
What’s New in the Classification, Diagnosis and Therapy of Myeloid Leukemias



Abstract
1. Introduction
2. Updates on the Classification, Diagnosis and Therapy of Acute Myeloid Leukemia (AML)
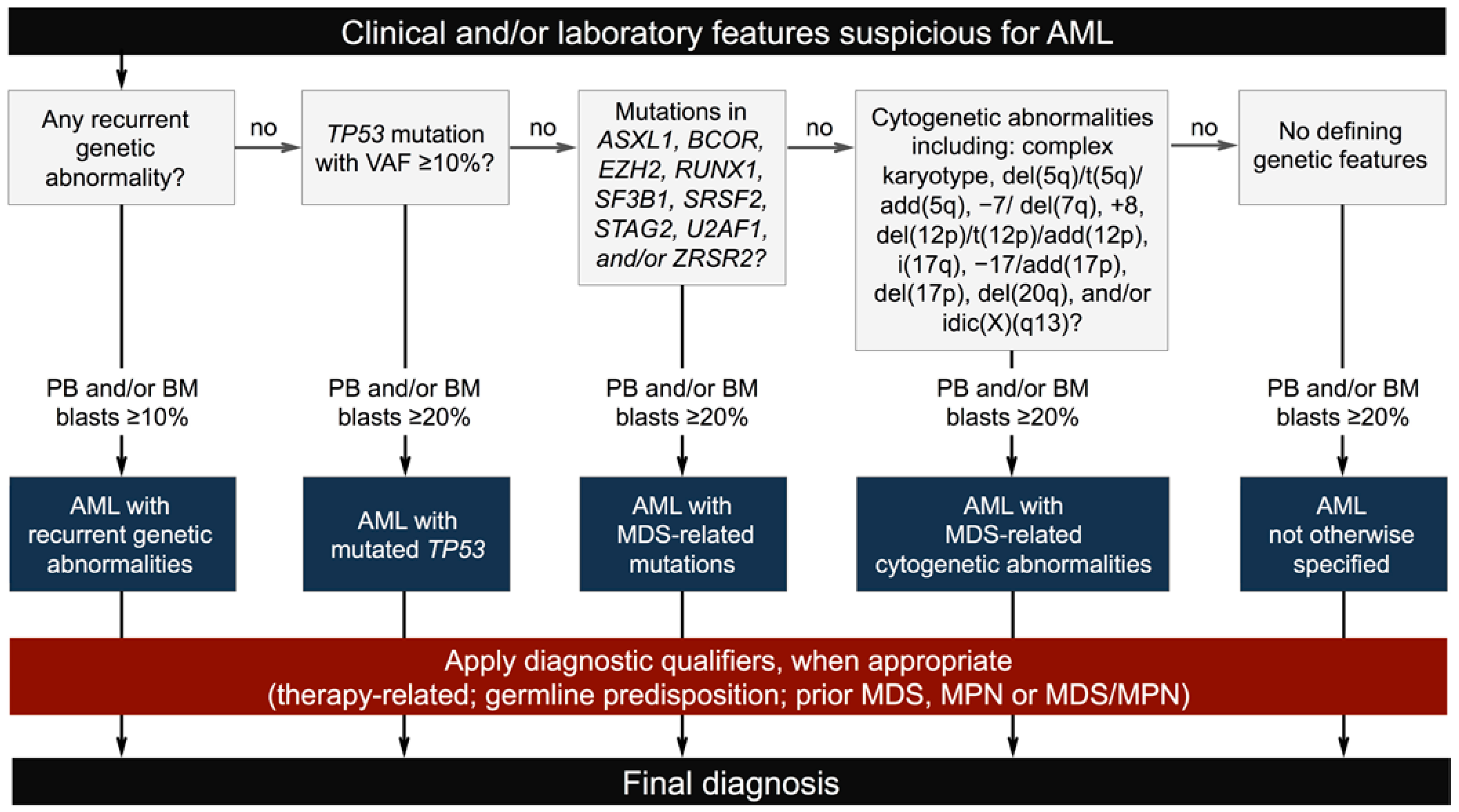
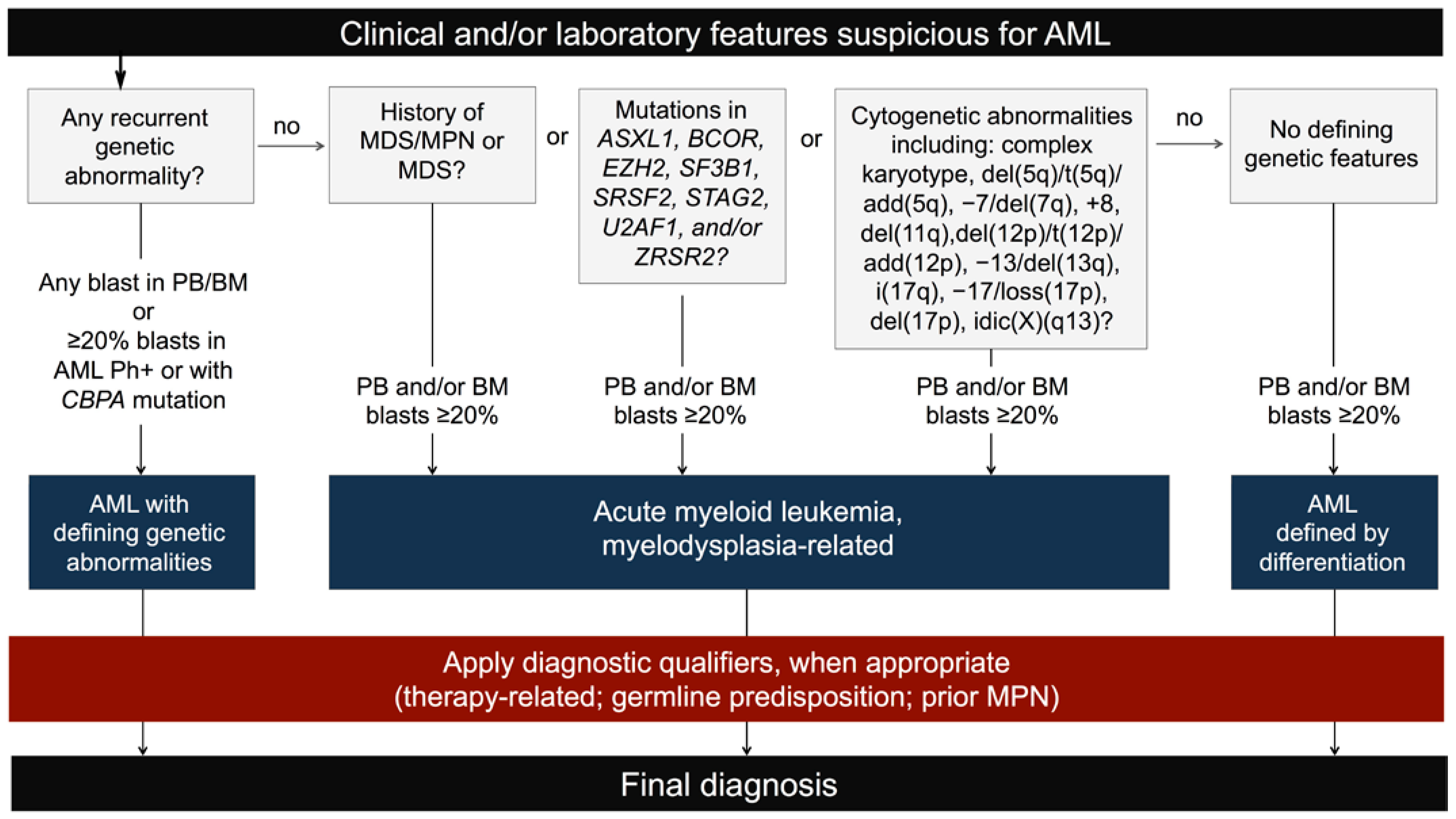
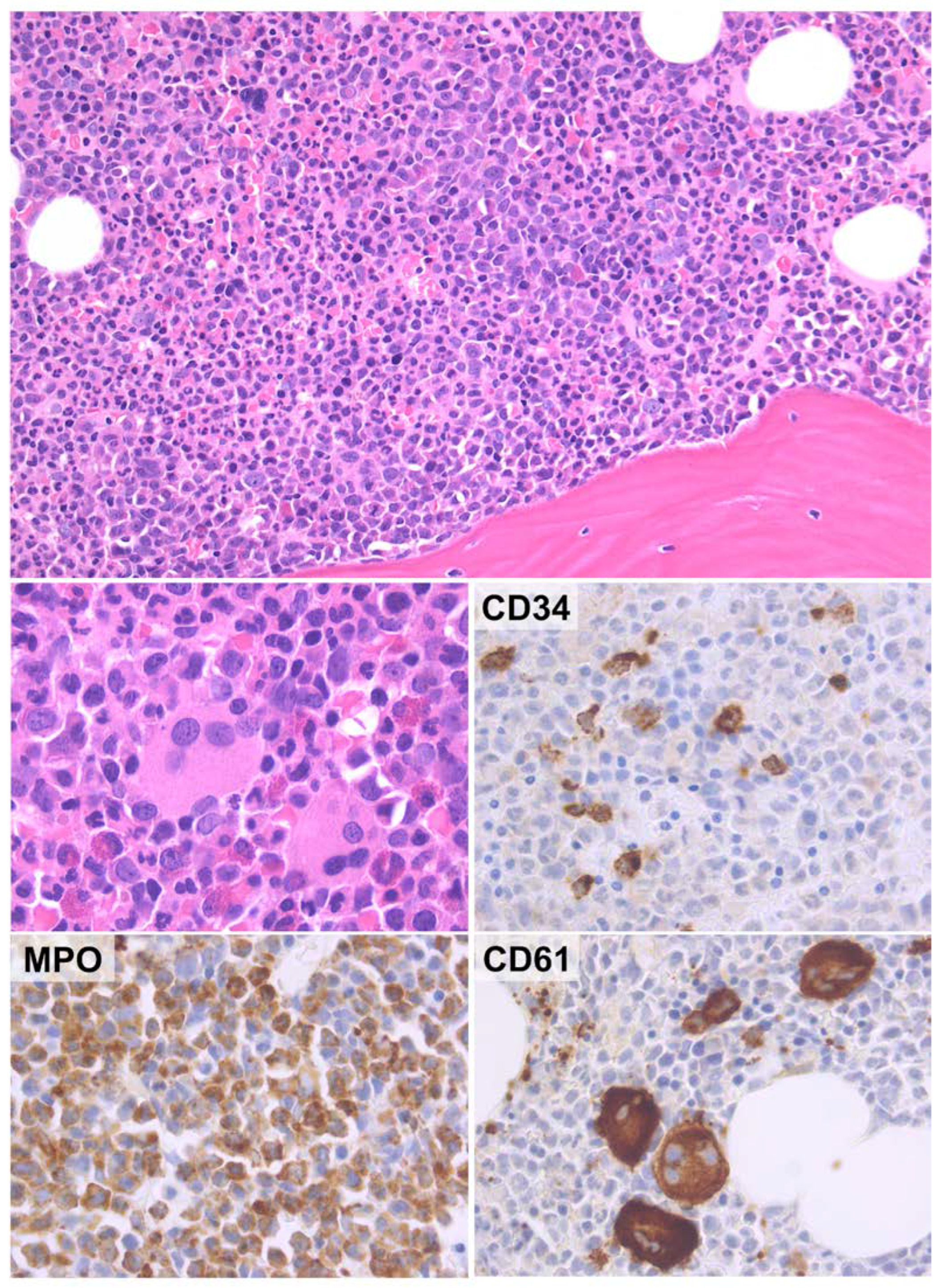
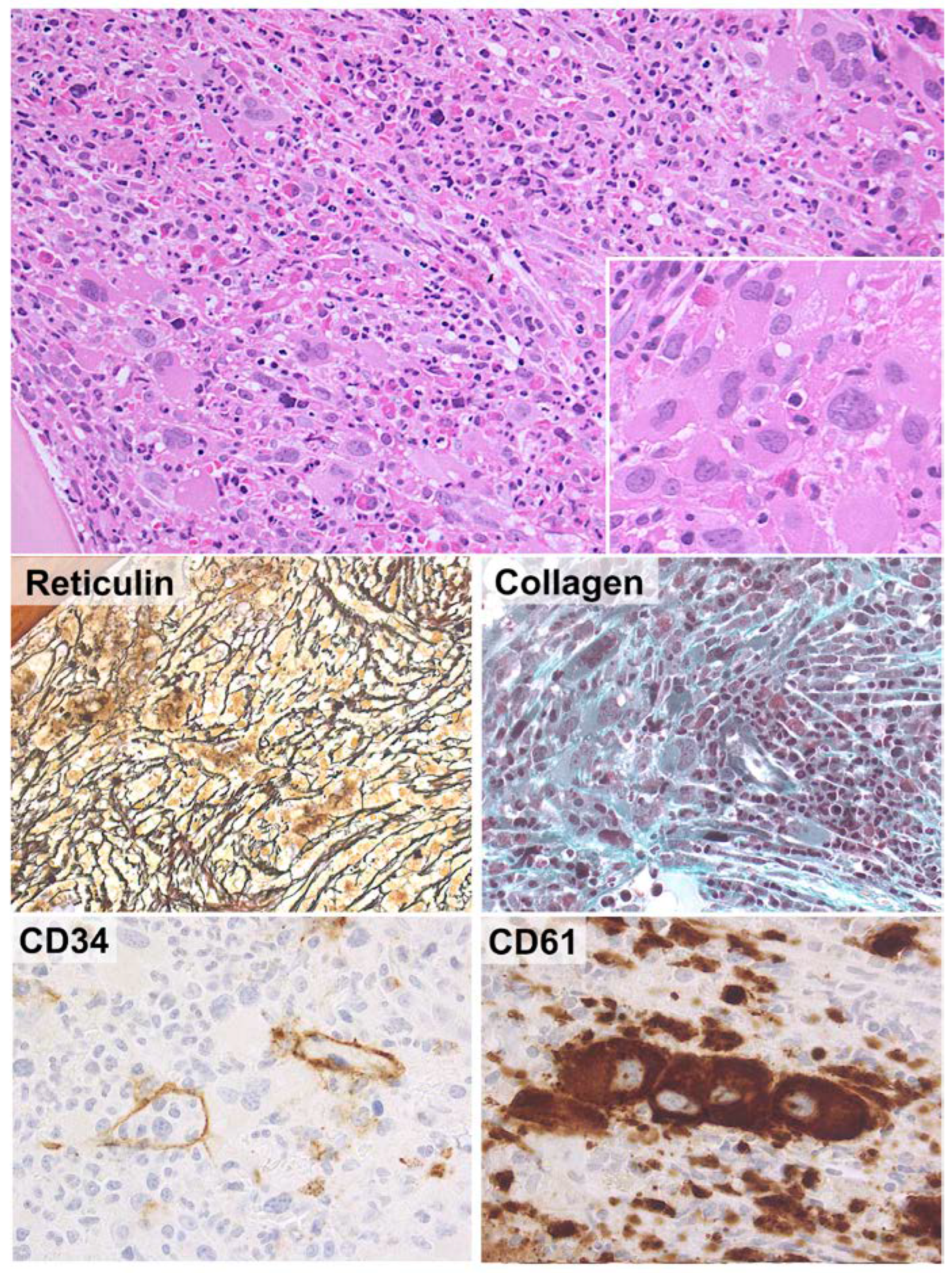
2.1. New Classification and Updates in the Diagnosis of Acute Myeloid Leukemia (AML)

2.2. Novelties in the Therapy of Acute Myeloid Leukemia (AML)
3. Updates on the Classification, Diagnosis and Therapy of Chronic Myelomonocytic Leukemia (CMML)
3.1. New Classification and Updates in the Diagnosis of Chronic Myelomonocytic Leukemia (CMML)
3.2. Novelties in the Therapy of Chronic Myelomonocytic Leukemia (CMML)
4. Updates on the Classification, Diagnosis and Therapy of Chronic Myeloid Leukemia (CML)
4.1. New Classification and Updates in the Diagnosis of Chronic Myeloid Leukemia (CML)
4.2. Novelties in the Therapy of Chronic Myeloid Leukemia (CML)
5. Updates on the Classification, Diagnosis and Therapy of Myeloid Neoplasms (MNs) with Eosinophilia
5.1. New Classification and Updates in the Diagnosis of Myeloid Neoplasms (MNs) with Eosinophilia
5.2. Novelties in the Therapy of Myeloid Neoplasms (MNs) with Eosinophilia
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kampen, K.R. The discovery and early understanding of leukemia. Leuk. Res. 2012, 36, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Geary, C.G. The story of chronic myeloid leukaemia. Br. J. Haematol. 2000, 110, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Fraenkel, A. Ueber acute Leukamie. Dtsch Med. Wochenschr. 1895, 21, 639–642. [Google Scholar] [CrossRef]
- Reschad, H.; Schilling-Torgau, V. Ueber eine neue Leukämie durch echte Uebergangsformen (Splenozytenleukämie) und ihre Bedeutung für die Selbständigkeit dieser Zellen. Münchener Med. Wchnschrift 1913, 60, 1981–1984. [Google Scholar]
- Neame, P.B.; Soamboonsrup, P.; Browman, G.P.; Meyer, R.M.; Benger, A.; Wilson, W.E.; McBride, J.A. Classifying Acute Leukemia by Immunophenotyping: A Combined FAB-Immunologic Classification of AML. Blood 1986, 68, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Vardiman, J.W.; Harris, N.L.; Brunning, R.D. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 2002, 100, 2292–2302. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Hasserjian, R.P.; Orazi, A.; Mathews, V.; Roberts, A.W.; Schiffer, C.A.; Roug, A.S.; Cazzola, M.; Döhner, H.; Tefferi, A. Classification of myeloid neoplasms/acute leukemia: Global perspectives and the international consensus classification approach. Am. J. Hematol. 2022, 97, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Martelli, M.P. Comparison of the International Consensus and 5th WHO edition classifications of adult myelodysplastic syndromes and acute myeloid leukemia. Am. J. Hematol. 2023, 98, 481–492. [Google Scholar] [CrossRef]
- Weinberg, O.K.; Porwit, A.; Orazi, A.; Hasserjian, R.P.; Foucar, K.; Duncavage, E.J.; Arber, D.A. The International Consensus Classification of acute myeloid leukemia. Virchows Arch. 2022, 482, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Tarlock, K.; Lamble, A.J.; Wang, Y.C.; Gerbing, R.B.; Ries, R.E.; Loken, M.R.; Brodersen, L.E.; Pardo, L.; Leonti, A.; Smith, J.L.; et al. CEBPA-bZip mutations are associated with favorable prognosis in de novo AML: A report from the Children’s Oncology Group. Blood 2021, 138, 1137–1147. [Google Scholar] [CrossRef]
- Taube, F.; Georgi, J.A.; Kramer, M.; Stasik, S.; Middeke, J.M.; Röllig, C.; Krug, U.; Krämer, A.; Scholl, S.; Hochhaus, A.; et al. CEBPA mutations in 4708 patients with acute myeloid leukemia: Differential impact of bZIP and TAD mutations on outcome. Blood 2022, 139, 87–103. [Google Scholar] [CrossRef]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Ok, C.Y.; Patel, K.P.; Garcia-Manero, G.; Routbort, M.J.; Peng, J.; Tang, G.; Goswami, M.; Young, O.C.; Singh, R.; Medeiros, L.J.; et al. TP53 mutation characteristics in therapy-related myelodysplastic syndromes and acute myeloid leukemia is similar to de novo diseases. J. Hematol. Oncol. 2015, 8, 45. [Google Scholar] [CrossRef]
- Grob, T.; Al Hinai, A.S.A.; Sanders, M.A.; Kavelaars, F.G.; Rijken, M.; Gradowska, P.L.; Biemond, B.J.; Breems, D.A.; Maertens, J.; Kooy, M.V.M.; et al. Molecular characterization of mutant TP53 acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood 2022, 139, 2347–2354. [Google Scholar] [CrossRef]
- Niparuck, P.; Police, P.; Noikongdee, P.; Siriputtanapong, K.; Limsuwanachot, N.; Rerkamnuaychoke, B.; Chuncharunee, S.; Siriboonpiputtana, T. TP53 mutation in newly diagnosed acute myeloid leukemia and myelodysplastic syndrome. Diagn. Pathol. 2021, 16, 100. [Google Scholar] [CrossRef]
- Fang, H.; Wang, S.A.; Khoury, J.D.; El Hussein, S.; Kim, D.H.; Tashakori, M.; Tang, Z.; Li, S.; Hu, Z.; Jelloul, F.Z.; et al. Pure erythroid leukemia is characterized by biallelic TP53 inactivation and abnormal p53 expression patterns in de novo and secondary cases. Haematologica 2022, 107, 2232–2237. [Google Scholar] [CrossRef] [PubMed]
- Pizzi, M.; Sbaraglia, M.; De Bartolo, D.; Santo, L.D.; Binotto, G.; Tosato, F.; Pravato, S.; Scapinello, G.; Martines, A.; Bonaldi, L.; et al. Relevance of bone marrow histology in challenging cases of Acute Myeloid Leukemia. Int. J. Lab. Hematol. 2021, 44, e107–e110. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Macijewski, K.; Weiss, T.; Bacher, U.; Schnittger, S.; Kern, W.; Kohlmann, A.; Klein, H.-U.; Vignetti, M.; Piciocchi, A.; et al. Multilineage dysplasia has no impact on biologic, clinicopathologic, and prognostic features of AML with mutated nucleophosmin (NPM1). Blood 2010, 115, 3776–3786. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Bennett, J.; Löffler, H.; Gassmann, W.; Andersen, J.W.; Tuzuner, N.; BÜChner, T. Acute myeloid leukemia with translocation (8;21). Cytomorphology, dysplasia and prognostic factors in 41 cases. AML Cooperative Group and ECOG. Leuk. Lymphoma 1996, 23, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Baer, C.; Walter, W.; Stengel, A.; Hutter, S.; Meggendorfer, M.; Kern, W.; Haferlach, C.; Haferlach, T. Molecular Classification of AML-MRC Reveals a Distinct Profile and Identifies MRC-like Patients with Poor Overall Survival. Blood 2019, 134, 2735. [Google Scholar] [CrossRef]
- Yates, J.W.; Wallace, H.J., Jr.; Ellison, R.R.; Holland, J.F. Cytosine arabinoside (NSC-63878) and daunorubicin (NSC-83142) therapy in acute nonlymphocytic leukemia. Cancer Chemother. Rep. 1973, 57, 485–488. [Google Scholar]
- Dombret, H.; Seymour, J.F.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.; Schuh, A.C.; Candoni, A.; et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015, 126, 291–299. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Thomas, X.G.; Dmoszynska, A.; Wierzbowska, A.; Mazur, G.; Mayer, J.; Gau, J.-P.; Chou, W.-C.; Buckstein, R.; Cermak, J.; et al. Multicenter, Randomized, Open-Label, Phase III Trial of Decitabine Versus Patient Choice, with Physician Advice, of Either Supportive Care or Low-Dose Cytarabine for the Treatment of Older Patients with Newly Diagnosed Acute Myeloid Leukemia. J. Clin. Oncol. 2012, 30, 2670–2677. [Google Scholar] [CrossRef]
- Liu, H. Emerging agents and regimens for AML. J. Hematol. Oncol. 2021, 14, 49. [Google Scholar] [CrossRef]
- Kayser, S.; Levis, M.J. Updates on targeted therapies for acute myeloid leukaemia. Br. J. Haematol. 2021, 196, 316–328. [Google Scholar] [CrossRef]
- Dinardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2018, 133, 7–17. [Google Scholar] [CrossRef]
- Dinardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Samra, B.; Konopleva, M.; Isidori, A.; Daver, N.; Dinardo, C. Venetoclax-Based Combinations in Acute Myeloid Leukemia: Current Evidence and Future Directions. Front. Oncol. 2020, 10, 562558. [Google Scholar] [CrossRef] [PubMed]
- Roboz, G.J.; Döhner, H.; Pocock, C.; Dombret, H.; Ravandi, F.; Jang, J.H.; Selleslag, D.; Mayer, J.; Martens, U.M.; Liesveld, J.; et al. Oral azacitidine preserves favorable level of fatigue and health-related quality of life for patients with acute myeloid leukemia in remission: Results from the phase 3, placebo-controlled QUAZAR AML-001 trial. Haematologica 2021, 106, 3240–3244. [Google Scholar] [CrossRef] [PubMed]
- Hunter, A.M.; Sallman, D.A. Current status and new treatment approaches in TP53 mutated AML. Best Pract. Res. Clin. Haematol. 2019, 32, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Rücker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.-M.; Holzmann, K.; Gaidzik, V.I.; et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2012, 119, 2114–2121. [Google Scholar] [CrossRef]
- Lehmann, S.; Bykov, V.J.; Ali, D.; Andrén, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in Vivo: A First-in-Human Study with p53-Targeting Compound APR-246 in Refractory Hematologic Malignancies and Prostate Cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar] [CrossRef]
- Maiti, A.; DiNardo, C.D.; Daver, N.G.; Rausch, C.R.; Ravandi, F.; Kadia, T.M.; Pemmaraju, N.; Borthakur, G.; Bose, P.; Issa, G.C.; et al. Triplet therapy with venetoclax, FLT3 inhibitor and decitabine for FLT3-mutated acute myeloid leukemia. Blood Cancer J. 2021, 11, 25. [Google Scholar] [CrossRef]
- Lachowiez, C.; Borthakur, G.; Loghavi, S.; Zeng, Z.; Kadia, T.M.; Masarova, L.; Dinardo, C.D. A phase Ib/II study of ivosidenib with venetoclax +/− azacitidine in IDH1-mutated myeloid malignancies. J. Clin. Oncol. 2021, 39, 7012. [Google Scholar] [CrossRef]
- Swaminathan, M.; Bourgeois, W.; Armstrong, S.; Wang, E.S. Menin Inhibitors in Acute Myeloid Leukemia-What Does the Future Hold? Cancer J. 2022, 28, 62–66. [Google Scholar] [CrossRef]
- Daver, N.G.; Vyas, P.; Kambhampati, S.; Al Malki, M.M.; Larson, R.; Asch, A.; Mannis, G.; Chai-Ho, W.; Tanaka, T.; Bradley, T.; et al. Tolerability and efficacy of the first-in-class anti-cd47 antibody magrolimab combined with azacitidine in frontline patient with tp53-mutated acute myeloid leukemia: Phase 1b results. Hemasphere 2022, 6, 33–34. [Google Scholar] [CrossRef]
- Brunner, A.M.; Esteve, J.; Porkka, K.; Knapper, S.; Traer, E.; Scholl, S.; Tovar, N. Efficacy and Safety of Sabatolimab (MBG453) in Combination with Hypomethylating Agents (HMAs) in Patients (Pts) with Very High/High-Risk Myelodysplastic Syndrome (vHR/HR-MDS) and Acute Myeloid Leukemia (AML): Final Analysis from a Phase Ib Study. Blood 2021, 138, 244. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Lasho, T. Evidence-Based Minireview: Myelodysplastic syndrome/myeloproliferative neoplasm overlap syndromes: A focused review. Hematology 2020, 2020, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S.; Arber, D.A.; Bueso-Ramos, C.; Hasserjian, R.P.; Orazi, A. Advances in myelodysplastic/myeloproliferative neoplasms. Virchows Arch. 2022, 482, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Rudelius, M.; Weinberg, O.K.; Niemeyer, C.M.; Shimamura, A.; Calvo, K.R. The International Consensus Classification (ICC) of hematologic neoplasms with germline predisposition, pediatric myelodysplastic syndrome, and juvenile myelomonocytic leukemia. Virchows Arch. 2022, 482, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Pophali, P.A.; Timm, M.M.; Mangaonkar, A.A.; Shi, M.; Reichard, K.; Tefferi, A.; Pavelko, K.; Villasboas, J.C.; Jevremovic, D.; Patnaik, M.M. Practical limitations of monocyte subset repartitioning by multiparametric flow cytometry in chronic myelomonocytic leukemia. Blood Cancer J. 2019, 9, 65. [Google Scholar] [CrossRef]
- Murali, A.; Cross, D.; Mollee, P. The use of monocyte subset repartitioning by flow cytometry for diagnosis of chronic myelomonocytic leukaemia. Blood Cancer J. 2021, 11, 6. [Google Scholar] [CrossRef]
- Geyer, J.T.; Tam, W.; Liu, Y.-C.; Chen, Z.; Wang, S.A.; Bueso-Ramos, C.; Oak, J.; Arber, D.A.; Hsi, E.; Rogers, H.J.; et al. Oligomonocytic chronic myelomonocytic leukemia (chronic myelomonocytic leukemia without absolute monocytosis) displays a similar clinicopathologic and mutational profile to classical chronic myelomonocytic leukemia. Mod. Pathol. 2017, 30, 1213–1222. [Google Scholar] [CrossRef]
- Calvo, X.; Garcia-Gisbert, N.; Parraga, I.; Gibert, J.; Florensa, L.; Andrade-Campos, M.; Merchan, B.; Garcia-Avila, S.; Montesdeoca, S.; Fernández-Rodríguez, C.; et al. Oligomonocytic and overt chronic myelomonocytic leukemia show similar clinical, genomic, and immunophenotypic features. Blood Adv. 2020, 4, 5285–5296. [Google Scholar] [CrossRef]
- Itzykson, R.; Fenaux, P.; Bowen, D.; Cross, N.C.P.; Cortes, J.; De Witte, T.; Germing, U.; Onida, F.; Padron, E.; Platzbecker, U.; et al. Diagnosis and Treatment of Chronic Myelomonocytic Leukemias in Adults: Recommendations from the European Hematology Association and the European LeukemiaNet. Hemasphere 2018, 2, e150. [Google Scholar] [CrossRef]
- Carr, R.M.; Vorobyev, D.; Lasho, T.; Marks, D.L.; Tolosa, E.J.; Vedder, A.; Almada, L.L.; Yurcheko, A.; Padioleau, I.; Alver, B.; et al. RAS mutations drive proliferative chronic myelomonocytic leukemia via a KMT2A-PLK1 axis. Nat. Commun. 2021, 12, 2901. [Google Scholar] [CrossRef]
- Loghavi, S.; Sui, D.; Wei, P.; Garcia-Manero, G.; Pierce, S.; Routbort, M.J.; Jabbour, E.J.; Pemmaraju, N.; Kanagal-Shamanna, R.; Gur, H.D.; et al. Validation of the 2017 revision of the WHO chronic myelomonocytic leukemia categories. Blood Adv. 2018, 2, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Xicoy, B.; Triguero, A.; Such, E.; García, O.; Jiménez, M.-J.; Arnán, M.; Bernal, T.; Diaz-Beya, M.; Valcárcel, D.; Pedro, C.; et al. The division of chronic myelomonocytic leukemia (CMML)-1 into CMML-0 and CMML-1 according to 2016 World Health Organization (WHO) classification has no impact in outcome in a large series of patients from the Spanish group of MDS. Leuk. Res. 2018, 70, 34–36. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zuo, Z.; Fu, B.; Oki, Y.; Tang, G.; Goswami, M.; Priyanka, P.; Muzzafar, T.; Medeiros, L.J.; Luthra, R.; et al. Chronic myelomonocytic leukemia with nucleophosmin (NPM1) mutation. Eur. J. Haematol. 2015, 96, 65–71. [Google Scholar] [CrossRef]
- Vallapureddy, R.; Lasho, T.L.; Hoversten, K.; Finke, C.M.; Ketterling, R.; Hanson, C.; Gangat, N.; Tefferi, A.; Patnaik, M.M. Nucleophosmin 1 (NPM1) mutations in chronic myelomonocytic leukemia and their prognostic relevance. Am. J. Hematol. 2017, 92, E614–E618. [Google Scholar] [CrossRef] [PubMed]
- Wudhikarn, K.; Loghavi, S.; Mangaonkar, A.A.; Al-Kali, A.; Binder, M.; Carr, R.; Reichard, K.; Finke, C.; Howard, M.; Gangat, N.; et al. SF3B1-mutant CMML defines a predominantly dysplastic CMML subtype with a superior acute leukemia-free survival. Blood Adv. 2020, 4, 5716–5721. [Google Scholar] [PubMed]
- Valent, P.; Orazi, A.; Savona, M.R.; Patnaik, M.M.; Onida, F.; Van de Loosdrecht, A.A.; Haase, D.; Haferlach, T.; Elena, C.; Pleyer, L.; et al. Proposed diagnostic criteria for classical chronic myelomonocytic leukemia (CMML), CMML variants and pre-CMML conditions. Haematologica 2019, 104, 1935–1949. [Google Scholar] [CrossRef]
- Renneville, A.; Patnaik, M.M.; Chan, O.; Padron, E.; Solary, E. Increasing recognition and emerging therapies argue for dedicated clinical trials in chronic myelomonocytic leukemia. Leukemia 2021, 35, 2739–2751. [Google Scholar] [CrossRef]
- De Witte, T.; Bowen, D.; Robin, M.; Malcovati, L.; Niederwieser, D.; Yakoub-Agha, I.; Kröger, N. Allogeneic hematopoietic stem cell transplantation for MDS and CMML: Recommendations from an international expert panel. Blood 2017, 129, 1753–1762. [Google Scholar] [CrossRef]
- Wattel, E.; Guerci, A.; Hecquet, B.; Economopoulos, T.; Copplestone, A.; Mahé, B.; Couteaux, M.E.; Resegotti, L.; Voglova, V.; Foussard, C.; et al. A randomized trial of hydroxyurea versus VP16 in adult chronic myelomonocytic leukemia. Groupe Français des Myélodysplasies and European CMML Group. Blood 1996, 88, 2480–2487. [Google Scholar] [CrossRef]
- Silverman, L.R.; Demakos, E.P.; Peterson, B.L.; Kornblith, A.B.; Holland, J.C.; Odchimar-Reissig, R.; Stone, R.M.; Nelson, D.; Powell, B.L.; DeCastro, C.M.; et al. Randomized Controlled Trial of Azacitidine in Patients with the Myelodysplastic Syndrome: A Study of the Cancer and Leukemia Group B. J. Clin. Oncol. 2002, 20, 2429–2440. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Kadia, T.; DiNardo, C.; Daver, N.; Borthakur, G.; Jabbour, E.; Garcia-Manero, G.; Konopleva, M.; Ravandi, F. Acute myeloid leukemia: Current progress and future directions. Blood Cancer J. 2021, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Griffiths, E.A.; Steensma, D.P.; Roboz, G.J.; Wells, R.A.; McCloskey, J.; Odenike, O.; DeZern, A.E.; Yee, K.; Busque, L.; et al. Oral cedazuridine/decitabine for MDS and CMML: A phase 2 pharmacokinetic/pharmacodynamic randomized crossover study. Blood 2020, 136, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.; Itzykson, R.; Renneville, A.; De Renzis, B.; Dreyfus, F.; Laribi, K.; Bouabdallah, K.; Vey, N.; Toma, A.; Recher, C.; et al. Molecular predictors of response to decitabine in advanced chronic myelomonocytic leukemia: A phase 2 trial. Blood 2011, 118, 3824–3831. [Google Scholar] [CrossRef] [PubMed]
- Wijermans, P.; Rüter, B.; Baer, M.; Slack, J.; Saba, H.; Lübbert, M. Efficacy of decitabine in the treatment of patients with chronic myelomonocytic leukemia (CMML). Leuk. Res. 2008, 32, 587–591. [Google Scholar] [CrossRef]
- Santini, V.; Allione, B.; Zini, G.; Gioia, D.; Lunghi, M.; Poloni, A.; Cilloni, D.; Sanna, A.; Masiera, E.; Ceccarelli, M.; et al. A phase II, multicentre trial of decitabine in higher-risk chronic myelomonocytic leukemia. Leukemia 2017, 32, 413–418. [Google Scholar] [CrossRef]
- Unnikrishnan, A.; Papaemmanuil, E.; Beck, D.; Deshpande, N.P.; Verma, A.; Kumari, A.; Woll, P.S.; Richards, L.A.; Knezevic, K.; Chandrakanthan, V.; et al. Integrative Genomics Identifies the Molecular Basis of Resistance to Azacitidine Therapy in Myelodysplastic Syndromes. Cell Rep. 2017, 20, 572–585. [Google Scholar] [CrossRef]
- Merlevede, J.; Droin, N.; Qin, T.; Meldi, K.; Yoshida, K.; Morabito, M.; Chautard, E.; Auboeuf, D.; Fenaux, P.; Braun, T.; et al. Mutation allele burden remains unchanged in chronic myelomonocytic leukaemia responding to hypomethylating agents. Nat. Commun. 2016, 7, 10767. [Google Scholar] [CrossRef]
- Pleyer, L.; Leisch, M.; Kourakli, A.; Padron, E.; Maciejewski, J.P.; Cirici, B.X.; Kaivers, J.; Ungerstedt, J.; Heibl, S.; Patiou, P.; et al. Outcomes of patients with chronic myelomonocytic leukaemia treated with non-curative therapies: A retrospective cohort study. Lancet Haematol. 2021, 8, e135–e148. [Google Scholar] [CrossRef]
- Itzykson, R.; Santini, V.; Thepot, S.; Ades, L.; Chaffaut, C.; Giagounidis, A.; Morabito, M.; Droin, N.; Lübbert, M.; Sapena, R.; et al. Decitabine Versus Hydroxyurea for Advanced Proliferative Chronic Myelomonocytic Leukemia: Results of a Randomized Phase III Trial within the EMSCO Network. J. Clin. Oncol. 2022. online ahead of print. [Google Scholar] [CrossRef]
- Gianelli, U.; Thiele, J.; Orazi, A.; Gangat, N.; Vannucchi, A.M.; Tefferi, A.; Kvasnicka, H.M. International Consensus Classification of myeloid and lymphoid neoplasms: Myeloproliferative neoplasms. Virchows Arch. 2022, 482, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Binotto, G.; Bertorelle, R.; Bonaldi, L.; Frison, L.; Vianello, F.; Pizzi, M. Chronic Myeloid Leukemia with Myelofibrosis-Like Features. Clues of Accelerated Phase? Int. J. Surg. Pathol. 2019, 27, 771–772. [Google Scholar] [CrossRef]
- How, J.; Venkataraman, V.; Hobbs, G.S. Blast and accelerated phase CML: Room for improvement. Hematology 2021, 2021, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Iriyama, N.; Tokuhira, M.; Takaku, T.; Ishikawa, M.; Nakazato, T.; Sugimoto, K.; Fujita, H.; Kimura, Y.; Fujioka, I.; et al. The EUTOS long-term survival score predicts disease-specific mortality and molecular responses among patients with chronic myeloid leukemia in a practice-based cohort. Cancer Med. 2020, 9, 8931–8939. [Google Scholar] [CrossRef]
- Rowley, J.D. A New Consistent Chromosomal Abnormality in Chronic Myelogenous Leukaemia identified by Quinacrine Fluorescence and Giemsa Staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef]
- Huang, X.; Cortes, J.; Kantarjian, H. Estimations of the increasing prevalence and plateau prevalence of chronic myeloid leukemia in the era of tyrosine kinase inhibitor therapy. Cancer 2012, 118, 3123–3127. [Google Scholar] [CrossRef] [PubMed]
- Hochhaus, A.; Larson, R.A.; Guilhot, F.; Radich, J.P.; Branford, S.; Hughes, T.P.; Baccarani, M.; Deininger, M.W.; Cervantes, F.; Fujihara, S.; et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N. Engl. J. Med. 2017, 376, 917–927. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.G.; Guilhot, F.; Larson, R.A.; Gathmann, I.; Baccarani, M.; Cervantes, F.; Cornelissen, J.J.; Fischer, T.; Hochhaus, A.; Hughes, T.; et al. Imatinib Compared with Interferon and Low-Dose Cytarabine for Newly Diagnosed Chronic-Phase Chronic Myeloid Leukemia. N. Engl. J. Med. 2003, 348, 994–1004. [Google Scholar] [CrossRef]
- Tokarski, J.S.; Newitt, J.A.; Chang, C.Y.J.; Cheng, J.D.; Wittekind, M.; Kiefer, S.E.; Kish, K.; Lee, F.Y.; Borzillerri, R.; Lombardo, L.J.; et al. The Structure of Dasatinib (BMS-354825) Bound to Activated ABL Kinase Domain Elucidates Its Inhibitory Activity against Imatinib-Resistant ABL Mutants. Cancer Res. 2006, 66, 5790–5797. [Google Scholar] [CrossRef]
- Weisberg, E.; Manley, P.; Breitenstein, W.; Brüggen, J.; Cowan-Jacob, S.W.; Ray, A.; Griffin, J.D. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 2005, 7, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Gambacorti-Passerini, C.; Deininger, M.W.; Mauro, M.J.; Chuah, C.; Kim, D.-W.; Dyagil, I.; Glushko, N.; Milojkovic, D.; Le Coutre, P.; et al. Bosutinib versus Imatinib for Newly Diagnosed Chronic Myeloid Leukemia: Results from the Randomized BFORE Trial. J. Clin. Oncol. 2018, 36, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Scalzulli, E.; Carmosino, I.; Bisegna, M.L.; Martelli, M.; Breccia, M. CML Resistant to 2nd-Generation TKIs: Mechanisms, Next Steps, and New Directions. Curr. Hematol. Malign-Rep. 2022, 17, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.P.; Mauro, M.J.; Cortes, J.E.; Minami, H.; Rea, D.; DeAngelo, D.J.; Breccia, M.; Goh, Y.-T.; Talpaz, M.; Hochhaus, A.; et al. Asciminib in Chronic Myeloid Leukemia after ABL Kinase Inhibitor Failure. N. Engl. J. Med. 2019, 381, 2315–2326. [Google Scholar] [CrossRef] [PubMed]
- Narlı Özdemir, Z.; Kılıçaslan, N.A.; Yılmaz, M.; Eşkazan, A.E. Guidelines for the treatment of chronic myeloid leukemia from the NCCN and ELN: Differences and similarities. Int. J. Hematol. 2023, 117, 3–15. [Google Scholar] [CrossRef]
- Ono, T. Which Tyrosine Kinase Inhibitors Should Be Selected as the First-Line Treatment for Chronic Myelogenous Leukemia in Chronic Phase? Cancers 2021, 13, 5116. [Google Scholar] [CrossRef] [PubMed]
- Yohanan, B.; George, B. Current Management of Chronic Myeloid Leukemia Myeloid Blast Phase. Clin. Med. Insights Oncol. 2022, 16, 1–9. [Google Scholar] [CrossRef]
- Mahon, F.-X.; Réa, D.; Guilhot, J.; Guilhot, F.; Huguet, F.; Nicolini, F.; Legros, L.; Charbonnier, A.; Guerci, A.; Varet, B.; et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: The prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010, 11, 1029–1035. [Google Scholar] [CrossRef]
- Latagliata, R.; Romano, A.; Mancini, M.; Breccia, M.; Carmosino, I.; Vozella, F.; Montagna, C.; Volpicelli, P.; De Angelis, F.; Petrucci, L.; et al. Discontinuation of alpha-interferon treatment in patients with chronic myeloid leukemia in long-lasting complete molecular response. Leuk. Lymphoma 2015, 57, 99–102. [Google Scholar] [CrossRef]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020, 34, 966–984. [Google Scholar] [CrossRef]
- Deininger, M.W.; Shah, N.P.; Altman, J.K.; Berman, E.; Bhatia, R.; Bhatnagar, B.; Sundar, H. Chronic Myeloid Leukemia, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2020, 18, 1385–1415. [Google Scholar] [CrossRef]
- Stuckey, R.; López-Rodríguez, J.F.; Sánchez-Sosa, S.; Segura-Díaz, A.; Sánchez-Farías, N.; Bilbao-Sieyro, C.; Gómez-Casares, M.T. Predictive indicators of successful tyrosine kinase inhibitor discontinuation in patients with chronic myeloid leukemia. World J. Clin. Oncol. 2020, 11, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Tzankov, A.; Reichard, K.K.; Hasserjian, R.P.; Arber, D.A.; Orazi, A.; Wang, S.A. Updates on eosinophilic disorders. Virchows Arch. 2022, 482, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.A.; Hasserjian, R.P.; Tam, W.; Tsai, A.; Geyer, J.T.; George, T.; Foucar, K.; Rogers, H.J.; Hsi, E.D.; Rea, B.A.; et al. Bone marrow morphology is a strong discriminator between chronic eosinophilic leukemia, not otherwise specified and reactive idiopathic hypereosinophilic syndrome. Haematologica 2017, 102, 1352–1360. [Google Scholar] [CrossRef] [PubMed]
- Morsia, E.; Reichard, K.; Pardanani, A.; Tefferi, A.; Gangat, N. WHO defined chronic eosinophilic leukemia, not otherwise specified (CEL, NOS): A contemporary series from the Mayo Clinic. Am. J. Hematol. 2020, 95, E172–E174. [Google Scholar] [CrossRef]
- Gerds, A.T.; Gotlib, J.; Bose, P.; Deininger, M.W.; Dunbar, A.; Elshoury, A.; George, T.I.; Gojo, I.; Gundabolu, K.; Hexner, E.; et al. Myeloid/Lymphoid Neoplasms with Eosinophilia and TK Fusion Genes, Version 3.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2020, 18, 1248–1269. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.-Q.; Qin, T.-J.; Xu, Z.-F.; Zhang, Y.; Ai, X.F.; Li, B.; Xiao, Z.J. Long-term outcomes of imatinib in patients with FIP1L1/PDGFRA associated chronic eosinophilic leukemia: Experience of a single center in China. Oncotarget 2016, 7, 33229. [Google Scholar] [CrossRef] [PubMed]
- Jawhar, M.; Naumann, N.; Schwaab, J.; Baurmann, H.; Casper, J.; Dang, T.-A.; Dietze, L.; Döhner, K.; Hänel, A.; Lathan, B.; et al. Imatinib in myeloid/lymphoid neoplasms with eosinophilia and rearrangement of PDGFRB in chronic or blast phase. Ann. Hematol. 2017, 96, 1463–1470. [Google Scholar] [CrossRef]
- Lierman, E.; Michaux, L.; Beullens, E.; Pierre, P.; Marynen, P.; Cools, J.; Vandenberghe, P. FIP1L1-PDGFRα D842V, a novel panresistant mutant, emerging after treatment of FIP1L1-PDGFRα T674I eosinophilic leukemia with single agent sorafenib. Leukemia 2009, 23, 845–851. [Google Scholar] [CrossRef]
- Sadovnik, I.; Lierman, E.; Peter, B.; Herrmann, H.; Suppan, V.; Stefanzl, G.; Haas, O.; Lion, T.; Pickl, W.; Cools, J.; et al. Identification of Ponatinib as a potent inhibitor of growth, migration, and activation of neoplastic eosinophils carrying FIP1L1-PDGFRA. Exp. Hematol. 2014, 42, 282–293.e4. [Google Scholar] [CrossRef]
- Schwaab, J.; Naumann, N.; Luebke, J.; Jawhar, M.; Somervaille, T.C.P.; Williams, M.S.; Frewin, R.; Jost, P.J.; Lichtenegger, F.S.; La Rosée, P.; et al. Response to tyrosine kinase inhibitors in myeloid neoplasms associated with PCM1-JAK2, BCR-JAK2 and ETV6-ABL1 fusion genes. Am. J. Hematol. 2020, 95, 824–833. [Google Scholar] [CrossRef]
- Verstovsek, S.; Vannucchi, A.M.; Rambaldi, A.; Gotlib, M.J.R.; Mead, A.J.; Hochhaus, A.; Kiladjian, J.-J.; Boluda, J.C.H.; Asatiani, E.; Lihou, B.C.; et al. Interim Results from Fight-203, a Phase 2, Open-Label, Multicenter Study Evaluating the Efficacy and Safety of Pemigatinib (INCB054828) in Patients with Myeloid/Lymphoid Neoplasms with Rearrangement of Fibroblast Growth Factor Receptor 1 (FGFR1). Blood 2018, 132, 690. [Google Scholar] [CrossRef]
- Hernández-Boluda, J.-C.; Pereira, A.; Zinger, N.; Gras, L.; Martino, R.; Nikolousis, E.; Finke, J.; Chinea, A.; Rambaldi, A.; Robin, M.; et al. Allogeneic hematopoietic cell transplantation in patients with myeloid/lymphoid neoplasm with FGFR1-rearrangement: A study of the Chronic Malignancies Working Party of EBMT. Bone Marrow Transplant. 2022, 57, 416–422. [Google Scholar] [CrossRef]
- Yao, J.; Xu, L.; Aypar, U.; Meyerson, H.J.; Londono, D.; Gao, Q.; Baik, J.; Dietz, J.; Benayed, R.; Sigler, A.; et al. Myeloid/lymphoid neoplasms with eosinophilia/basophilia and ETV6-ABL1 fusion: Cell-of-origin and response to tyrosine kinase inhibition. Haematologica 2020, 106, 614–618. [Google Scholar] [CrossRef]
- Falchi, L.; Mehrotra, M.; Newberry, K.J.; Lyle, L.M.; Lu, G.; Patel, K.P.; Luthra, R.; Popat, U.; Verstovsek, S. Professor of Medicine ETV6–FLT3 fusion gene-positive, eosinophilia-associated myeloproliferative neoplasm successfully treated with sorafenib and allogeneic stem cell transplant. Leukemia 2014, 28, 2090–2092. [Google Scholar] [CrossRef] [PubMed]
- Munthe-Kaas, M.C.M.; Forthun, R.B.; Brendehaug, A.M.; Eek, A.K.M.; Høysæter, T.B.; Osnes, L.T.M.; Prescott, T.M.; Spetalen, S.M.; Hovland, R. Partial Response to Sorafenib in a Child with a Myeloid/Lymphoid Neoplasm, Eosinophilia, and a ZMYM2-FLT3 Fusion. J. Pediatr. Hematol. 2020, 43, e508–e511. [Google Scholar] [CrossRef] [PubMed]
- Rumi, E.; Milosevic, J.D.; Selleslag, D.; Casetti, I.; Lierman, E.; Pietra, D.; Cavalloni, C.; Bellini, M.; Milanesi, C.; Dambruoso, I.; et al. Efficacy of ruxolitinib in myeloid neoplasms with PCM1-JAK2 fusion gene. Ann. Hematol. 2015, 94, 1927–1928. [Google Scholar] [CrossRef]
- Rizzuto, G.; Leoncin, M.; Imbergamo, S.; Taurino, D.; Mico, M.C.; Tosi, M.; Michelato, A.; Buklijas, K.; Spinelli, O.; Lussana, F.; et al. Sequential allogeneic transplantation and ruxolitinib maintenance for a synchronous PCM1-JAK2 positive myeloid sarcoma and acute B-lymphoblastic leukemia. Clin. Case Rep. 2022, 10, e05212. [Google Scholar] [CrossRef]
- Duncavage, E.J.; Bagg, A.; Hasserjian, R.P.; DiNardo, C.D.; Godley, L.A.; Iacobucci, I.; Jaiswal, S.; Malcovati, L.; Vannucchi, A.M.; Patel, K.P.; et al. Genomic profiling for clinical decision making in myeloid neoplasms and acute leukemia. Blood 2022, 140, 2228–2247. [Google Scholar] [CrossRef]
- Pizzi, M. Crossing the Borders: An Integrated Approach to Myeloproliferative Neoplasms and Mastocytoses. Cancers 2021, 13, 1492. [Google Scholar] [CrossRef]
- Orazi, A. Histopathology in the Diagnosis and Classification of Acute Myeloid Leukemia, Myelodysplastic Syndromes, and Myelodysplastic/Myeloproliferative Diseases. Pathobiology 2007, 74, 97–114. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| International Consensus Classification (ICC) | 2022 WHO Classification | ||
|---|---|---|---|
| AML subtypes | Blasts * | AML subtypes | Blasts * |
| AML with recurrent genetic abnormalities | AML with defining genetic abnormalities | ||
| Acute promyelocytic leukemia with t(15;17) (q24.1;q21.2)/PML::RARA | ≥10% | Acute promyelocytic leukemia with PML::RARA fusion | no threshold |
| Acute promyelocytic leukemia with other RARA rearrangements | |||
| AML with t(8;21)(q22;q22.1)/RUNX1::RUNX1T1 | ≥10% | AML with t(8;21)(q22;q22.1)/RUNX1::RUNX1T1 fusion | no threshold |
| AML with inv(16)(p13.1;q22) or t(16;16) (p13.1;q22)/CBFB::MYH11 | ≥10% | AML with CBFB::MYH11 fusion | no threshold |
| AML with t(9;11)(p21.3;q23.3)/MLLT3::KTM2A | ≥10% | AML with KTM2A rearrangement | no threshold |
| AML with other KMT2A rearrangements | |||
| AML with t (6;9)(p22.3;q34.1)/DEK::NUP214 | ≥10% | AML with DEK::NUP214 fusion | no threshold |
| AML with inv(3)(q21.3q;26.2) or t(3;3)(q21.3;q26.2)/GATA2::MECOM | ≥10% | AML with MECOM rearrangements | no threshold |
| AML with other MECOM rearrangements | |||
| AML with other rare recurring translocations | ≥10% | AML with other defined genetic alterations | no threshold |
| AML with t(1;3)(p36.3;q21.3)/PRDM16::RPN1 | AML with NPM1::MLF1 | ||
| AML with t(3;5)(q25.3;q35.1)/NPM1::MLF1 | AML with KAT6A::CREBBP | ||
| AML with t(8;16)(p11.2;p13.3)/KAT6A::CREBB | AML with MNX1::ETV6 | ||
| AML with t(1;22)(p13.3;q13.1)/RBM15::MRTF1 | AML with FUS::ERG | ||
| AML with t(5;11)(q35.2;p15.4/NUP98::NSD1 | AML with RUNX1T3(CBFA2T3)::GLIS2 | ||
| AML with t(11;12)(p15.4;p13.3)/NUP98::KMD5A | |||
| AML with NUP98 and other partners | |||
| AML with t(7;12)(q36.3;p13.2)/ETV6::MNX1 | |||
| AML with t(10;11)(p12.3;q14.2)/PICALM::MLLT10 | |||
| AML with t(16;21)(p11.2;q22.2)/FUS::ERG | |||
| AML with t(16;21)(q24.3;q22.1)/RUNX1::CBFA2T3 | |||
| AML with inv(16)(p13.3q24.3)/CBFA2T3::GLIS2 | |||
| AML with t(9;22)(q34.1;q11.2)/BCR::ABL1 | ≥20% | AML with BCR:: ABL1 fusion | ≥20% |
| AML with mutated NPM1 | ≥10% | AML with NPM1 mutation | no threshold |
| AML with in-frame bZIP CEBPA mutations | ≥10% | AML with CEBPA mutation | ≥20% |
| AML with mutated TP53 ** | ≥20% | - | |
| AML with myelodysplasia-related gene mutations § | ≥20% | AML, myelodysplasia-related | ≥20% |
| AML with myelodysplasia-related cytogenetic abnormalities # | |||
| AML, not otherwise specified | ≥20% | AML, defined by differentiation | ≥20% |
| Myeloid sarcoma | n.a | Myeloid sarcoma | n.a |
| International Consensus Classification (ICC) Criteria | 2022 WHO Classification Criteria |
|---|---|
| MDS with del(5q) | MDS with low blasts and isolated 5q deletion |
| MDS with mutated SF3B1 | MDS with low blasts and SF3B1 mutation |
| MDS with mutated TP53 a | MDS with biallelic TP53 inactivation a |
| MDS not otherwise specified (MDS-NOS) b - MDS-NOS, with single lineage dysplasia - MDS-NOS, with multilineage dysplasia - MDS-NOS, without dysplasia c | MDS with low blasts b - with single lineage dysplasia (optional) - with multilineage dysplasia (optional) |
| - | MDS, hypoplastic d |
| MDS with excess blasts e | MDS with increased blasts 1 e |
| MDS/AML f - MDS/AML with MDS-related cytogenetic abnormalities - MDS/AML with MDS-related gene mutations - MDS/AML with mutated TP53 - MDS/AML not otherwise specified | MDS with increased blasts 2 f |
| MDS with increased blast and fibrosis g |
| Monocytosis defined as monocytes ≥ 0.5 × 109/L and ≥10% of the WBC |
| Cytopenia a |
| Blasts (including promonocytes) < 20% of nucleated cells in PB and BM |
| Presence of clonality |
| Abnormal cytogenetics and/or |
| ≥1 myeloid neoplasm-associated gene mutation (VAF ≥ 10%) b |
| In cases without evidence of clonality: |
| monoctyes ≥ 1.0 × 109/L and ≥10% of the WBC with ≥1 of the following |
| -increased blasts (including promonocytes) c |
| -morphologic dysplasia |
| -abnormal immunophenotype consistent with CMML |
| BM examination consistent with CMML (hypercellularity due to myeloid proliferation often with increased monocytes) and lacking diagnostic features of AML, MPN or other conditions associated with monocytosis |
| No BCR::ABL1 fusion or genetic abnormalities consistent with M/LN-eo-TK |
| Persistent monocytosis defined as monocytes ≥ 0.5 × 109/L and ≥10% of the WBC |
| Absence or presence of cytopenia a |
| Presence of ≥1 myeloid neoplasm-associated gene mutation (VAF ≥ 2%) b |
| No significant dysplasia, increased blasts (including promonocytes) or morphologic findings of CMML on BM examination c |
| No criteria for a myeloid or other hematopoietic neoplasm are fulfilled |
| No reactive conditions that would explain monocytosis are detected |
| Prerequisite Criteria |
|---|
| 1. Persistent monocytosis, defined as monocytes ≥ 0.5 × 109/L and ≥10% of the WBC |
| 2. Blasts a < 20% of nucleated cells in PB and BM |
| 3. Not meeting diagnostic criteria of CML or other MPN |
| 4. Not meeting diagnostic criteria of M/LN-eo-TK |
| Supporting Criteria |
| 1. Dysplasia involving ≥ 1 myeloid lineage b |
| 2. Acquired clonal cytogenetic or molecular abnormality |
| 3. Abnormal partitioning of PB monocyte subsets c |
| Diagnostic Requirements A diagnosis of CMML is posed if all prerequisite criteria are present together with: (a) ≥1 supporting criteria, if monocytosis is ≥1 × 109/L (b) both supporting criteria #1 and #2, if monocytosis is 0.5 − 1.0 × 109/L |
| Subtyping Criteria - Myelodysplastic CMML: WBC < 13 × 109/L - Myeloproliferative CMML: WBC ≥ 13 × 109/L |
| Subgrouping Criteria (based on percentage of blasts and promonocytes) - CMML-1: <5% in PB and <10% in BM - CMML-2: 5–19% in PB and 10–19% in BM |
| Diagnostic Criteria of Accelerated Phase CML (Any of the Following) |
|---|
|
- 10–19% blasts in BM or PB - ≥20% basophils in PB - Additional clonal cytogenetic abnormalities in Philadelphia-positive cells a |
| Diagnostic criteria of blast phase CML (any of the following) |
|
- ≥20% blasts in BM or PB - Myeloid sarcoma b - >5% morphologically apparent lymphoblasts warrants consideration of lymphoblastic crisis |
| At Diagnosis (Any of the Following) |
|---|
|
- High ELTS score - 10–19% blasts in BM and/or PB a - ≥20% basophils in PB - Additional clonal cytogenetic abnormalities in Philadelphia-positive cells b - Clusters of small megakaryocytes (including micromegakaryocytes) with increased reticulin and/or collagen fibrosis. |
| Emerging on treatment (any of the following) |
|
- Failure to achieve a complete hematological response to the first TKI - Any indication of resistance to 2 sequential TKIs (excluding explicable causes, such as kinase domain mutations resistant - Development of new additional chromosomal abnormalities - Occurrence of compound mutations in the BCR::ABL1 fusion gene during TKI therapy |
| International Consensus Classification (ICC) Criteria | 2022 WHO Classification Criteria |
|---|---|
| Eosinophilia defined as eosinophils ≥1.5 × 109/L and ≥10% of the WBC | Eosinophilia defined as eosinophils >1.5 × 109/L on at least 2 occasions over an interval of at least 4 weeks |
| Blasts < 20% of nucleated cells in PB and BM, not meeting any other diagnostic criteria for other AML | Not meeting criteria for any other myeloid or lymphoid neoplasm |
| No tyrosine kinase fusions including BCR::ABL1, other ABL1, PDGFRA, PDGFRB, FGFR1, JAK2, FLT3 fusions | |
| Not meeting criteria for other well-defined MPN, CMML or SM a | |
| BM examination showing increased cellularity and dysplastic megakaryocytes with/without dysplastic features in other lineages and often significant fibrosis, associated with esoinophilic infiltrate or | BM examination showing increased cellularity and dysplastic megakaryocytes with/without dysplastic features in other lineages and often significant fibrosis, associated with eosinophilic infiltrate |
| ≥5% blasts in the BM and/or ≥2% blasts in the PB | |
| Demonstration of a clonal cytogenetic abnormality and/or somatic mutation b | Demonstration of a clonal cytogenetic abnormality and/or somatic mutation b |
| International Consensus Classification (ICC) | 2022 WHO Classification |
|---|---|
| M/LN with PDGFRA rearrangement a | M/LN with PDGFRA rearrangement a |
| M/LN with PDGFRB rearrangement b | M/LN with PDGFRB rearrangement b |
| M/LN with FGFR1 rearrangement c | M/LN with FGFR1 rearrangement c |
| M/LN with JAK2 rearrangement d | M/LN with JAK2 rearrangement d |
| M/LN with FLT3 rearrangement e | M/LN with FLT3 rearrangement e |
| M/LN with ETV6::ABL1 | M/LN with ETV6::ABL1 rearrangement |
| M/LN with other TK fusions (ETV6::FGFR2 fusion; ETV6::LYN fusion; ETV6::NTRK3 fusion; RANBP2::ALK fusion; BCR::RET fusion; FGFR1OP::RET fusion) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pizzi, M.; Gurrieri, C.; Orazi, A. What’s New in the Classification, Diagnosis and Therapy of Myeloid Leukemias. Hemato 2023, 4, 112-134. https://doi.org/10.3390/hemato4020011
Pizzi M, Gurrieri C, Orazi A. What’s New in the Classification, Diagnosis and Therapy of Myeloid Leukemias. Hemato. 2023; 4(2):112-134. https://doi.org/10.3390/hemato4020011
Chicago/Turabian StylePizzi, Marco, Carmela Gurrieri, and Attilio Orazi. 2023. "What’s New in the Classification, Diagnosis and Therapy of Myeloid Leukemias" Hemato 4, no. 2: 112-134. https://doi.org/10.3390/hemato4020011
APA StylePizzi, M., Gurrieri, C., & Orazi, A. (2023). What’s New in the Classification, Diagnosis and Therapy of Myeloid Leukemias. Hemato, 4(2), 112-134. https://doi.org/10.3390/hemato4020011