1. Introduction
In our previous study, the anti-inflammatory and antioxidant functions of tuna oil (TO) in cigarette smoke (CS) exposure-induced lung inflammation in mice were demonstrated. At the genus level, the abundance of
Lacticaseibacillus in the CS-induced model group was reduced compared with the blank control group. The abundance of
Lacticaseibacillus increased in the experimental group with different doses of fish oil when compared with the CS-induced pneumonic mice. At the species level, the abundance of
Lacticaseibacillus rhamnosus (
L. rhamnosus) increased after treatment with different doses of TO [
1]. The anti-inflammatory properties of
Lacticaseibacillus have been shown to enhance the biosynthesis of serum IL-10, while concurrently inhibiting the production of TNF-α, IL-6, and interleukin-12 (IL-12). This dual action contributes to the subsequent mitigation of inflammatory responses in animals [
2,
3].
L. rhamnosus is a widely studied
Lacticaseibacillus in both domestic and international research. Additionally, it is a facultative anaerobic, non-spore-forming, Gram-positive bacteria with long or short rods. It is classified within the genus
Lacticaseibacillus under the
rhamnose subspecies. It is acid-resistant, resistant to pancreatic juice and bile salts, and resistant to a variety of antibiotics and other biological characteristics [
4].
L. rhamnosus is a symbiotic microorganism in the intestinal system of humans and animals. Its advantage lies in its high intestinal adhesion rate and strong colonisation. Additionally, it is beneficial for improving the host’s systemic immune response, and is often used to enhance the body’s immunity and disease prevention and treatment [
5].
L. rhamnosus earned the prestigious designation of being generally recognised as safe (GRAS) by the United States Food and Drug Administration. It has also been included in the European Food Safety Authority’s Qualified presumption of safety (QPS) registry [
6,
7]. A growing body of evidence demonstrates that
L. rhamnosus and its metabolites can modulate immune cells, such as M1 macrophages and T lymphocytes, effectively suppressing the production of pro-inflammatory cytokines including tumour necrosis factor-alpha (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), and interleukin-2 (IL-2). Concurrently, these microbial components enhance the generation of anti-inflammatory cytokines such as interleukin-10 (IL-10), thereby regulating inflammatory responses and maintaining immune homeostasis through this dual mechanism of cytokine balance modulation [
8,
9,
10]. However, recent research has shown that probiotics’ function is highly strain-specific. Their biological effect cannot be estimated at the species level and necessitates a strain-by-strain assessment [
11]. This is one of the reasons why researchers continue to screen new probiotic strains with potential health benefits.
Comparative genomic analysis stands as a staple in bioinformatics, serving as a powerful tool to unravel the intricate connections between bacterial strains and their respective origins. It also facilitates the assessment of gene distribution within specific species, thereby shedding light on the phenotypic traits exhibited by the organisms under study [
12,
13]. Simultaneously, compared with other types of molecular biology technology, the research results of genomics have wider coverage and deeper analysis. With the development of technology in recent years, whole-genome sequencing (WGS) technology based on all genetic materials emerged. This method can fully mine the gene information of microorganisms, annotate their functional characteristics, and predict the relevant metabolic pathways, laying the foundation for studying the classification relationship and genetic progress of microorganisms [
14]. In the early 21st century, Bolotin et al. first sequenced the whole genome of the first
Lacticaseibacillus lactic IL1403 (
L. lactic IL1403), thereby establishing the comprehensive availability of in-depth genetic and physical maps for the
L. lactic IL1403 genome [
15]. Following this seminal work, an array of
Lacticaseibacillus species have undergone extensive sequencing and in-depth exploration, expanding our understanding of these microorganisms [
16]. All the genetic material information of strains and for classifying the coding genes were obtained through WGS, which plays an essential part in the comprehensive analysis of
Lacticaseibacillus bacteria. Therefore, in this study, the probiotic-related genes and probiotic characteristics of
L. rhamnosus strains were isolated from the guts of mice with smoking-induced pneumonia relieved by fish oil and were deeply understood through WGS to better understand their potential biological functions and information and explore their possible anti-inflammatory mechanism.
4. Discussion
The earliest isolated
Lacticaseibacillus rhamnosus GG (LGG) has been recognised as a quintessential probiotic strain. Fundamentally, the probiotic properties of various strains are intimately tied to their genome, with their growth characteristics and nutritional functions being dictated and shaped by the intricate guidance of their gene sequences.
L. rhamnosus is a non-toxic probiotic with no side effects and has biological functions such as regulating the gut microbiota, preventing and treating diarrhhoea, toxin elimination, and enhancing the body’s immunity, which is of high value and developmental prospects [
30,
31]. However, strains derived from various sources exhibit distinct characteristics, and WGS enables us to comprehend these differences more thoroughly. For probiotics used in disease relief or treatment, higher safety is a prerequisite. Sequencing analysis revealed that the
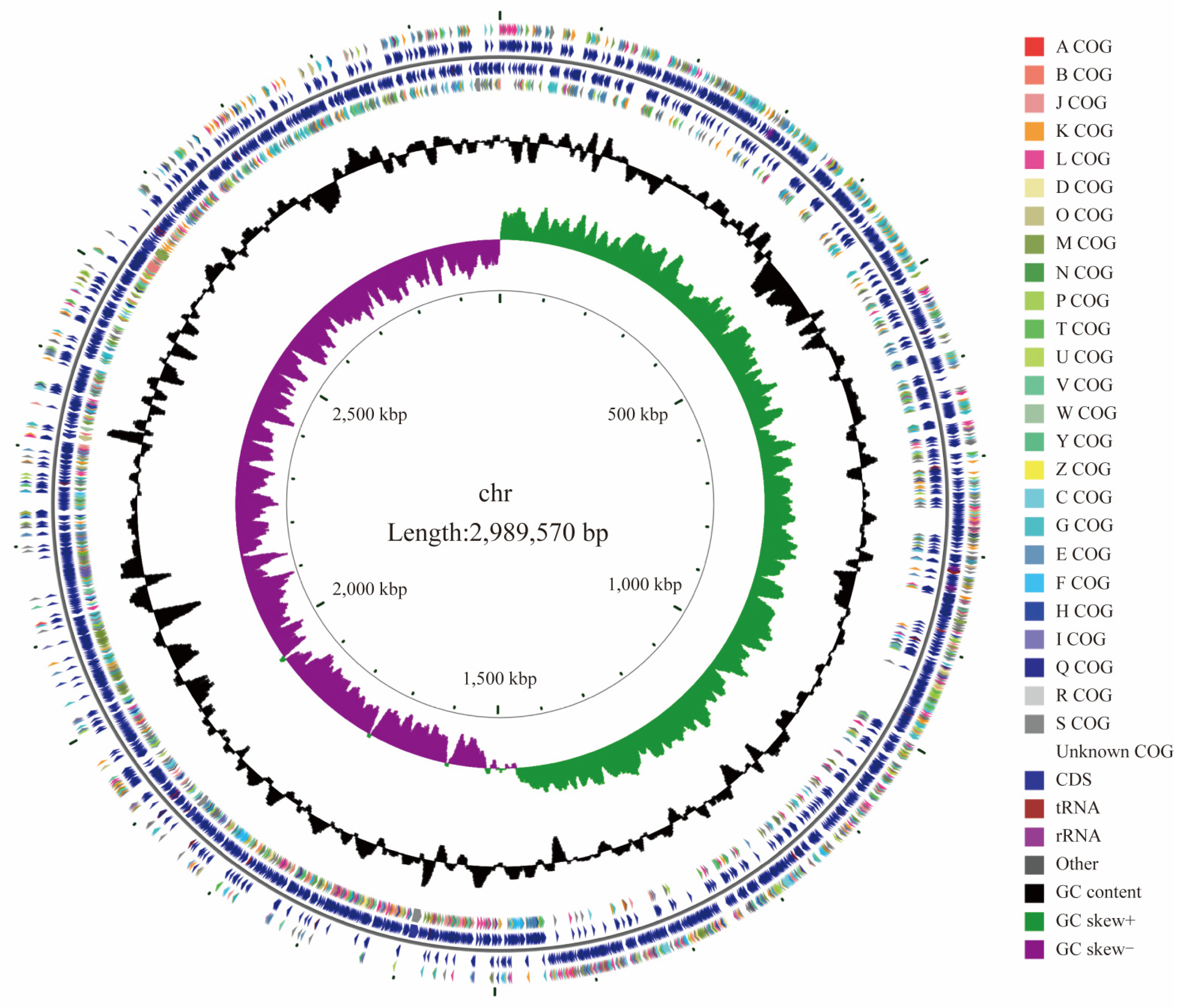
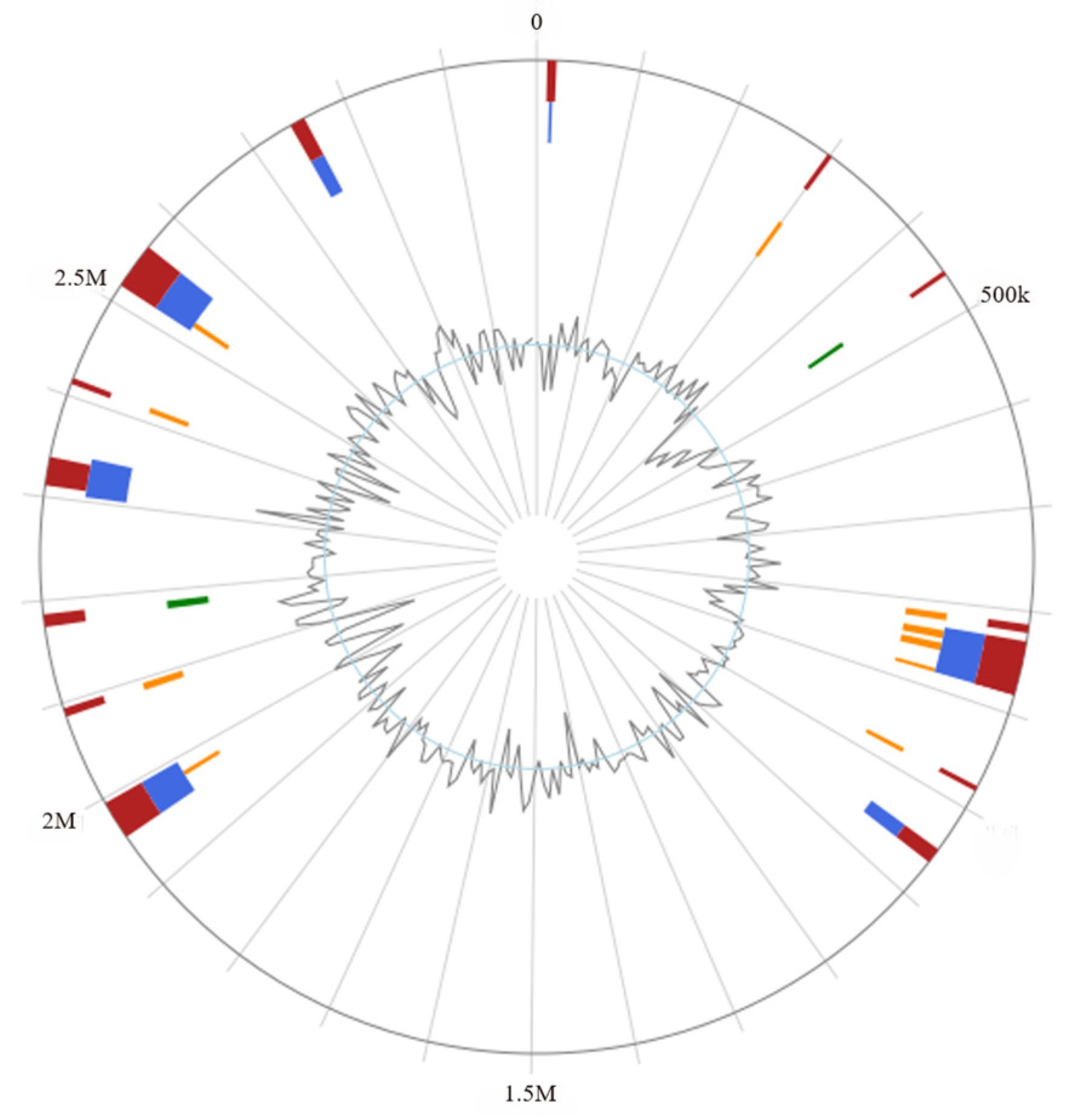
L. rhamnosus CP-1 genome lacks genes encoding transmissible and infectious virulence factors. Given that these resistance genes do not possess the potential for horizontal transfer to other strains, they pose no significant safety concerns. In line with the classification by Ochman and Davalos, the genome size of
L. rhamnosus CP-1 is considered medium-sized, which is typically associated with a robust metabolic capacity, high tolerance levels, and the ability to adapt to a variety of ecological niches [
32,
33].
L. rhamnosus CP-1 is devoid of plasmids, thereby eliminating the potential for the transfer of antibiotic resistance and virulence genes among bacteria via plasmid-mediated mechanisms. This characteristic makes it a safer option when compared to other strains that carry plasmids. Referring to the information available in public databases,
L. rhamnosus CP-1 exhibits a distinct genetic profile compared to the
Lacticaseibacillus rhamnosus 1.0320 (
L. rhamnosus 1.0320) previously known. While
L. rhamnosus 1.0320 boasts a genome length of 2.90 Mb and contains 2736 genes,
L. rhamnosus CP-1 has a higher number of operons for fermentation OFRs and dedicated carbohydrate utilisation proteins. This genetic advantage positions
L. rhamnosus CP-1 to more effectively engage in glycolysis and other probiotic functions [
12]. In addition, according to the results of genome alignment, it was indicated that the unique evolutionary status of
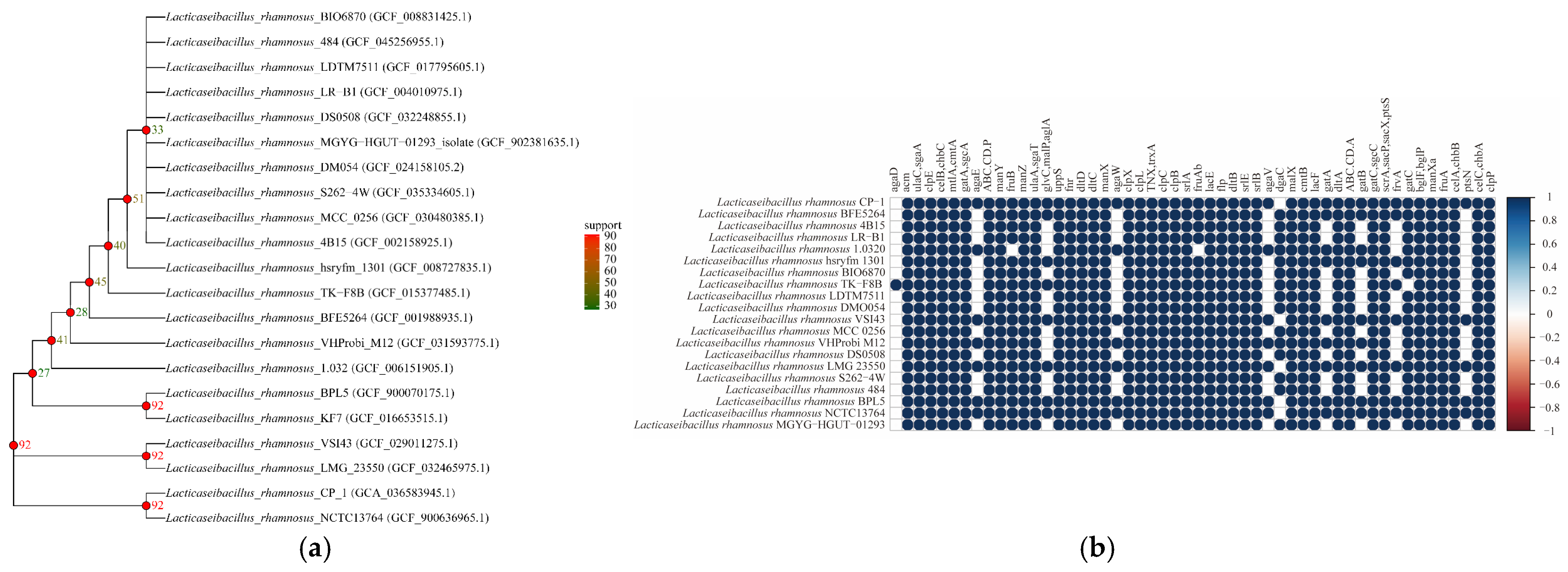
L. rhamnosus CP-1 was revealed, which formed a close association cluster with a specific closely related strain on the core gene evolutionary tree, indicating that
L. rhamnosus CP-1 is an important genetic resource for
L. rhamnosus, as it not only reflects the high genetic similarity with some strains, but also clarifies the genetic boundaries with other strains, and this study provided a solid theoretical basis for further study on the phylogeny, population differentiation, and evolution of functional genes of the strain. At the same time, functional gene cluster analysis showed that strain zero exhibited strong correlation and consistent genetic characteristics with
L. rhamnosus CP-1 at many gene loci (such as agaD, ACMA, etc.). A small number of genetic loci showed weak correlation, indicating that there were differences in genetic characteristics. Most of these genes are related to the synthesis of sugar-specific components of glucose, sorbose, cellobiose, fructose, and lactose, which can produce SCFAs through related metabolic pathways, and they are also involved in the regulation of glucose metabolism and play a variety of anti-inflammatory and antibacterial-related physiological functions. This genetic difference may affect the functional characteristics of strains, which provides important clues for further study of genetic diversity and functional differences between strains.
Furthermore, ongoing research into the molecular underpinnings of the functional disparities within
L. rhamnosus encompasses a range of gene-associated processes. These include genes are linked to fimbriae proteins, carbohydrate transport and metabolism, the biosynthesis of extracellular polysaccharides, bacteriocin production, restrictive modification systems, and bacterial defence mechanisms such as the CRISPR-Cas system. Regarding prebiotic functions, the majority of these are associated with carbohydrate metabolism, surface carbohydrate modifications, and surface proteins.
L. rhamnosus is capable of fermenting a diverse array of carbohydrates, thereby harnessing metabolic energy from these substrates [
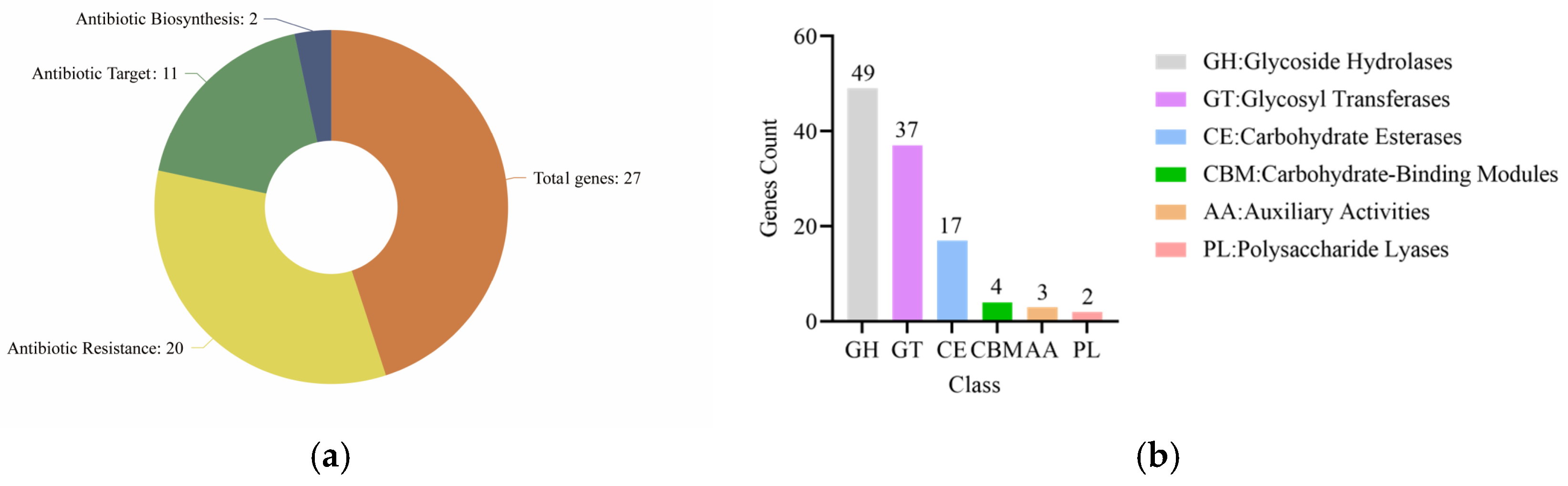
34]. According to the CAZy database annotations, glycosidase genes were found to be the most abundant, and glucose, fucose, lactose, galactose, raffinose, and other sugars could be used. In contrast, coenzyme genes are annotated in the least number, yet they represent a broad class of redox-active enzymes that are indispensable for the intricate processes of carbohydrate metabolism [
35]. The sugar-specific component transporter genes of glucose, sorbose, cellobiose, fructose, and lactose in the PTS system were annotated in the TCDB database. They are closely related to carbohydrate transport and catabolism. The diversity of carbohydrate metabolism genes enables probiotics to efficiently degrade dietary fibers (e.g., cellulose, pectin, and inulin) that are indigestible to the host, converting them into short-chain fatty acids (SCFAs, including acetate, propionate, and butyrate). These metabolites serve not only as crucial energy substrates for intestinal epithelial cells, but also exert multiple physiological functions: lowering intestinal pH to inhibit pathogenic bacterial proliferation, activating host immune signalling pathways (e.g., G protein-coupled receptors GPR41/43) to modulate local inflammatory responses and barrier functions, and systemically influencing host metabolism through circulatory transport [
36]. Furthermore, the superior carbohydrate utilisation capacity grants probiotics ecological competitiveness in the gut niche, allowing them to outcompete pathogens through nutrient sequestration, antimicrobial secretion, and adhesion-mediated colonisation, thereby maintaining microbial homeostasis. Certain genes may additionally participate in metabolising mucin-derived oligosaccharides within the intestinal mucus layer, facilitating mucus renewal and enhancing barrier integrity, while pathway byproducts such as B vitamins supplement host nutritional requirements. The synergistic effects of these functions not only enhance probiotic adaptability in the complex gut environment, but also extend their health benefits through systemic metabolic regulation (e.g., SCFA-mediated modulation of hepatic and adipose tissue metabolism), potentially ameliorating metabolic disorders, including obesity and diabetes [
16,
34,
35,
36]. Notably, the enrichment of such genes may reflect co-evolutionary adaptations between probiotics and hosts during long-term symbiosis. However, comprehensive validation integrating transcriptomic/proteomic analyses and in vivo models remains essential to elucidate the functional activity and physiological contributions of these genetic elements. It is important to note that bacteriocins constitute a class of compounds that are synthesised within bacterial ribosomes and possess antibacterial properties. Typically, these bacteriocins do not exhibit antibacterial activity against the bacteria that produce them. The osmotic component of the antimicrobial peptide ABC transporter, various bacteriocin immunoproteins, and glycosidase lysozyme were annotated in the whole genome of
L. rhamnosus CP-1. Meanwhile, in the CAZy database, four genes encoding GH25 lysozyme (EC 3.2.1.17) were annotated. Lysozyme, the seminal antibacterial peptide to be identified, stands out as a natural enzyme that possesses potent antimicrobial properties. It selectively targets and disrupts microorganisms, particularly Gram-positive bacteria, by selectively hydrolysing the 1,4-beta linkages that bind N-acetylmuramic acid to N-acetylglucosamine within the bacterial cell wall structure [
37]. These findings suggest that
L. rhamnosus CP-1 has possible potential antibacterial activity at the molecular level, but the specific antibacterial properties need to be further verified by in vitro related antibacterial experiments.
Notably, hyaluronan lyase plays a crucial role in degrading hyaluronic acid into smaller hyaluronan oligosaccharides. These oligosaccharides are known for their anti-inflammatory, antioxidant, and inhibitory effects on the proliferation of pathogenic cells [
38]. Chondroitin lyase, along with certain hyaluronic acids, is capable of cleaving chondroitin sulfate into its constituent chondroitin sulfate oligosaccharides. These oligosaccharides exert inhibitory actions on inflammation and demonstrate potent antioxidant properties [
39]. Various biomedical and pharmaceutical applications have also shown the antimicrobial activity of hyaluronic acid, and most of the hyaluronidase-producing microorganisms are Gram-positive bacteria [
40]. Moreover, the EfaA gene that may play an adhesin role in endocarditis, the GroEL gene involved in the adhesion or invasion of various target cells or tissues, and clpC, clpE, and clpP genes, which are related to stress survival, respond to stress and protect proteins from excessive damage, also were annotated in the whole genome of
L. rhamnosus CP-1 [
41,
42,
43,
44,
45,
46,
47,
48]. The gene encoding the hyaluronic acid capsule, cpsA, uppS, and gndA genes encoding the capsule, as well as the white gene encoding LPS, were also annotated. The hyaluronic acid capsule not only prevents phagocytosis by discouraging C3b binding, but also camouflages the bacteria as ‘self’ to the immune system, contributing to host immune evasion [
49,
50,
51]. Moreover, LPS, apart from its role in immunity, is also involved in inflammatory signalling pathways [
52,
53]. These discoveries indicate that
L. rhamnosus CP-1 may have anti-inflammatory potential at the molecular level.
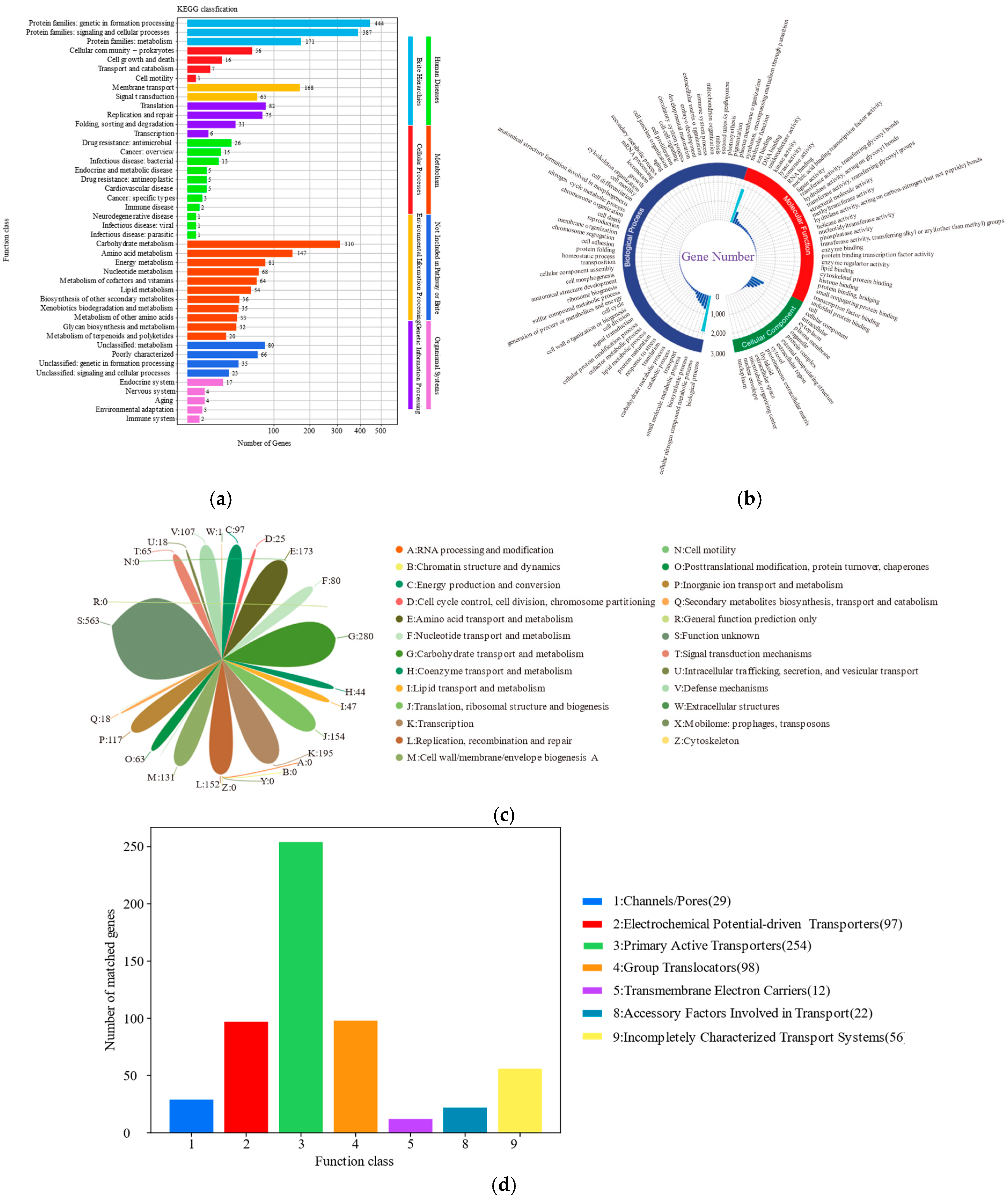
The antimicrobial peptide ABC transporter permease component has been meticulously annotated within the KEGG database, while the ABC-type antimicrobial peptide transporter, which serves as the primary active component in the transport mechanism, has been documented in the TCDB. AMPs are short peptides consisting of 10 to 50 amino acids with a wide range of antimicrobial activities. They serve as crucial immune effector molecules, playing pivotal roles in initiating and modulating the host’s immune defence system to combat pathogenic bacteria [
54]. The GlnP and GlnQ genes that contribute to bacterial survival in an acidic environment, as well as the clpL, and oppABCDF genes that contribute to acid and bile tolerance and bile salt resistance, were annotated in this
L. rhamnosus CP-1 genome [
55,
56]. Activation of dltA gene expression protects LTA-expressing Gram-positive bacteria from the innate immune antimicrobial defence. The dltB and dltD genes may play significant roles in both human immune responses and anti-inflammatory processes [
57]. Cellular metabolism can lead to excessive production of reactive oxygen species (ROS) and reactive nitrogen species (RNS), which induce oxidative stress and subsequently destroy macromolecules, and the clpC, clpB, and clpX genes respond to stress and protect proteins from excessive damage [
58]. Among the metabolic pathways annotated in the KEGG database, terpenoid skeletons are involved in the biosynthesis of 10 genes, while keto-glycan units contribute to the biosynthesis of 5 genes. Medicinal plants, which are rich in compounds such as terpenoids, flavonoids, and phenols, have been shown to be effective in alleviating acute lung injury (ALI) or chronic obstructive pulmonary disease (COPD). Certain diterpenoids and triterpenoids demonstrated inhibitory activity against inflammatory diseases, with mechanistic studies indicating that their modulatory effects primarily occur through dual inhibition of the mitogen-activated protein kinase (MAPK) cascade and nuclear factor kappa-B (NF-κB) transcriptional activation pathways. Flavonoids, a class of polyphenolic compounds, exhibit biological activities including antihepatotoxic, anti-inflammatory, and anti-ulcer properties. Furthermore, numerous flavonoid derivatives have been demonstrated to possess therapeutic efficacy against pulmonary inflammation. As well-established anti-inflammatory phytochemicals, specific flavonoids have shown suppressive effects in various animal models of inflammation. The underlying mechanism of this inhibition may involve the attenuation of oxidative stress, suppression of NF-κB activation, and inhibition of epidermal growth factor receptor (EGFR) phosphorylation [
59,
60]. Additionally,
L. rhamnosus CP-1 was isolated from the stools of mice with high-DHA tuna oil-relieved smoking-induced pneumoni [
1]. In summary, whole-genome sequencing revealed that the
L. rhamnosus CP-1 strain isolated in this study exhibits inherent acid and bile tolerance at the molecular level and possesses potential antimicrobial and anti-inflammatory properties at the genetic level. The subsequent phase may involve conducting specific in vivo or in vitro anti-inflammatory experiments based on the characteristics demonstrated at the molecular level. For instance, animal treatment model experiments could be implemented to further validate the anti-inflammatory functions associated with this strain.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}