (e.g., phenotypes) [
66,
67,
68]. GSEA was performed using (i.e., the priori-defined set of genes) the 758 genes of the pan-cancer panel of immuno-oncology and translational research as predictors, which includes clinically relevant actionable genes and pathways. The predicted variables (i.e., phenotypes) were the overall survival outcome, such as dead vs. alive, and the
MYC,
BCL2, and
BCL6 expression (high vs. low groups, the same as the neural networks). In the GSEA, the genes are ranked based on their rank metric score and running enrichment score (ES). The ES reflects the degree to which a gene set is overrepresented at the top or bottom of a ranked list of genes [
66,
67,
68], for example, the gene expression profile of patients who died.
Figure 11.
Gene set enrichment analysis (GSEA). Gene set enrichment analysis (GSEA) is a computational method that determines whether an a priori defined set of genes shows statistically significant, concordant differences between two biological states (e.g., phenotypes) [
66,
67,
68]. GSEA was performed using (i.e., the priori-defined set of genes) the 758 genes of the pan-cancer panel of immuno-oncology and translational research as predictors, which includes clinically relevant actionable genes and pathways. The predicted variables (i.e., phenotypes) were the overall survival outcome, such as dead vs. alive, and the
MYC,
BCL2, and
BCL6 expression (high vs. low groups, the same as the neural networks). In the GSEA, the genes are ranked based on their rank metric score and running enrichment score (ES). The ES reflects the degree to which a gene set is overrepresented at the top or bottom of a ranked list of genes [
66,
67,
68], for example, the gene expression profile of patients who died.
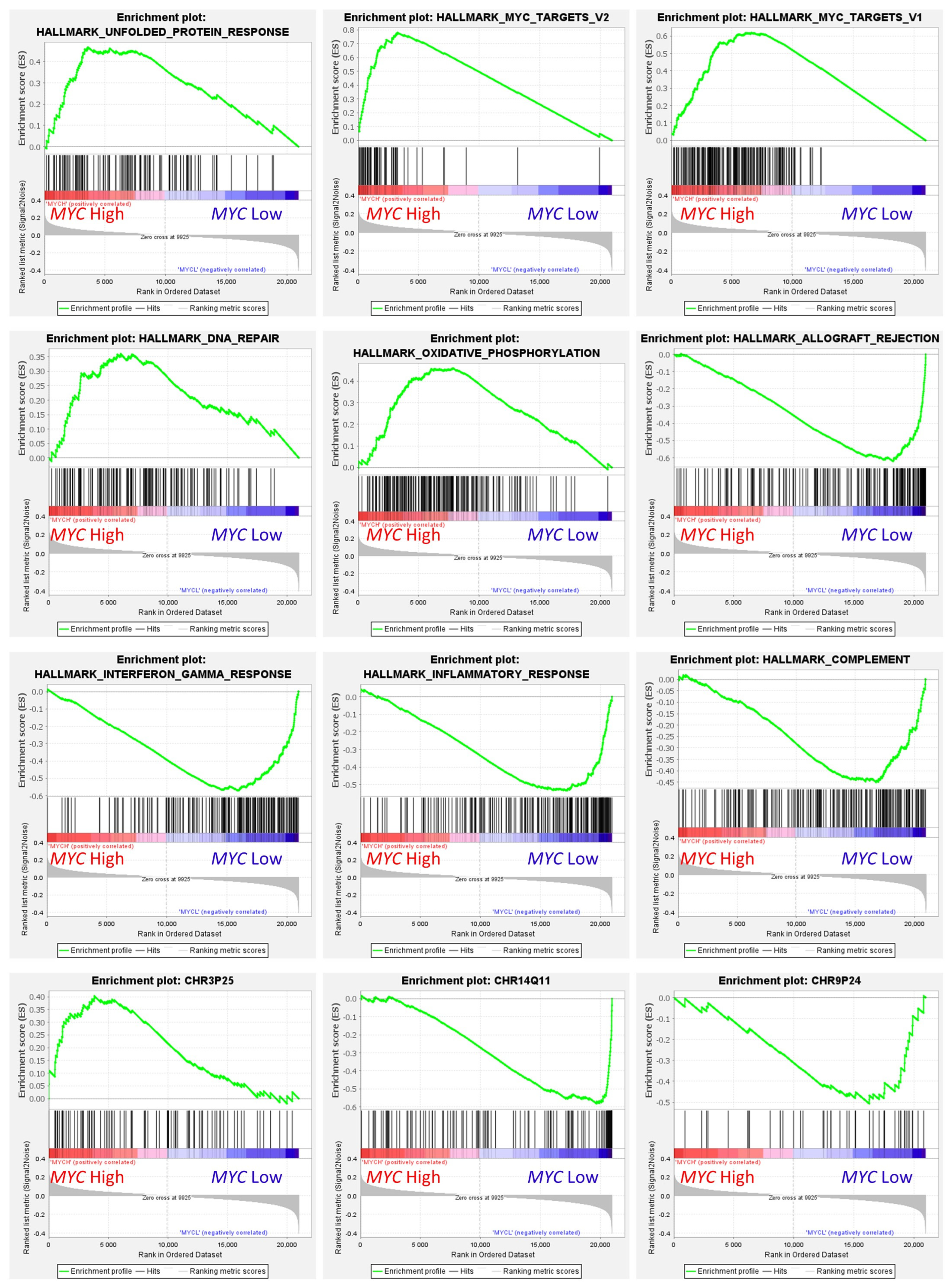
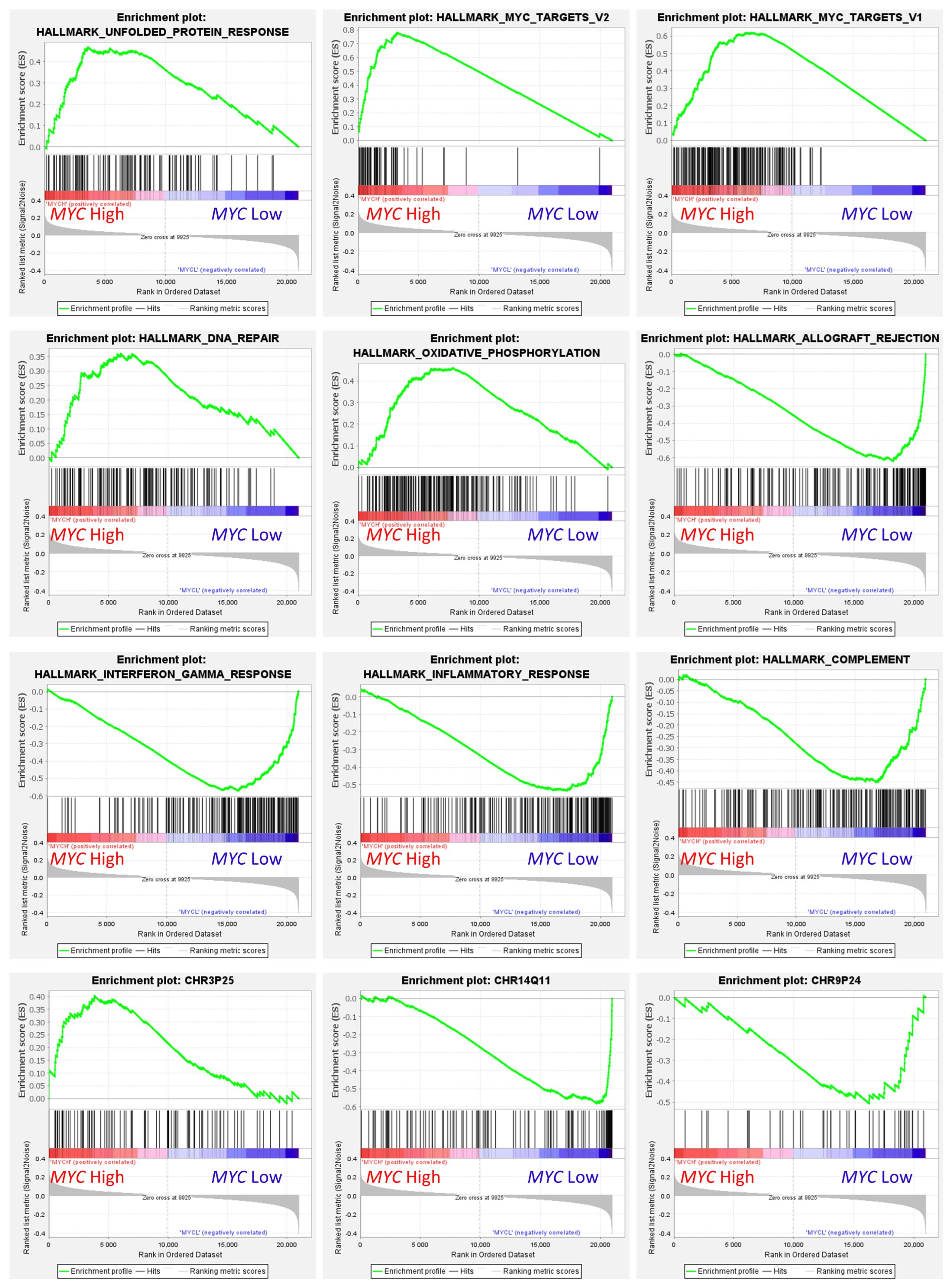
Figure 12.
Gene set enrichment analysis (GSEA) on MYC expression groups. GSEA analysis was performed using as predictors several gene sets of the Molecular signatures database (MSigDB), including hallmark (H), positional (C1), and curated (C2) gene sets. The predicted variable (phenotype) was the MYC expression (high vs. low groups), the same as the neural networks. This figure shows some of the most relevant GSEA plots. High MYC expression correlated with high expression of genes associated with protein response, MYC targets, DNA repair, oxidative phosphorylation pathways, and chromosome 3p25 locus-associated genes. Low MYC expression correlated with allograft rejection, interferon gamma response, inflammatory response, complement, and chromosome 1q11 and 8p24.
Figure 12.
Gene set enrichment analysis (GSEA) on MYC expression groups. GSEA analysis was performed using as predictors several gene sets of the Molecular signatures database (MSigDB), including hallmark (H), positional (C1), and curated (C2) gene sets. The predicted variable (phenotype) was the MYC expression (high vs. low groups), the same as the neural networks. This figure shows some of the most relevant GSEA plots. High MYC expression correlated with high expression of genes associated with protein response, MYC targets, DNA repair, oxidative phosphorylation pathways, and chromosome 3p25 locus-associated genes. Low MYC expression correlated with allograft rejection, interferon gamma response, inflammatory response, complement, and chromosome 1q11 and 8p24.
Table 1.
Applications of artificial intelligence in the medical field.
Table 1.
Applications of artificial intelligence in the medical field.
- 1.
Disease detection
|
- 1.1.
Using DNA methylation analysis, a neural network was used for the early detection of hepatocellular carcinoma [ 13].
|
- 1.2.
Several biochemical parameters from the cerebrospinal fluid were evaluated using Raman spectroscopy and convolutional neural networks for the early diagnosis of Alzheimer’s disease. The study achieved a good classification accuracy of around 90% and a good correlation with the clinical dementia rating score [ 14].
|
- 1.3.
Several machine learning techniques and artificial neural networks were used to predict heart disease at an early stage using clinical, biochemical, and ECG data. In this study, the highest accuracy was achieved using random forests [ 15].
|
- 1.4.
A machine learning algorithm was used to classify patients with coronary disease based on 31 features. The classification had an acceptable accuracy for the identification of severe disease [ 16].
|
- 2.
Personalized medicine
|
- 2.1.
A proof-of-concept analysis based on machine learning algorithms was used to classify patients with similar clinical and echocardiographic parameters to optimize the rate of responders to specific cardiac resynchronization therapies [ 17].
|
- 2.2.
Several machine learning algorithms were used to identify patients with a higher probability of major depression and anxiety disorder and who would benefit from digital psychiatric interventions [ 18].
|
- 2.3.
The Leukemia Artificial Intelligence Program (LEAP) used a machine learning method for the optimal treatment of tyrosine kinase inhibitors in patients with chronic myeloid leukemia [ 19].
|
- 3.
Medical imaging
|
- 3.1.
Mammogram images were evaluated by two radiologists using an AI-assisted method. The study showed that both methods provided comparable results [ 20].
|
- 3.2.
Convolutional neural networks were used to detect lung nodules on chest computed tomography in patients with complex lung disease. The accuracy of the neural network was similar to that of experienced radiologists [ 21].
|
- 4.
Clinical trials
|
- 4.1.
A randomized, controlled clinical trial (NCT0438756) used an AI-based system to assist in the evaluation of mammography images. The study concluded that AI-supported screening was comparable to standard double reading [ 22].
|
- 4.2.
The clinical trial NCT03954548 compared the evaluation of colonoscopy between a deep learning-assisted method and the standard method in patients undergoing colorectal cancer screening or surveillance. The study found that the AI method had a 2-fold lower misrate of colorectal cancer [ 23].
|
- 4.3.
The clinical trial ChiCTR1800018403 used an AI-based system to evaluate endoscopic images for the early detection of gastric cancer. The study showed that the deep convolutional neural network and deep reinforcement learning method correctly predicted cancer lesions but with less performance than the human-based method [ 24]
|
Table 2.
Applications of artificial intelligence in hematological neoplasia.
Table 2.
Applications of artificial intelligence in hematological neoplasia.
- 1.
Molecular pathology
|
- 1.1.
More than 130 genetic markers, gene expression, and microenvironment data were used to classify the seven most frequent non-Hodgkin B-cell lymphomas (B-NHLs) [ 25].
|
- 1.2.
A supervised machine learning method used the expression of 6817 genes to predict the overall survival of patients with diffuse large B-cell lymphoma [ 8].
|
- 1.3.
A series of 123 cases of mantle cell lymphoma were analyzed using gene expression data and several machine learning and artificial neural networks. This research highlighted pathogenic genes and immune–oncology pathways [ 26].
|
- 1.4.
Based on the RNA expression of 1408 genes, next-generation profiling, and machine learning (geometric mean naïve Bayesian algorithm), several diagnostic entities, including carcinomas and lymphoma, were classified with good performance [ 27].
|
- 1.5.
The prognosis of diffuse large B-cell lymphoma was predicted using a feedforward neural network in a series of 414 cases and gene expression data, which correlated to other prognostic markers, including MYC and BCL2 [ 28].
|
- 1.6.
Several mature B-cell neoplasms were analyzed using gene expression, immunohistochemical markers, machine learning, and neural networks. The study managed to classify the patients according to their lymphoma subtype and predict their survival. Pan-cancer analysis was also performed [ 29].
|
- 1.7.
Based on 730 immune–oncology genes, overall survival and cell-of-origin subtypes were predicted in a series of 106 cases of diffuse large B-cell lymphoma. The analysis included several machine learning and neural networks [ 30].
|
- 1.8.
Targeted RNA sequencing data obtained from a next-generation sequencing analysis platform were used to classify 418 cases of diffuse large B-cell lymphoma using AI and to predict the survival of the patients [ 31].
|
- 2.
Medical imaging
|
- 2.1.
A total of 31 variables were used by an artificial neural network to predict the 5-year recurrence after treatment of 114 patients with Hodgkin’s lymphoma [ 32].
|
- 2.2.
A discrimination method that includes convolution and a neural network combined with the least absolute shrinkage and selection operator (LASSO) model was used to analyze the computed tomography data of 276 patients with enlarged cervical lymph nodes. The accuracy of this method was above 86% for lymphoma cases [ 33].
|
- 2.3.
The data of 5275 patients with lung and breast cancer and non-Hodgkin lymphoma were analyzed using an AI-based tool to create a predictive model of risk stratification and early disease detection [ 34].
|
- 2.4.
AI was used to analyze PET/CT images of 382 cases of diffuse large B-cell lymphoma (DLBCL) using only 2 maximum-intensity projection (MIP) images, and it correlated with the prognosis of the patients [ 35].
|
- 2.5.
The focal skeleton/bone marrow uptake (BMU) of FDG-PET/CT images was analyzed using an AI-based method in 201 patients with Hodgkin’s lymphoma [ 36].
|
- 2.6.
Anterior segment optical coherence tomography (AS-OCT) images were used to classify between vitreoretinal lymphoma and uveitis in 28 patients using the xgboost python function with good performance (AUC 0.84–0.94) [ 37].
|
- 3.
Histological and cytological images
|
- 3.1.
Several artificial intelligence-based tools (Phyton SciPy package) were used to model several morphological biomarkers (nuclear size, cell density, and cell distance) to distinguish 125 tissue samples of chronic lymphocytic leukemia (CLL) progression to accelerated CLL (aCLL) or transformation to diffuse large B-cell lymphoma (Richter transformation; RT). The performance of the method was moderate, with an area under the curve (AUC) ranging from 0.66 to 0.94 [ 38].
|
- 3.2.
A series of 287 samples from several hospitals were used to predict MYC rearrangement using histological slides of diffuse large B-cell lymphoma. The analysis had a good sensitivity of 0.93 but a low specificity of 0.52 [ 39].
|
- 3.3.
Neural networks were used to differentiate between diffuse large B-cell lymphoma and Burkitt lymphoma in a series of 70 cases, including 10,818 images [ 40].
|
- 3.4.
Hematoxylin and eosin (H&E) images of 388 cases were analyzed by AI to classify the samples into diffuse large B-cell lymphoma, follicular lymphoma, and reactive lymphoid tissue with high accuracy [ 41].
|
- 3.5.
The images of 629 patients with non-Hodgkin lymphoma were analyzed using a convolutional neural network to stratify the patients according to different lymphoma subtypes. The algorithm had an accuracy of 96% [ 42].
|
- 3.6.
Histological images of chronic lymphocytic leukemia were analyzed using AI in proliferation centers to identify the accelerated phase and Richter transformation based on nuclear characteristics [ 43].
|
- 3.7.
Blood films from 591 samples were used to identify circulating abnormal cells (leukemic and dysplastic cells) [ 44].
|
Table 3.
Confusion matrix.
Table 3.
Confusion matrix.
| Confusion Matrix | True Class |
|---|
| Positive | Negative |
|---|
| Predicted class | Positive | True Positive (TP) | False Positive (FP) |
| Negative | False Negative (FN) | True Negative (TN) |
Table 4.
Cutpoints of MYC, BCL2, and BCL6 genes.
Table 4.
Cutpoints of MYC, BCL2, and BCL6 genes.
| Gene | Cutpoint | Distribution |
|---|
| MYC | ≤12.0 | 176/233 (75.5%) |
| | 12.01+ | 57/233 (24.5%) |
| BCL2 | ≤10.28 | 117/233 (50.2%) |
| | 10.29+ | 116/233 (49.8%) |
| BCL6 | ≤12.37 | 67/233 (28.8%) |
| | 12.38+ | 166/233 (71.2%) |
Table 5.
Neural network characteristics.
Table 5.
Neural network characteristics.
| | MYC | BCL2 | BCL6 | MYC, BCL2, and BCL6 |
|---|
| Training set | 161/233 (69.1%) | 173/233 (74.2%) | 161/233 (69.1%) | 159/233 (68.2%) |
| Testing set | 72/233 (30.9%) | 60/233 (35.8%) | 72/233 (30.9%) | 74/233 (31.8%) |
| Input layer | | | | |
| Units | 757 | 757 | 758 | 756 |
| Rescaling | Standardized | Standardized | Standardized | Standardized |
| Hidden layer | | | | |
| Number | 1 | 1 | 1 | 1 |
| Units | 9 | 9 | 10 | 11 |
| Activation function | Hyperbolic tangent | Hyperbolic tangent | Hyperbolic tangent | Hyperbolic tangent |
| Output layer | | | | |
| Predicted variables | 1 | 1 | 1 | 3 |
| Units | 2 | 2 | 2 | 6 |
| Activation function | Softmax | Softmax | Softmax | Softmax |
| Error function | Cross-entropy | Cross-entropy | Cross-entropy | Cross-entropy |
| Classification percentage correct | | | | |
| Training set | 86.3% | 82.2% | 88.2% | 76.7%, 81.1%, and 83.6% |
| Testing set | 88.9% | 63.3% | 86.1% | 83.8%, 67.6%, and 77.0% |
| Area under the curve (AUC) | 0.925 | 0.783 | 0.939 | 0.81, 0.86, and 0.86 |
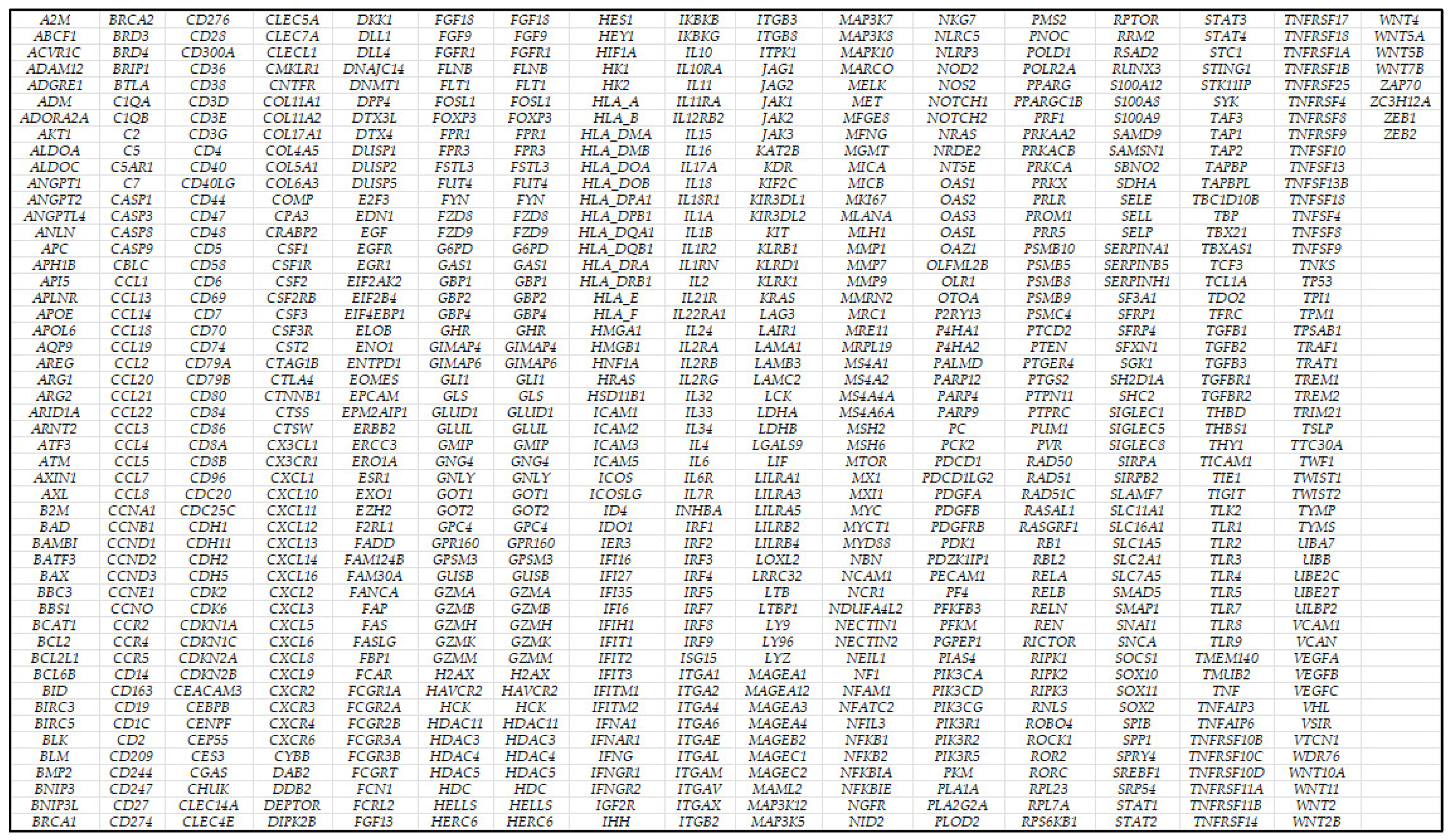
| First and most relevant predictors | PSMC4, NCAM1, SOX10, PTPRC, PSMB10, C5AR1, IL6, CBLC, FCGR3B, and MTOR | PSMC4, CNTFR, PSMB10, TNFAIP3, MLH1, CXCR2, FADD, CD7, AREG, and TBXAS1 | RAD51, SMAP1, HRAS, SFRP1, LAG3, BTLA, TICAM1, BCL2L1, G6PD, and ICAM2 | NCAM1, CCND1, MMRN2, RAD51, TIGIT, THY1, BTLA, ITGA2, HCK, and SFRP1 |
Table 6.
Confusion matrix for MYC prediction.
Table 6.
Confusion matrix for MYC prediction.
| Training | Predicted | Testing | Predicted |
|---|
| Observed | Low | High | Observed | Low | High |
|---|
| Low | 115 | 7 | Low | 50 | 4 |
| High | 15 | 24 | High | 4 | 14 |
Table 7.
Confusion matrix for BCL2 prediction.
Table 7.
Confusion matrix for BCL2 prediction.
| Training | Predicted | Testing | Predicted |
|---|
| Observed | Low | High | Observed | Low | High |
|---|
| Low | 74 | 16 | Low | 20 | 7 |
| High | 30 | 53 | High | 15 | 18 |
Table 8.
Confusion matrix for BCL6 prediction.
Table 8.
Confusion matrix for BCL6 prediction.
| Training | Predicted | Testing | Predicted |
|---|
| Observed | Low | High | Observed | Low | High |
|---|
| Low | 31 | 15 | Low | 17 | 4 |
| High | 4 | 111 | High | 6 | 45 |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}