
Leveraging the Activated Monomer Mechanism to Create Grafted Polymer Networks in Epoxide–Acrylate Hybrid Photopolymerizations



Abstract
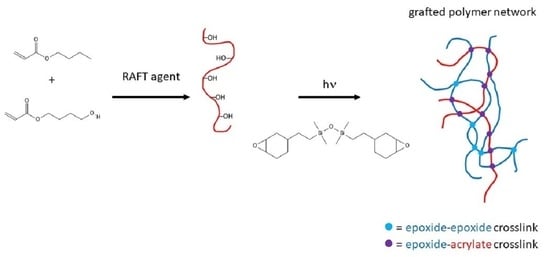
1. Introduction
2. Materials and Methods
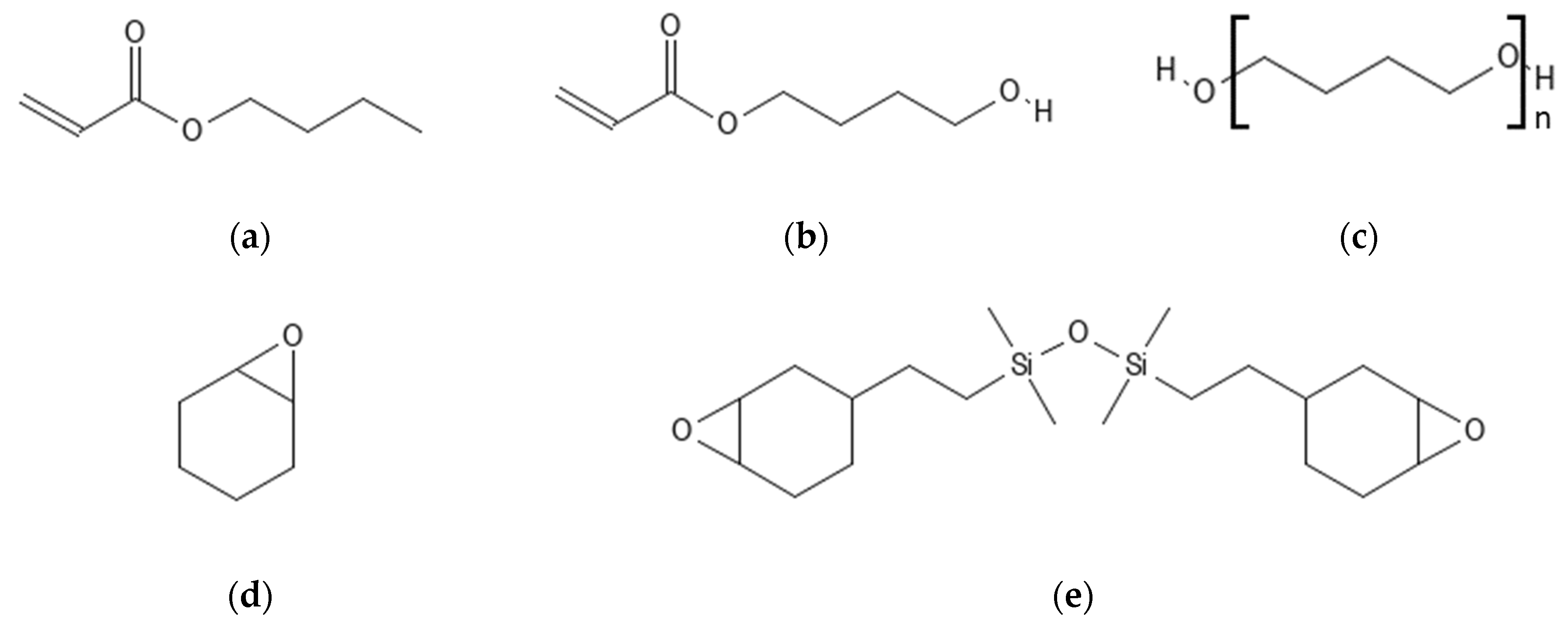
2.1. Materials
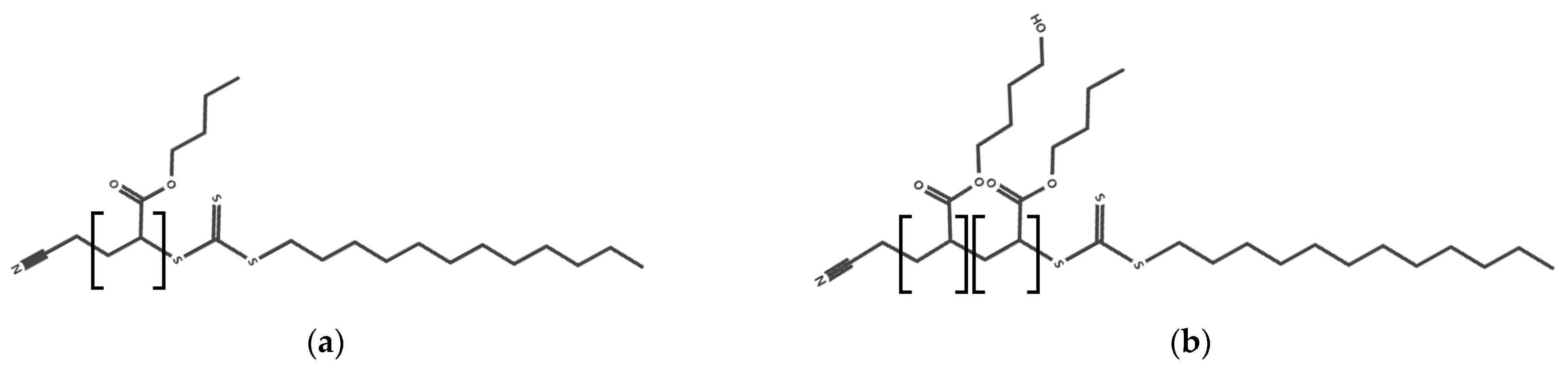
2.1.1. RAFT Agent Synthesis
2.1.2. RAFT Polymer Synthesis
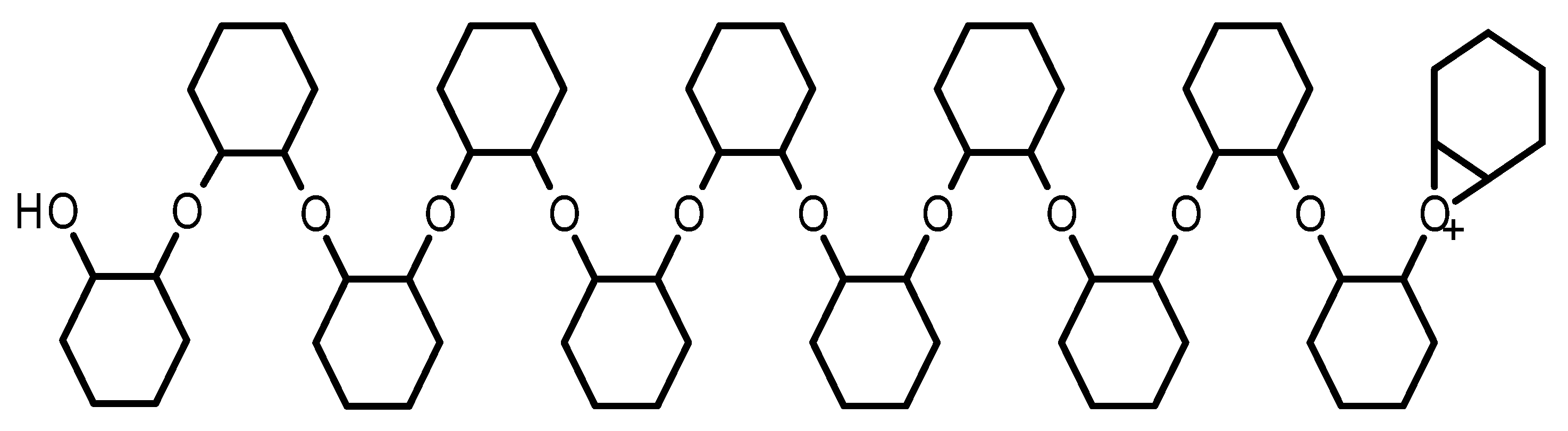
2.1.3. Linear Epoxide Thermal Polymerization
2.1.4. Cross-Linked Epoxide Photopolymerization
2.2. Analytical Methods
2.2.1. Gel Permeation Chromatography
2.2.2. Dynamic Mechanical Analysis
3. Results and Discussion
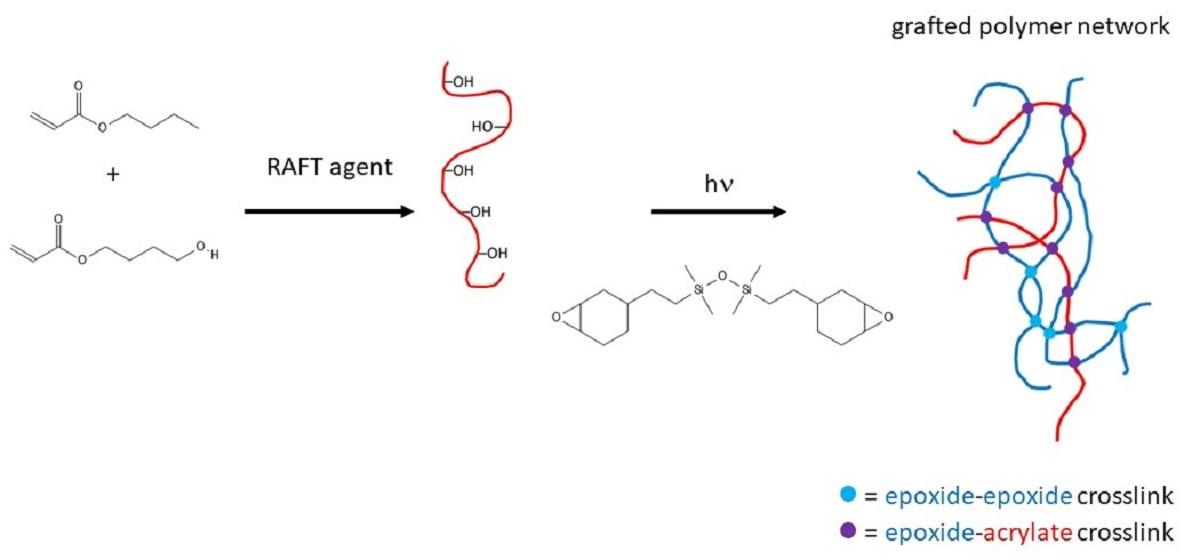
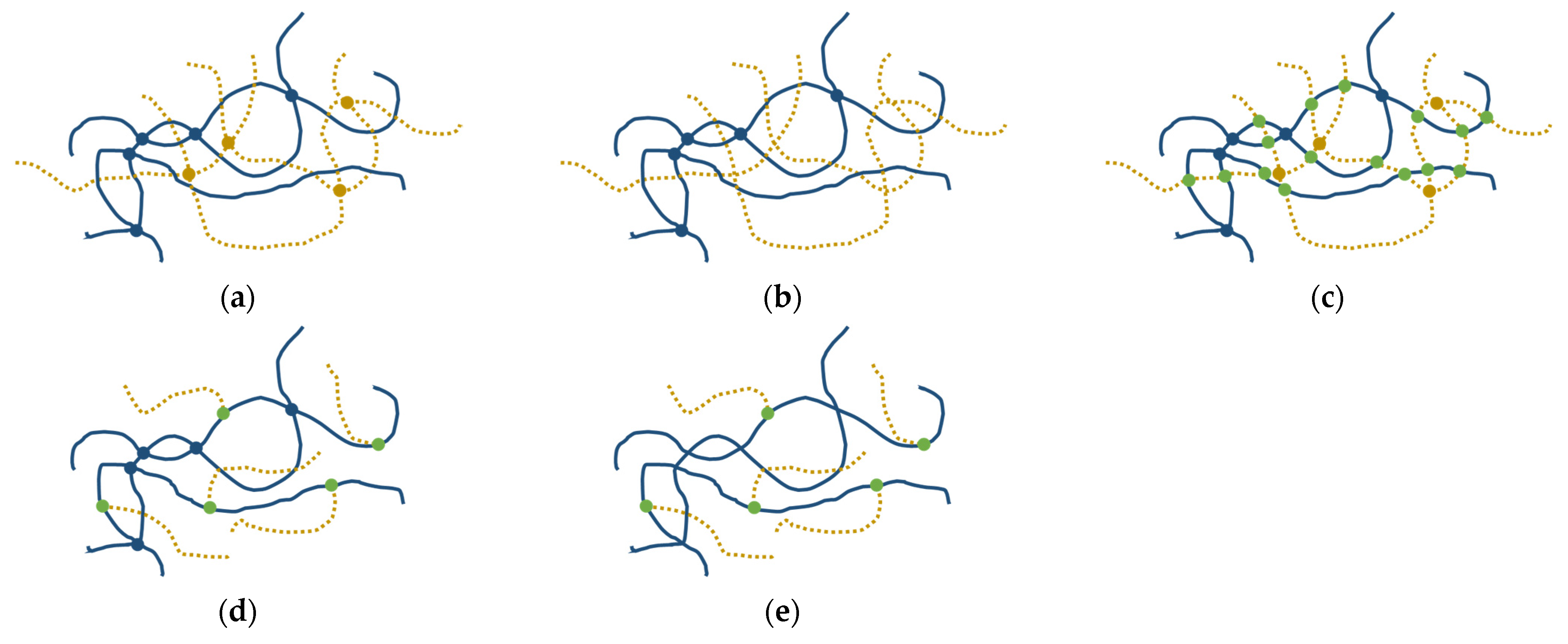
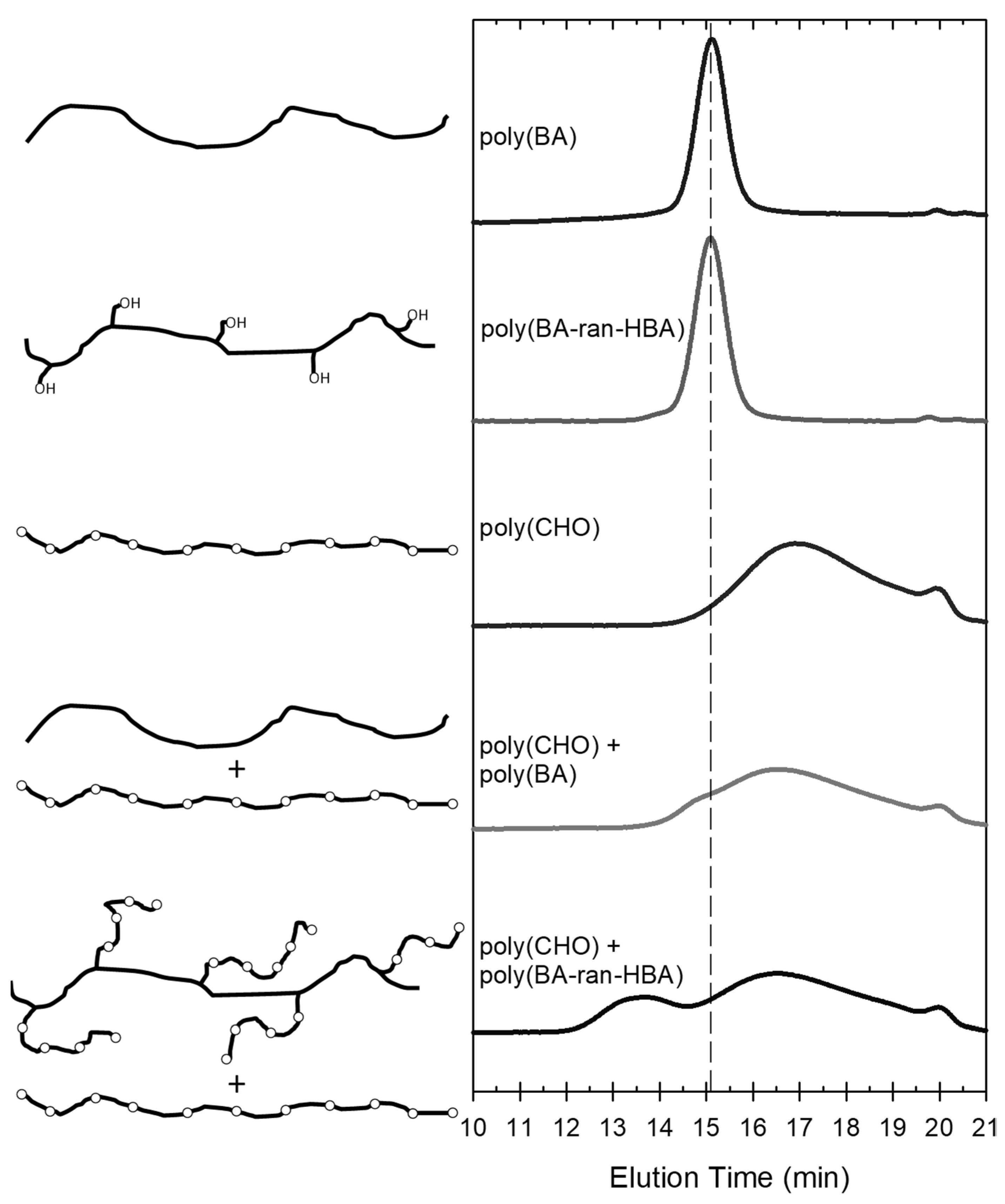
3.1. Demonstration of Grafting
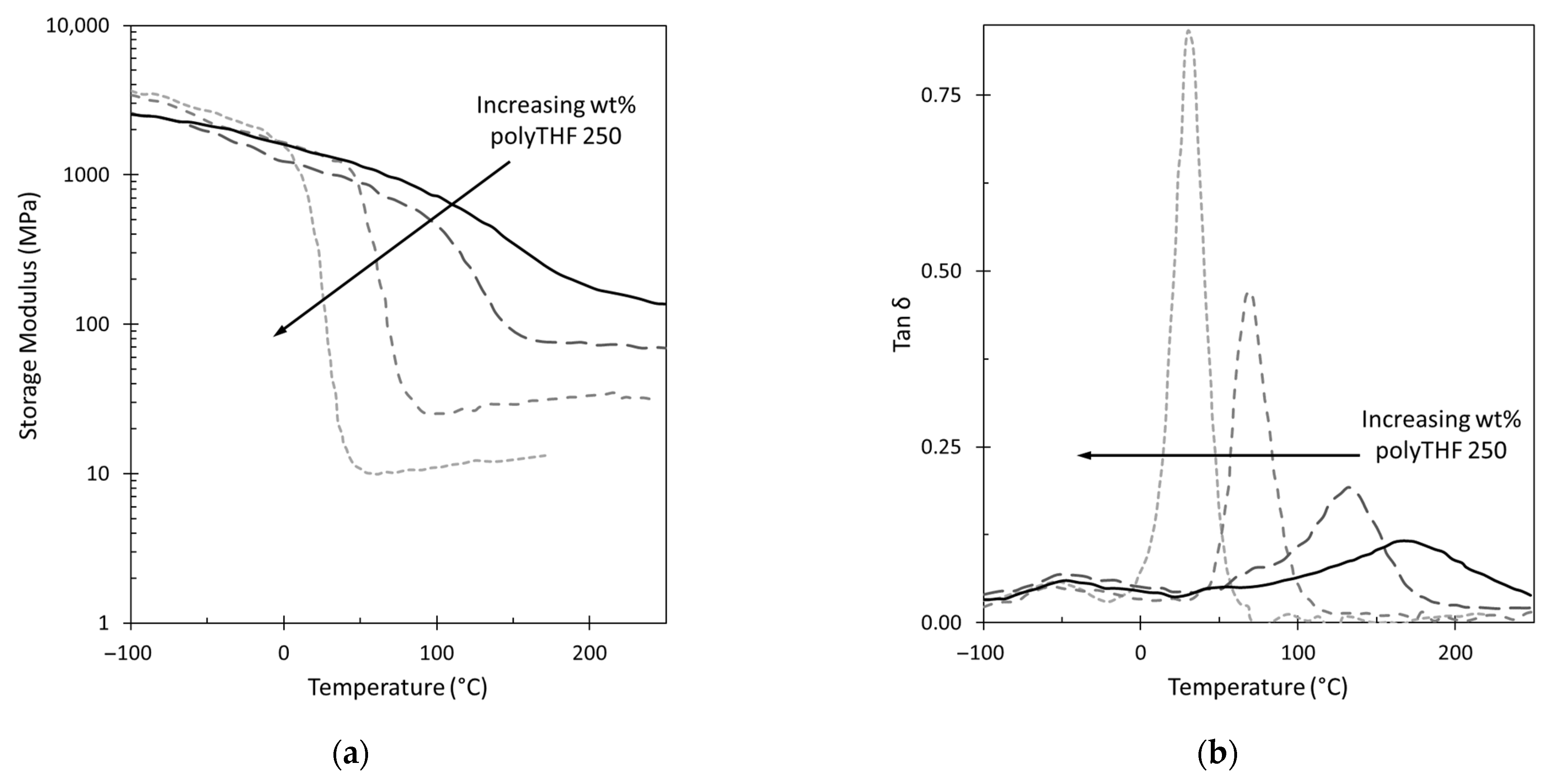
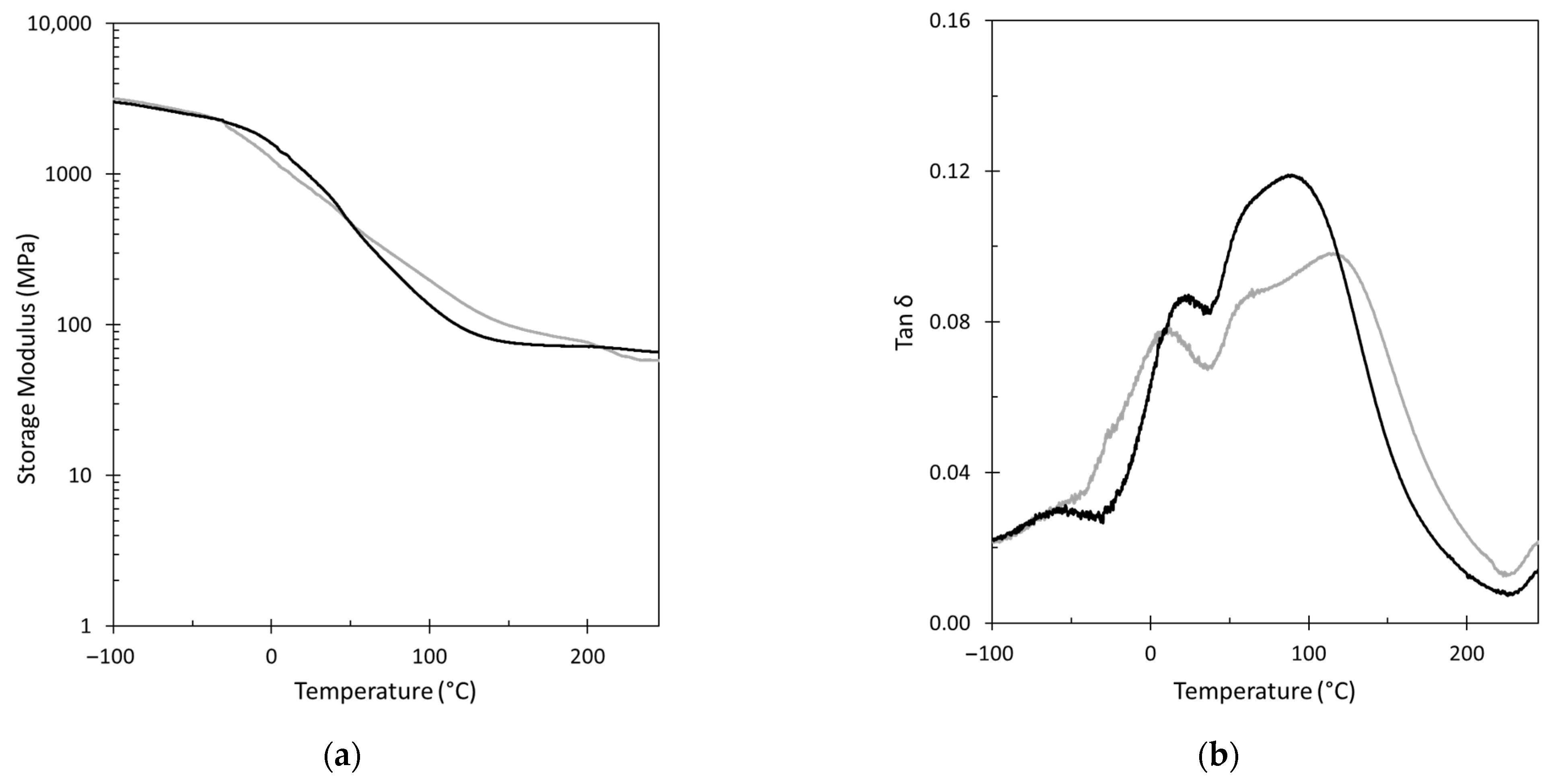
3.2. GPN Physical Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oxman, J.D.; Jacobs, D.W.; Trom, M.C.; Sipani, V.; Ficek, B.; Scranton, A.B. Evaluation of initiator systems for controlled and sequentially curable free-radical/cationic hybrid photopolymerizations. J. Polym. Sci. A Polym. Chem. 2005, 43, 1747–1756. [Google Scholar] [CrossRef]
- Odian, G. Principles of Polymerization, 4th ed.; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2004. [Google Scholar]
- Salim, M.S. Overview of UV curable coatings. In Radiation Curing of Polymers II; Randell, D.R., Ed.; The Royal Society of Cambridge: Cambridge, UK, 1991; pp. 3–21. [Google Scholar]
- Fouassier, J. Photoinitiation, Photopolymerization, and Photocuring: Fundamentals and Applications; Hanser/Gardner Publication Inc.: Cincinnati, OH, USA, 1995. [Google Scholar]
- Pappas, S.P. Radiation curing—A personal perspective. In Radiation Curing: Science and Technology; Pappas, S.P., Ed.; Plenum Press: New York, NY, USA, 1992; pp. 1–20. [Google Scholar]
- Dillman, B.; Jessop, J.L.P. Chain transfer agents in cationic photopolymerization of a bis-cycloaliphatic epoxide monomer: Kinetic and physical property effects. J. Polym. Sci. A Polym. Chem. 2013, 51, 2058–2067. [Google Scholar] [CrossRef]
- Crivello, J.V.; Liu, S. Photoinitiated cationic polymerization of epoxy alcohol monomers. J. Polym. Sci. A Polym. Chem. 2000, 38, 389–401. [Google Scholar] [CrossRef]
- Biedron, T.; Kubisa, P.; Szymanski, R.; Penczek, S. Kinetics of polymerization by activated monomer mechanism. Makromol. Chem. Macromol. Symp. 1990, 32, 155–168. [Google Scholar] [CrossRef]
- Dillman, B.F. The Kinetics and Physical Properties of Epoxides, Acrylates, and Hybrid Epoxy-Acrylate Photopolymerization Systems. Ph.D. Thesis, University of Iowa, Iowa City, IA, USA, 2013. [Google Scholar]
- Schissel, S.M.; Jessop, J.L.P. Enhancing epoxide kinetics and tuning polymer properties using hydroxyl-containing (meth) acrylates in hybrid photopolymerizations. Polymer 2019, 161, 78–91. [Google Scholar] [CrossRef]
- Ajiboye, G.; Jessop, J.L.P. Synergistic effect of hydroxyl-containing acrylates in epoxide acrylate hybrid photopolymerizations. In Proceedings of the 2012 RadTech UV&EB Technology & Conference, Chicago, IL, USA, 30 April–2 May 2012; Available online: https://www.radtech.org/proceedings/2012/papers/Session%209%20-%20Chemistry/GAjiboye_UIowa.pdf (accessed on 7 March 2023).
- Ge, X.; Ye, Q.; Song, L.; Misra, A.; Spencer, P. Visible-light initiated free-radical/cationic ring-opening hybrid photopolymerization of methacrylate/epoxy: Polymerization kinetics, crosslinking structure, and dynamic mechanical properties. Macromol. Chem. Phys. 2015, 216, 856–872. [Google Scholar] [CrossRef] [PubMed]
- Decker, C. Photoinitiated crosslinking polymerization. Prog. Polym. Sci. 1996, 21, 593–650. [Google Scholar] [CrossRef]
- Davidson, C.L.; Feilzer, A.J. Polymerization shrinkage and polymerization shrinkage stress in polymer-based restoratives. J. Dentistry 1997, 25, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Anseth, K.S.; Bowman, C.N.; Peppas, N.A. Polymerization kinetics and volume relaxation behavior of photopolymerized multifunctional monomers producing highly crosslinked networks. J. Polym. Sci. A Polym. Chem. 1994, 32, 139–147. [Google Scholar] [CrossRef]
- Andrzejewska, E. Photopolymerization kinetics of multifunctional monomers. Prog. Polym. Sci. 2001, 26, 605–665. [Google Scholar] [CrossRef]
- Rajaraman, S.S.K.; Mowers, W.A.; Crivello, J.V. Interaction of epoxy and vinyl ethers during photoinitiated cationic polymerization. J. Polym. Sci. A Polym. Chem. 1999, 37, 4007–4018. [Google Scholar] [CrossRef]
- Nelson, E.W.; Jacobs, J.L.; Scranton, A.B.; Anseth, K.S.; Bowman, C.N. Photo-differential scanning calorimetry studies of cationic polymerizations of divinyl ethers. Polymer 1995, 36, 4651–4656. [Google Scholar] [CrossRef]
- Hoppe, C.C.; Ficek, B.A.; Eom, H.S.; Scranton, A.B. Cationic photopolymerization of epoxides containing carbon black nanoparticles. Polymer 2010, 51, 6151–6160. [Google Scholar] [CrossRef]
- Cai, Y.; Jessop, J.L.P. Decreased oxygen inhibition in photopolymerized acrylate/epoxide hybrid polymer coatings as demonstrated by Raman spectroscopy. Polymer 2006, 47, 6560–6566. [Google Scholar] [CrossRef]
- Cai, Y.; Jessop, J.L.P. Effect of water concentration on photopolymerized acrylate/epoxide hybrid polymer coatings as demonstrated by Raman spectroscopy. Polymer 2009, 50, 5406–5413. [Google Scholar] [CrossRef]
- Sangermano, M.; Carbonaro, W.; Malucelli, G.; Priola, A. UV-cured interpenetrating acrylic-epoxy polymer networks: Preparation and characterization. Macromol. Mat. Eng. 2008, 293, 515–520. [Google Scholar] [CrossRef]
- Dean, K.; Cook, W.D. Effect of curing sequence on the photopolymerization and thermal curing kinetics of dimethacrylate/epoxy interpenetrating polymer networks. Macromolecules 2002, 35, 7942–7954. [Google Scholar] [CrossRef]
- Sperling, L.H. Interpenetrating polymer networks: An overview. In Advances in Chemistry; Sperling, L.H., Klempner, D., Utracki, L.A., Eds.; American Chemical Society: Washington, DC, USA, 1994; Volume 239, pp. 3–38. [Google Scholar] [CrossRef]
- Hasa, E.; Scholte, J.P.; Jessop, J.L.P.; Stansbury, J.W.; Guymon, C.A. Kinetically controlled photoinduced phase separation for hybrid radical/cationic systems. Macromolecules 2019, 52, 2975–2986. [Google Scholar] [CrossRef]
- Cook, W.D.; Chen, F.; Ooi, S.K.; Moorhoff, C.; Knott, R. Effect of curing order on the curing kinetics and morphology of bisGMA/DGEBA interpenetrating polymer networks. Polym. Int. 2006, 55, 1027–1039. [Google Scholar] [CrossRef]
- Dean, K.; Cook, W.D.; Zipper, M.D.; Burchill, P. Curing behaviour of IPNs formed from model VERs and epoxy systems I amine cured epoxy. Polymer 2001, 42, 1345–1359. [Google Scholar] [CrossRef]
- Jansen, B.J.P.; Rastogi, S.; Meijer, H.E.H.; Lemstra, P.J. Rubber-modified glassy amorphous polymers prepared via chemically induced phase separation. 4. Comparison of properties of semi-and full-IPNs, and copolymers of acrylate−aliphatic epoxy systems. Macromolecules 1999, 32, 6290–6297. [Google Scholar] [CrossRef][Green Version]
- Biedron, T.; Brzezinska, K.; Kubisa, P.; Penczek, S. Macromonomers by activated polymerization of oxiranes. Synthesis and polymerization. Polym. Int. 1995, 36, 73–80. [Google Scholar] [CrossRef]
- Yagci, Y. Photoinitiated cationic polymerization of unconventional monomers. Macromol. Symp. 2006, 240, 93–101. [Google Scholar] [CrossRef]
- Lin, J.; Wu, X.; Zheng, C.; Zhang, P.; Li, Q.; Wang, W.; Yang, Z. A novolac epoxy resin modified polyurethane acylates polymer grafted network with enhanced thermal and mechanical properties. J. Polym. Res. 2014, 21, 435. [Google Scholar] [CrossRef]
- Touhsaent, R.E.; Thomas, D.A.; Sperling, L.H. Epoxy/acrylic simultaneous interpenetrating networks. J. Polym. Sci. Polym. Symp. 1974, 46, 175–190. [Google Scholar] [CrossRef]
- Scarito, P.R.; Sperling, L.H. Effect of grafting on phase volume fraction, composition, and mechanical behavior: Epoxy/poly (n-butyl acrylate) simultaneous interpenetrating networks. Polym. Eng. Sci. 1979, 19, 297–303. [Google Scholar] [CrossRef]
- Dong, T.V.; Tinh, V.D.C.; Kim, D. Ether-free sulfonated poly (fluorene biphenyl indole) membranes and ionomer binders for proton exchange membrane fuel cells. J. Power Sources 2023, 556, 232418. [Google Scholar] [CrossRef]
- Jeon, Y.; Tinh, V.D.C.; Thuc, V.D.; Kim, D. Ether-free polymer based bipolar electrolyte membranes without an interlayer catalyst for water electrolysis with durability at a high current density. Chem. Eng. J. 2023, 459, 141467. [Google Scholar] [CrossRef]
- Fijten, M.W.; Paulus, R.M.; Schubert, U.S. Systematic parallel investigation of RAFT polymerizations for eight different (meth) acrylates: A basis for the designed synthesis of block and random copolymers. J. Polym. Sci. A Polym. Chem. 2005, 43, 3831–3839. [Google Scholar] [CrossRef]
- Chiefari, J.; Chong, Y.K.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T.P.; Mayadunne, R.T.; Meijs, G.F.; Moad, C.L.; Moad, G.; et al. Living free-radical polymerization by reversible addition-fragmentation chain transfer: The RAFT process. Macromolecules 1998, 31, 5559–5562. [Google Scholar] [CrossRef]
- Perrier, S. 50th anniversary perspective: RAFT polymerization—A user guide. Macromolecules 2017, 50, 7433–7447. [Google Scholar] [CrossRef]
- Gu, J.; Narang, S.C.; Pearce, E.M. Curing of epoxy resins with diphenyliodonium salts as thermal initiators. J. Appl. Polym. Sci. 1985, 30, 2997–3007. [Google Scholar] [CrossRef]
- Lu, H.; Lovell, L.G.; Bowman, C.N. Exploiting the heterogeneity of cross-linked photopolymers to create high-Tg polymers from polymerizations performed at ambient conditions. Macromolecules 2001, 34, 8021–8025. [Google Scholar] [CrossRef]
- Anseth, K.S.; Bowman, C.N.; Peppas, N.A. Dynamic mechanical studies of the glass transition temperature of photopolymerized multifunctional acrylates. Polym. Bull. 1993, 31, 229–233. [Google Scholar] [CrossRef]
- Poly(tetrahydrofuran). Available online: https://polymerdatabase.com/polymers/polytetrahydrofuran.html (accessed on 7 March 2023).
- Menard, K.P. Dynamic Mechanical Analysis: A Practical Introduction, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Murayama, T. Dynamic Mechanical Analysis of Polymeric Materials; Elsevier Scientific Publishing: New York, NY, USA, 1978. [Google Scholar]
- Thermal Transitions of Homopolymers: Glass Transition & Melting Point. Available online: https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/materials-science-and-engineering/polymer-synthesis/thermal-transitions-of-homopolymers (accessed on 7 March 2023).
- Deane, O.J.; Jennings, J.; Neal, T.J.; Musa, O.M.; Fernyhough, A.; Armes, S.P. Synthesis and aqueous solution properties of shape-shifting stimulus-responsive diblock copolymer nano-objects. Chem. Mat. 2021, 33, 7767–7779. [Google Scholar] [CrossRef]
- Hiemenz, P.C.; Lodge, T.P. Polymer Chemistry, 2nd ed.; CRC Press: New York, NY, USA, 2007; p. 494. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | CHO, g | Cationic PI, g | Polyacrylate, g | CHCl3, mL |
|---|---|---|---|---|
| Neat epoxide | 5.033 | 0.378 | – | 10 |
| Epoxide + poly(BA) (10:1) | 5.050 | 0.374 | 0.516 | 10 |
| Epoxide + poly(BA-ran-HBA) (10:1) | 5.051 | 0.370 | 0.500 | 10 |
| Formulation | Diepoxide, g | Cationic PI, g | Polyacrylate. g | PolyTHF 250, g |
|---|---|---|---|---|
| Neat epoxide | 5.002 | 0.195 | – | – |
| Epoxide + poly(BA) (10:3) | 5.002 | 0.193 | 1.489 | – |
| Epoxide + poly(BA-ran-HBA) (10:3) | 5.016 | 0.193 | 1.507 | – |
| Epoxide + polyTHF (10:0.7) | 5.008 | 0.189 | – | 0.330 |
| Epoxide + polyTHF (10:1.5) | 5.009 | 0.190 | – | 0.814 |
| Epoxide + polyTHF (10:3) | 5.023 | 0.189 | – | 1.629 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dillman, B.F.; Schissel, S.M.; Jessop, J.L.P. Leveraging the Activated Monomer Mechanism to Create Grafted Polymer Networks in Epoxide–Acrylate Hybrid Photopolymerizations. Macromol 2024, 4, 104-116. https://doi.org/10.3390/macromol4010005
Dillman BF, Schissel SM, Jessop JLP. Leveraging the Activated Monomer Mechanism to Create Grafted Polymer Networks in Epoxide–Acrylate Hybrid Photopolymerizations. Macromol. 2024; 4(1):104-116. https://doi.org/10.3390/macromol4010005
Chicago/Turabian StyleDillman, Brian F., Sage M. Schissel, and Julie L. P. Jessop. 2024. "Leveraging the Activated Monomer Mechanism to Create Grafted Polymer Networks in Epoxide–Acrylate Hybrid Photopolymerizations" Macromol 4, no. 1: 104-116. https://doi.org/10.3390/macromol4010005
APA StyleDillman, B. F., Schissel, S. M., & Jessop, J. L. P. (2024). Leveraging the Activated Monomer Mechanism to Create Grafted Polymer Networks in Epoxide–Acrylate Hybrid Photopolymerizations. Macromol, 4(1), 104-116. https://doi.org/10.3390/macromol4010005