
Lipopolysaccharide-Induced Immunological Tolerance in Monocyte-Derived Dendritic Cells



{kind=link}
Abstract
1. Introduction
2. Function of LPS
2.1. Virulence and Toxicity
2.2. Biological Activity of Lipopolysaccharide
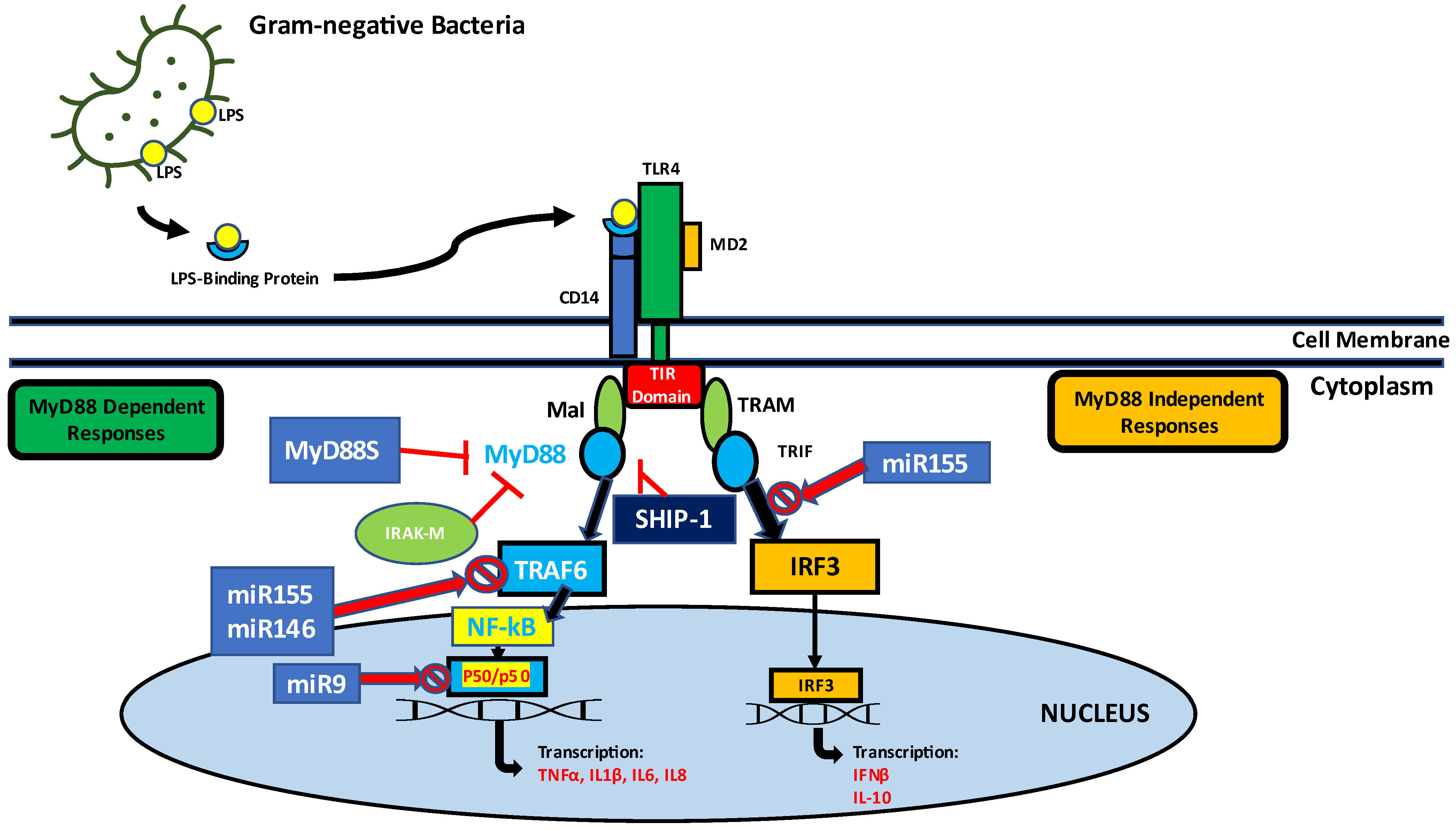
3. Lipopolysaccharide Signaling and Immune Activation Mechanisms in Higher Organisms
3.1. Lipopolysaccharide Detoxification Mechanisms in Higher Animals
3.2. Host-Microbe Interactions (Lipopolysaccharide Activity) in Invertebrates—Insects
3.3. Expression of Genes and Signaling Action Induced by Lipopolysaccharide in Vertebrates and Invertebrates
4. LPS in Inflammatory-Mediated Pathogenesis
4.1. Role of LPS from the Gut Microbiome in Inflammatory Conditions
4.2. Bacterial LPS-Induced Lung Injury and Pathologies
4.3. LPS-induced Meningococcal Inflammatory Disorders
4.4. LPS-induced Periodontal Inflammatory-Mediated Pathologies
4.5. LPS and Neuroinflammation
4.6. LPS Aggravates Inherited Retinal Dystrophy
4.7. LPS-Induced Perinatal Impact of Maternal Inflammation
5. The Function of LPS in Immune Suppression
5.1. Endotoxin Tolerance
5.2. Role of LPS from the Gut Microbiome as an Anti-Inflammatory Agent
5.3. Role of LPS in CTB-INS-mediated Tolerance
5.3.1. CTB-INS Vaccines
5.3.2. CTB-INS Vaccine Stimulation of Dendritic Cell Maturation
5.3.3. The Role of LPS in CTB-INS-Mediated Tolerance
6. Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bertani, B.; Ruiz, N. Function and Biogenesis of Lipopolysaccharides. EcoSal Plus 2018, 8. [Google Scholar] [CrossRef]
- Alexander, T.E.; Smith, I.M.; Lipsky, Z.W.; Lozeau, L.D.; Camesano, T.A. Role of lipopolysaccharides and lipoteichoic acids on C-Chrysophsin-1 interactions with model Gram-positive and Gram-negative bacterial membranes. Biointerphases 2020, 15, 031007. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, K.; Schromm, A.B.; Weindl, G.; Heinbockel, L.; Correa, W.; Mauss, K.; de Tejada, G.M.; Garidel, P. An update on endotoxin neutralization strategies in Gram-negative bacterial infections. Expert Rev. Anti-Infect. Ther. 2020, 19, 495–517. [Google Scholar] [CrossRef] [PubMed]
- Giordano, N.P.; Cian, M.B.; Dalebroux, Z.D. Outer Membrane Lipid Secretion and the Innate Immune Response to Gram-Negative Bacteria. Infect. Immun. 2020, 88, e00920-19. [Google Scholar] [CrossRef] [PubMed]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.-Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987–995. [Google Scholar] [CrossRef]
- Rosadini, C.V.; Kagan, J.C. Early innate immune responses to bacterial LPS. Curr. Opin. Immunol. 2016, 44, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Kagan, J.C. A Cross-Disciplinary perspective on the innate immune responses to bacterial lipopolysaccharide. Mol. Cell 2014, 54, 212–223. [Google Scholar] [CrossRef]
- Kent, L.W.; Rahemtulla, F.; Hockett, R.D.; Gilleland, R.C.; Michalek, S.M. Effect of lipopolysaccharide and inflammatory cytokines on Interleukin-6 production by healthy human gingival fibroblasts. Infect. Immun. 1998, 66, 608–614. [Google Scholar] [CrossRef]
- Choi, J.; Moon, S.; Bae, H.; Kim, Y.-W.; Lee, D.; Kim, S.; Seo, Y.; Wang, H.S.; Choi, Y.W.; Lee, M.W.; et al. Alnus Sibirica Extracts Suppress the Expression of Inflammatory Cytokines Induced by Lipopolysaccharides, Tumor Necrosis Factor-α, and Interferon-γ in Human Dermal Fibroblasts. Molecules 2019, 24, 2883. [Google Scholar] [CrossRef]
- Bonham, K.; Orzalli, M.H.; Hayashi, K.; Wolf, A.I.; Glanemann, C.; Weninger, W.; Iwasaki, A.; Knipe, D.M.; Kagan, J.C. A promiscuous lipid-binding protein diversifies the subcellular sites of toll-like receptor signal transduction. Cell 2014, 156, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Ge, M.; Shen, S.; Yang, L.; Jin, T.; Cao, D.; Xu, H.; Zheng, X.; Qiu, S.; Wang, K.; et al. Activation of NFKB-JMJD3 signaling promotes bladder fibrosis via boosting bladder smooth muscle cell proliferation and collagen accumulation. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 2403–2410. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.; Kim, M.-J.; Lee, S.; Chun, E.; Lee, K.-Y. Inhibition of TRAF6 ubiquitin-ligase activity by PRDX1 leads to inhibition of NFKB activation and autophagy activation. Autophagy 2018, 14, 1347–1358. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhou, S.; Ge, Y.; Lu, M.; Liu, Z.; Gong, R. Valproate hampers podocyte acquisition of immune phenotypes via intercepting the GSK3β facilitated NFkB activation. Oncotarget 2017, 8, 88332–88344. [Google Scholar] [CrossRef][Green Version]
- Van Acker, T.; Eyckerman, S.; Walle, L.V.; Gerlo, S.; Goethals, M.; Lamkanfi, M.; Bovijn, C.; Tavernier, J.; Peelman, F. The small GTPase Arf6 is essential for the Tram/Trif pathway in TLR4 signaling. J. Biol. Chem. 2014, 289, 1364–1376. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Liu, X.; Wang, N.; Cao, H.; Lu, Y.; Zhou, H.; Zheng, J. Inhibition of clathrin/dynamin-dependent internalization interferes with LPS-mediated TRAM–TRIF-dependent signaling pathway. Cell. Immunol. 2012, 274, 121–129. [Google Scholar] [CrossRef]
- Zhang, S.; Yuquan, W.; Guo, Q.; Li, R.; Li, G.-B.; Tan, S.; Li, X.; Wei, Y.; Wu, M. Annexin A2 binds to endosomes and negatively regulates TLR4-triggered inflammatory responses via the TRAM-TRIF pathway. Sci. Rep. 2015, 5, 15859. [Google Scholar] [CrossRef]
- Hilliard, A.; Mendonca, P.; Soliman, K.F. Involvement of NFƙB and MAPK signaling pathways in the preventive effects of Ganoderma lucidum on the inflammation of BV-2 microglial cells induced by LPS. J. Neuroimmunol. 2020, 345, 577269. [Google Scholar] [CrossRef]
- Warren, H.S.; Fitting, C.; Hoff, E.; Adib-Conquy, M.; Beasley-Topliffe, L.; Tesini, B.; Liang, X.; Valentine, C.; Hellman, J.; Hayden, D.; et al. Resilience to bacterial infection: Difference between species could be due to proteins in serum. J. Infect. Dis. 2010, 201, 223–232. [Google Scholar] [CrossRef]
- Reid, R.R.; Prodeus, A.P.; Khan, W.; Hsu, T.; Rosen, F.S.; Carroll, M.C. Endotoxin shock in antibody-deficient mice: Unraveling the role of natural antibody and complement in the clearance of lipopolysaccharide. J. Immunol. 1997, 159, 970–975. [Google Scholar]
- Boes, M.; Prodeus, A.P.; Schmidt, T.; Carroll, M.C.; Chen, J. A critical role of natural immunoglobulin M in immediate defense against systemic bacterial infection. J. Exp. Med. 1998, 188, 2381–2386. [Google Scholar] [CrossRef]
- Zhou, F.; Zhang, G.-X.; Rostami, A. LPS-treated bone marrow-derived dendritic cells induce immune tolerance through modulating differentiation of CD4+ regulatory T cell subpopulations mediated by 3G11 and CD127. Immunol. Res. 2016, 65, 630–638. [Google Scholar] [CrossRef]
- Hayashi, T.; Gray, C.S.; Chan, M.; Tawatao, R.I.; Ronacher, L.; McGargill, M.A.; Datta, S.K.; Carson, D.A.; Corr, M. Prevention of autoimmune disease by induction of tolerance to Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 2009, 106, 2764–2769. [Google Scholar] [CrossRef]
- Mbongue, J.; Nicholas, D.; Firek, A.; Langridge, W. The role of dendritic cells in tissue-specific autoimmunity. J. Immunol. Res. 2014, 2014. [Google Scholar] [CrossRef]
- Joffre, O.; Nolte, M.A.; Spörri, R.; Sousa, C.R.E. Inflammatory signals in dendritic cell activation and the induction of adaptive immunity. Immunol. Rev. 2009, 227, 234–247. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Akira, S. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 2009, 388, 621–625. [Google Scholar] [CrossRef]
- Steinman, R.M.; Banchereau, J. Taking dendritic cells into medicine. Nature 2007, 449, 419–426. [Google Scholar] [CrossRef]
- Galkina, E.; Ley, K. Immune and Inflammatory Mechanisms of Atherosclerosis. Annu. Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef]
- Oyoshi, M.K.; He, R.; Kumar, L.; Yoon, J.; Geha, R.S. Cellular and molecular mechanisms in atopic dermatitis. Adv. Immunol. 2009, 102, 135–226. [Google Scholar]
- Said, E.A.; Dupuy, F.P.; Trautmann, L.; Zhang, Y.; Shi, Y.; El-Far, M.; Hill, B.J.; Noto, A.; Ancuta, P.; Peretz, Y.; et al. Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat. Med. 2010, 16, 452–459. [Google Scholar] [CrossRef]
- Agarwal, S.; Piesco, N.; Johns, L.; Riccelli, A. Differential Expression of IL-1β, TNF-α, IL-6, and IL-8 in Human Monocytes in Response to Lipopolysaccharides from Different Microbes. J. Dent. Res. 1995, 74, 1057–1065. [Google Scholar] [CrossRef]
- Bryn, T.; Yaqub, S.; Mahic, M.; Henjum, K.; Aandahl, E.M.; Taskén, K. LPS-activated monocytes suppress T-cell immune responses and induce FOXP3+ T cells through a COX-2-PGE2-dependent mechanism. Int. Immunol. 2008, 20, 235–245. [Google Scholar] [CrossRef]
- Adib-Conquy, M.; Adrie, C.; Moine, P.; Asehnoune, K.; Fitting, C.; Pinsky, M.R.; Dhainaut, J.F.; Cavaillon, J.M. NF-kappaB expression in mononuclear cells of patients with sepsis resembles that observed in lipopolysaccharide tolerance. Am. J. Respir. Crit. Care Med. 2000, 162, 1877–1883. [Google Scholar] [CrossRef]
- López-Collazo, E.; del Fresno, C. Pathophysiology of endotoxin tolerance: Mechanisms and clinical consequences. Crit. Care 2013, 17, 242. [Google Scholar] [CrossRef]
- Randolph, G.J.; Jakubzick, C.; Qu, C. Antigen presentation by monocytes and monocyte-derived cells. Curr. Opin. Immunol. 2008, 20, 52–60. [Google Scholar] [CrossRef]
- Morin, J.; Faideau, B.; Gagnerault, M.; Lepault, F.; Boitard, C.; Boudaly, S. Passive transfer of flt-3L-derived dendritic cells delays diabetes development in NOD mice and associates with early production of interleukin (IL)-4 and IL-10 in the spleen of recipient mice. Clin. Exp. Immunol. 2003, 134, 388–395. [Google Scholar] [CrossRef]
- Kim, N.-S.; Torrez, T.; Langridge, W. LPS enhances CTB-INSULIN induction of IDO1 and IL-10 synthesis in human dendritic cells. Cell. Immunol. 2019, 338, 32–42. [Google Scholar] [CrossRef]
- Klaska, I.P.; Muckersie, E.; Martin-Granados, C.; Christofi, M.; Forrester, J.V. Lipopolysaccharide-primed heterotolerant dendritic cells suppress experimental autoimmune uveoretinitis by multiple mechanisms. Immunology 2016, 150, 364–377. [Google Scholar] [CrossRef]
- Braun-Fahrländer, C.; Riedler, J.; Herz, U.; Eder, W.; Waser, M.; Grize, L.; Maisch, S.; Carr, D.; Gerlach, F.; Bufe, A.; et al. Environmental exposure to endotoxin and its relation to asthma in school-age children. N. Engl. J. Med. 2002, 347, 869–877. [Google Scholar] [CrossRef]
- Bashir, M.E.H.; Louie, S.; Shi, H.N.; Nagler-Anderson, C. Toll-Like receptor 4 signaling by intestinal microbes influences susceptibility to food allergy. J. Immunol. 2004, 172, 6978–6987. [Google Scholar] [CrossRef]
- Vatanen, T.; Kostic, A.D.; d’Hennezel, E.; Siljander, H.; Franzosa, E.A.; Yassour, M.; Kolde, R.; Vlamakis, H.; Arthur, T.D.; Hämäläinen, A.M.; et al. Variation in Microbiome LPS Immunogenicity Contributes to Autoimmunity in Humans. Cell 2016, 165, 1551. [Google Scholar] [CrossRef]
- Mbongue, J.C.; Nicholas, D.A.; Zhang, K.; Kim, N.S.; Hamilton, B.N.; Larios, M.; Zhang, G.; Umezawa, K.; Firek, A.F.; Langridge, W.H. Induction of indoleamine 2, 3-dioxygenase in human dendritic cells by a cholera toxin B subunit-proinsulin vaccine. PLoS ONE 2015, 10, e0118562. [Google Scholar] [CrossRef]
- Kim, N.-S.; Mbongue, J.C.; Nicholas, D.A.; Esebanmen, G.E.; Unternaehrer, J.J.; Firek, A.F.; Langridge, W.H.R. Chimeric Vaccine Stimulation of Human Dendritic Cell Indoleamine 2, 3-Dioxygenase Occurs via the Non-Canonical NF-κB Pathway. PLoS ONE 2016, 11, e0147509. [Google Scholar] [CrossRef]
- Zariri, A.; van der Ley, P. Biosynthetically engineered lipopolysaccharide as vaccine adjuvant. Expert Rev. Vaccines 2015, 14, 861–876. [Google Scholar] [CrossRef]
- Chilton, P.M.; Hadel, D.M.; To, T.T.; Mitchell, T.C.; Darveau, R.P. Adjuvant activity of naturally occurring monophosphoryl lipopolysaccharide preparations from mucosa-associated bacteria. Infect. Immun. 2013, 81, 3317–3325. [Google Scholar] [CrossRef]
- Raetz, C.R.; Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef]
- Gaspar, J.A.; Thomas, J.A.; Marolda, C.L.; Valvano, M.A. Surface expression of O-specific lipopolysaccharide in Escherichia coli requires the function of the TolA protein. Mol. Microbiol. 2000, 38, 262–275. [Google Scholar] [CrossRef]
- Whitfield, C. Biosynthesis and Assembly of Capsular Polysaccharides in Escherichia coli. Annu. Rev. Biochem. 2006, 75, 39–68. [Google Scholar] [CrossRef]
- Vinés, E.D.; Marolda, C.L.; Balachandran, A.; Valvano, M.A. Defective O-Antigen polymerization in tolA and pal mutants of Escherichia coli in response to Extracytoplasmic stress. J. Bacteriol. 2005, 187, 3359–3368. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Diamond, G. The role of cationic antimicrobial peptides in innate host defences. Trends Microbiol. 2000, 8, 402–410. [Google Scholar] [CrossRef]
- Papo, N.; Shai, Y. A molecular mechanism for lipopolysaccharide protection of gram-negative bacteria from antimicrobial peptides. J. Biol. Chem. 2005, 280, 10378–10387. [Google Scholar] [CrossRef]
- Surapaneni, K.M.; Vishnu Priya, V.; Mallika, J. Effect of pioglitazone, quercetin, and hydroxy citric acid on vascular endothelial growth factor messenger RNA (VEGF mRNA) expression in experimentally induced nonalcoholic steatohepatitis (NASH). Turk. J. Med. Sci. 2015, 45, 542–546. [Google Scholar] [PubMed]
- Nitkin, C.R.; Xia, S.; Menden, H.; Yu, W.; Xiong, M.; Heruth, D.P.; Ye, S.Q.; Sampath, V. FOSL1 is a novel mediator of endotoxin/lipopolysaccharide-induced pulmonary angiogenic signaling. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Mamat, U.; Wilke, K.; Bramhill, D.; Schromm, A.B.; Lindner, B.; Kohl, T.A.; Corchero, J.L.; Villaverde, A.; Schaffer, L.; Head, S.R.; et al. Detoxifying Escherichia coli for endotoxin-free production of recombinant proteins. Microb. Cell Factories 2015, 14, 57. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.A. The immune response of Drosophila. Nature 2003, 426, 33–38. [Google Scholar] [CrossRef]
- Royet, J. Drosophila melanogaster innate immunity: An emerging role for peptidoglycan recognition proteins in bacteria detection. Experientia 2004, 61, 537–546. [Google Scholar] [CrossRef]
- Hultmark, D. Immune reactions in Drosophila and other insects: A model for innate immunity. Trends Genet. 1993, 9, 178–183. [Google Scholar] [CrossRef]
- Lackie, A. Immune mechanisms in insects. Parasitol. Today 1988, 4, 98–105. [Google Scholar] [CrossRef]
- Cociancich, S.; Bulet, P.; Hetru, C.; Hoffmann, J. The inducible antibacterial peptides of insects. Parasitol. Today 1994, 10, 132–139. [Google Scholar] [CrossRef]
- Cociancich, S.; Dupont, A.; Hegy, G.; Lanot, R.; Holder, F.; Hetru, C.; Hoffmann, J.A.; Bulet, P. Novel inducible antibacterial peptides from a hemipteran insect, the sap-sucking bug Pyrrhocoris apterus. Biochem. J. 1994, 300, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, N.; Imamura, M.; Kadotani, T.; Yaoi, K.; Iwahana, H.; Sato, R. The lipopolysaccharide-binding protein participating in hemocyte nodule formation in the silkworm Bombyx mori is a novel member of the C-type lectin superfamily with two different tandem carbohydrate-recognition domains. FEBS Lett. 1999, 443, 139–143. [Google Scholar] [CrossRef]
- Wittwer, D.; Weise, C.; Götz, P.; Wiesner, A. LPS (Lipopolysaccharide)-activated immune responses in a hemocyte cell line from Estigmene acraea (Lepidoptera). Dev. Comp. Immunol. 1997, 21, 323–336. [Google Scholar] [CrossRef]
- Kato, Y.; Motoi, Y.; Taniai, K.; Kadono-Okuda, K.; Yamamoto, M.; Higashino, Y.; Shimabukuro, M.; Chowdhury, S.; Xu, J.; Sugiyama, M.; et al. Lipopolysaccharide-lipophorin complex formation in insect hemolymph: A common pathway of lipopolysaccharide detoxification both in insects and in mammals. Insect Biochem. Mol. Biol. 1994, 24, 547–555. [Google Scholar] [CrossRef]
- Kawabata, S.-I.; Nagayama, R.; Hirata, M.; Shigenaga, T.; Agarwala, K.L.; Saito, T.; Cho, J.; Nakajima, H.; Takagi, T.; Iwanaga, S. Tachycitin, a Small Granular Component in Horseshoe Crab Hemocytes, Is an Antimicrobial Protein with Chitin-Binding Activity. J. Biochem. 1996, 120, 1253–1260. [Google Scholar] [CrossRef]
- Kawabata, S.-I.; Saeki, K.; Iwanaga, S. Limulus kexin: A new type of Kex2-like endoprotease specifically expressed in hemocytes of the horseshoe crab. FEBS Lett. 1996, 386, 201–204. [Google Scholar] [CrossRef][Green Version]
- Kawabata, S.-I.; Tokunaga, F.; Kugi, Y.; Motoyama, S.; Miura, Y.; Hirata, M.; Iwanaga, S. Limulus factor D, a 43-kDa protein isolated from horseshoe crab hemocytes, is a serine protease homologue with antimicrobial activity. FEBS Lett. 1996, 398, 146–150. [Google Scholar] [CrossRef]
- Xu, W.-H.; Sato, Y.; Ikeda, M.; Yamashita, O. Molecular characterization of the gene encoding the precursor protein of diapause hormone and pheromone biosynthesis activating neuropeptide (DH-PBAN) of the silkworm, Bombyx mori and its distribution in some insects. Biochim. Biophys. Acta (BBA) Gene Struct. Expr. 1995, 1261, 83–89. [Google Scholar] [CrossRef]
- Xu, W.-H.; Sato, Y.; Ikeda, M.; Yamashita, O. Stage-dependent and temperature-controlled expression of the gene encoding the precursor protein of diapause hormone and pheromone biosynthesis activating neuropeptide in the silkworm, bombyx mori. J. Biol. Chem. 1995, 270, 3804–3808. [Google Scholar] [CrossRef]
- Sugiyama, M.; Kuniyoshi, H.; Kotani, E.; Taniai, K.; Kadono-Okuda, K.; Kato, Y.; Yamamoto, M.; Shimabukuro, M.; Chowdhury, S.; Xu, J.; et al. Characterization of a Bombyx mori cDNA encoding a novel member of the attacin family of insect antibacterial proteins. Insect Biochem. Mol. Biol. 1995, 25, 385–392. [Google Scholar] [CrossRef]
- Takeda, K.; Akira, S. Toll-like receptors in innate immunity. Int. Immunol. 2005, 17, 1–14. [Google Scholar] [CrossRef]
- Kang, D.; Liu, G.; Lundström, A.; Gelius, E.; Steiner, H. A peptidoglycan recognition protein in innate immunity conserved from insects to humans. Proc. Natl. Acad. Sci. USA 1998, 95, 10078–10082. [Google Scholar] [CrossRef] [PubMed]
- Fabrick, J.; Baker, J.; Kanost, M. cDNA cloning, purification, properties, and function of a β-1,3-glucan recognition protein from a pyralid moth, Plodiainterpunctella. Insect Biochem. Mol. Biol. 2003, 33, 579–594. [Google Scholar] [CrossRef]
- Ma, C.; Kanost, M. A β1,3-Glucan Recognition protein from an insect, manduca sexta, agglutinates microorganisms and activates the Phenoloxidase cascade. J. Biol. Chem. 2000, 275, 7505–7514. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, M.; Ashida, M. Purification of a beta-1,3-glucan recognition protein in the prophenoloxidase activating system from hemolymph of the silkworm, Bombyx mori. J. Biol. Chem. 1988, 263, 12056–12062. [Google Scholar] [CrossRef]
- Ashida, M.; Ochiai, M.; Niki, T. Immunolocalization of prophenoloxidase among hemocytes of the silkworm, Bombyx mori. Tissue Cell 1988, 20, 599–610. [Google Scholar] [CrossRef]
- Dimopoulos, G.; Richman, A.; Müller, H.M.; Kafatos, F.C. Molecular immune responses of the mosquito Anopheles gambiae to bacteria and malaria parasites. Proc. Natl. Acad. Sci. USA 1997, 94, 11508–11513. [Google Scholar] [CrossRef]
- Dimopoulos, G.; Seeley, D.; Wolf, A.; Kafatos, F.C. Malaria infection of the mosquito Anopheles gambiae activates immune-responsive genes during critical transition stages of the parasite life cycle. EMBO J. 1998, 17, 6115–6123. [Google Scholar] [CrossRef]
- Yeh, M.-S.; Lai, C.-Y.; Liu, C.-H.; Kuo, C.-M.; Cheng, W. A second proPO present in white shrimp Litopenaeus vannamei and expression of the proPOs during a Vibrio alginolyticus injection, molt stage, and oral sodium alginate ingestion. Fish Shellfish Immunol. 2009, 26, 49–55. [Google Scholar] [CrossRef]
- Yeh, M.-S.; Liu, C.-H.; Hung, C.-W.; Cheng, W. cDNA cloning, identification, tissue localisation, and transcription profile of a transglutaminase from white shrimp, Litopenaeus vannamei, after infection by Vibrio alginolyticus. Fish Shellfish Immunol. 2009, 27, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Litman, G.W.; Rast, J.P.; Fugmann, S.D. The origins of vertebrate adaptive immunity. Nat. Rev. Immunol. 2010, 10, 543–553. [Google Scholar] [CrossRef]
- Miller, J.S.; Nguyen, T.; Stanley-Samuelson, D.W. Eicosanoids mediate insect nodulation responses to bacterial infections. Proc. Natl. Acad. Sci. USA 1994, 91, 12418–12422. [Google Scholar] [CrossRef] [PubMed]
- Jomori, T.; Natori, S. Function of the lipopolysaccharide-binding protein of Periplaneta americana as an opsonin. FEBS Lett 1992, 296, 283–286. [Google Scholar] [CrossRef]
- Shigenaga, T.; Takayenoki, Y.; Kawasaki, S.; Seki, N.; Muta, T.; Toh, Y.; Ito, A.; Iwanaga, S. Separation of large and small granules from horseshoe crab (Tachypleus tridentatus) hemocytes and characterization of their components1. J. Biochem. 1993, 114, 307–316. [Google Scholar] [CrossRef]
- Marmaras, V.J.; Charalambidis, N.D.; Zervas, C. Immune response in insects: The role of phenoloxidase in defense reactions in relation to melanization and sclerotization. Arch. Insect Biochem. Physiol. 1996, 31, 119–133. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef]
- Amir, I.; Konikoff, F.M.; Oppenheim, M.; Gophna, U.; Half, E.E. Gastric microbiota is altered in oesophagitis and Barrett’s oesophagus and further modified by proton pump inhibitors. Env. Microbiol. 2014, 16, 2905–2914. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Pussinen, P.J.; Havulinna, A.S.; Lehto, M.; Sundvall, J.; Salomaa, V. Endotoxemia is associated with an increased risk of incident diabetes. Diabetes Care 2011, 34, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Candelli, M.; Franza, L.; Pignataro, G.; Ojetti, V.; Covino, M.; Piccioni, A.; Gasbarrini, A.; Franceschi, F. Interaction between Lipopolysaccharide and Gut Microbiota in Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2021, 22, 6242. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Hussey, S.E.; Sanchez-Avila, A.; Tantiwong, P.; Musi, N. Effect of lipopolysaccharide on inflammation and insulin action in human muscle. PLoS ONE 2013, 8, e63983. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Willment, A.; Patel, S.S.; Sun, X.; Song, M.; Mannery, Y.O.; Kosters, A.; McClain, C.J.; Vos, M.B. Fructose induced endotoxemia in pediatric nonalcoholic fatty liver disease. Int. J. Hepatol. 2014, 2014, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ahola, A.J.; Lassenius, M.I.; Forsblom, C.; Harjutsalo, V.; Lehto, M.; Groop, P.-H. Dietary patterns reflecting healthy food choices are associated with lower serum LPS activity. Sci. Rep. 2017, 7, 6511. [Google Scholar] [CrossRef] [PubMed]
- Fraser, G.E.; Cosgrove, C.M.; Mashchak, A.; Orlich, M.J.; Altekruse, S.F.; Mph, C.M.C.; Ms, A.D.M. Lower rates of cancer and all-cause mortality in an Adventist cohort compared with a US Census population. Cancer 2019, 126, 1102–1111. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.A.; Holscher, H.D. Microbiome-Mediated Effects of the Mediterranean Diet on Inflammation. Adv. Nutr. 2018, 9, 193–206. [Google Scholar] [CrossRef]
- Dong, Z.; Yuan, Y. Accelerated inflammation and oxidative stress induced by LPS in acute lung injury: Ιnhibition by ST1926. Int. J. Mol. Med. 2018, 41, 3405–3421. [Google Scholar] [CrossRef]
- Martí-Lliteras, P.; Regueiro, V.; Morey, P.; Hood, D.W.; Saus, C.; Sauleda, J.; Agustí, A.G.N.; Bengoechea, J.A.; Garmendia, J. Nontypeable Haemophilus influenzae Clearance by Alveolar Macrophages Is Impaired by Exposure to Cigarette Smoke. Infect. Immun. 2009, 77, 4232–4242. [Google Scholar] [CrossRef]
- Lugade, A.A.; Bogner, P.N.; Murphy, T.F.; Ethanavala, Y. The Role of TLR2 and Bacterial Lipoprotein in Enhancing Airway Inflammation and Immunity. Front. Immunol. 2011, 2, 10. [Google Scholar] [CrossRef]
- McClure, R.; Massari, P. TLR-Dependent Human Mucosal Epithelial Cell Responses to Microbial Pathogens. Front. Immunol. 2014, 5, 386. [Google Scholar] [CrossRef]
- Su, Y.-C.; Jalalvand, F.; Thegerström, J.; Riesbeck, K. The Interplay between Immune Response and Bacterial Infection in COPD: Focus upon Non-typeable Haemophilus influenzae. Front. Immunol. 2018, 9, 2530. [Google Scholar] [CrossRef]
- Leiva-Juárez, M.M.; Kolls, J.K.; Evans, S.E. Lung epithelial cells: Therapeutically inducible effectors of antimicrobial defense. Mucosal Immunol. 2017, 11, 21–34. [Google Scholar] [CrossRef]
- Hughes, B.M.; Burton, C.S.; Reese, A.; Jabeen, M.F.; Wright, C.; Willis, J.; Khoshaein, N.; Marsh, E.K.; Peachell, P.; Sun, S.C.; et al. Pellino-1 Regulates Immune Responses to Haemophilus influenzae in Models of Inflammatory Lung Disease. Front. Immunol. 2019, 10, 1721. [Google Scholar] [CrossRef]
- Rasaei, R.; Sarodaya, N.; Kim, K.-S.; Ramakrishna, S.; Hong, S.-H. Importance of Deubiquitination in Macrophage-Mediated Viral Response and Inflammation. Int. J. Mol. Sci. 2020, 21, 8090. [Google Scholar] [CrossRef]
- Matsui, K.; Tanaka, N.; Nishikawa, A. Lipopolysaccharide of Haemophilus influenzae induces Interleukin-5 mRNA expression in human peripheral blood mononuclear cells. J. Interf. Cytokine Res. 2001, 21, 439–443. [Google Scholar] [CrossRef]
- Øvstebø, R.; Olstad, O.K.; Brusletto, B.; Møller, A.S.; Aase, A.; Haug, K.B.F.; Brandtzaeg, P.; Kierulf, P. Identification of genes particularly sensitive to lipopolysaccharide (LPS) in human monocytes induced by wild-type versus LPS-deficient Neisseria meningitidis strains. Infect. Immun. 2008, 76, 2685–2695. [Google Scholar] [CrossRef]
- Cheng, R.; Liu, W.; Zhang, R.; Feng, Y.; Bhowmick, N.A.; Hu, T. Porphyromonas gingivalis-Derived Lipopolysaccharide Combines Hypoxia to Induce Caspase-1 Activation in Periodontitis. Front. Cell. Infect. Microbiol. 2017, 7, 474. [Google Scholar] [CrossRef]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS activates caspase-11: Implications in TLR4-independent endotoxic shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef]
- Liu, J.; Wang, X.; Zheng, M.; Luan, Q. Lipopolysaccharide from Porphyromonas gingivalis promotes autophagy of human gingival fibroblasts through the PI3K/Akt/mTOR signaling pathway. Life Sci. 2018, 211, 133–139. [Google Scholar] [CrossRef]
- Aquino-Martinez, R.; Rowsey, J.L.; Fraser, D.G.; Eckhardt, B.A.; Khosla, S.; Farr, J.N.; Monroe, D.G. LPS-induced premature osteocyte senescence: Implications in inflammatory alveolar bone loss and periodontal disease pathogenesis. Bone 2020, 132, 115220. [Google Scholar] [CrossRef]
- Blazkova, H.; Krejcikova, K.; Moudry, P.; Frisan, T.; Hodny, Z.; Bartek, J. Bacterial intoxication evokes cellular senescence with persistent DNA damage and cytokine signalling. J. Cell. Mol. Med. 2009, 14, 357–367. [Google Scholar] [CrossRef]
- Feng, X.; Feng, G.; Xing, J.; Shen, B.; Tan, W.; Huang, D.; Lu, X.; Tao, T.; Zhang, J.; Li, L.; et al. Repeated lipopolysaccharide stimulation promotes cellular senescence in human dental pulp stem cells (DPSCs). Cell Tissue Res. 2014, 356, 369–380. [Google Scholar] [CrossRef]
- Kim, C.O.; Huh, A.J.; Han, S.H.; Kim, J.M. Analysis of cellular senescence induced by lipopolysaccharide in pulmonary alveolar epithelial cells. Arch. Gerontol. Geriatr. 2012, 54, e35–e41. [Google Scholar] [CrossRef]
- Guerra, L.; Guidi, R.; Frisan, T. Do bacterial genotoxins contribute to chronic inflammation, genomic instability and tumor progression? FEBS J. 2011, 278, 4577–4588. [Google Scholar] [CrossRef]
- Martin, M.; Katz, J.; Vogel, S.N.; Michalek, S.M. Differential induction of endotoxin tolerance by lipopolysaccharides derived from Porphyromonas gingivalis and Escherichia coli. J. Immunol. 2001, 167, 5278–5285. [Google Scholar] [CrossRef]
- Zhang, J.; Yu, C.; Zhang, X.; Chen, H.; Dong, J.; Lu, W.; Song, Z.; Zhou, W. Porphyromonas gingivalis lipopolysaccharide induces cognitive dysfunction, mediated by neuronal inflammation via activation of the TLR4 signaling pathway in C57BL/6 mice. J. Neuroinflamm. 2018, 15, 1–14. [Google Scholar] [CrossRef]
- Sell, K.M.; Crowe, S.F.; Kent, S. Lipopolysaccharide induces memory-processing deficits in day-old chicks. Pharmacol. Biochem. Behav. 2001, 68, 497–502. [Google Scholar] [CrossRef]
- Sell, K.M.; Crowe, S.F.; Kent, S. Lipopolysaccharide induces biochemical alterations in chicks trained on the passive avoidance learning task. Physiol. Behav. 2003, 78, 679–688. [Google Scholar] [CrossRef]
- Charoensaensuk, V.; Chen, Y.C.; Lin, Y.H.; Ou, K.L.; Yang, L.Y.; Lu, D.Y. Induces Proinflammatory Cytokine Expression Leading to Apoptotic Death through the Oxidative Stress/NF-κB Pathway in Brain Endothelial Cells. Cells 2021, 10, 3033. [Google Scholar] [CrossRef]
- Wang, W.-Y.; Tan, M.-S.; Yu, J.-T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar]
- Tremblay, S.; Miloudi, K.; Chaychi, S.; Favret, S.; Binet, F.; Polosa, A.; Lachapelle, P.; Chemtob, S.; Sapieha, P. Systemic Inflammation Perturbs Developmental Retinal Angiogenesis and Neuroretinal Function. Investig. Opthalmology Vis. Sci. 2013, 54, 8125–8139. [Google Scholar] [CrossRef]
- Noailles, A.; Maneu, V.; Campello, L.; Lax, P.; Cuenca, N. Systemic inflammation induced by lipopolysaccharide aggravates inherited retinal dystrophy. Cell Death Dis. 2018, 9, 1–18. [Google Scholar] [CrossRef]
- Fernández-Sánchez, L.; Esquiva, G.; Pinilla, I.; Lax, P.; Cuenca, N. Retinal Vascular Degeneration in the Transgenic P23H Rat Model of Retinitis Pigmentosa. Front. Neuroanat. 2018, 12, 55. [Google Scholar] [CrossRef]
- Jackson, C.M.; Mukherjee, S.; Wilburn, A.N.; Cates, C.; Lewkowich, I.P.; Deshmukh, H.; Zacharias, W.J.; Chougnet, C.A. Pulmonary Consequences of Prenatal Inflammatory Exposures: Clinical Perspective and Review of Basic Immunological Mechanisms. Front. Immunol. 2020, 11, 1285. [Google Scholar] [CrossRef]
- Muk, T.; Jiang, P.-P.; Stensballe, A.; Skovgaard, K.; Sangild, P.T.; Nguyen, D.N. Prenatal Endotoxin Exposure Induces Fetal and Neonatal Renal Inflammation via Innate and Th1 Immune Activation in Preterm Pigs. Front. Immunol. 2020, 11, 565484. [Google Scholar] [CrossRef]
- Duncan, J.R.; Cock, M.L.; Scheerlinck, J.P.Y.; Westcott, K.T.; McLean, C.; Harding, R.; Rees, S.M. White matter injury after repeated endotoxin exposure in the preterm ovine fetus. Pediatr. Res. 2002, 52, 941–949. [Google Scholar] [CrossRef]
- Mallard, C.; Welin, A.-K.; Peebles, D.; Hagberg, H.; Kjellmer, I. White matter injury following systemic endotoxemia or asphyxia in the fetal sheep. Neurochem. Res. 2003, 28, 215–223. [Google Scholar] [CrossRef]
- Garnier, Y.; Berger, R.; Alm, S.; von Duering, M.U.; Coumans, A.B.; Michetti, F.; Bruschettini, M.; Lituania, M.; Hasaart, T.H.; Gazzolo, D. Systemic endotoxin administration results in increased S100B protein blood levels and periventricular brain white matter injury in the preterm fetal sheep. Eur. J. Obstet. Gynecol. Reprod. Biol. 2006, 124, 15–22. [Google Scholar] [CrossRef]
- Garnier, Y.; Frigiola, A.; Volti, G.L.; Florio, P.; Frulio, R.; Berger, R.; Alm, S.; von Duering, M.U.; Coumans, A.B.C.; Reis, F.M.; et al. Increased maternal/fetal blood S100B levels following systemic endotoxin administration and periventricular white matter injury in preterm fetal sheep. Reprod. Sci. 2009, 16, 758–766. [Google Scholar] [CrossRef]
- Coumans, A.B.C.; Middelanis, J.; Garnier, Y.; Vaihinger, H.-M.; Leib, S.L.; Von Duering, M.U.; Hasaart, T.H.M.; Jensen, A.; Berger, R. Intracisternal application of endotoxin enhances the susceptibility to subsequent hypoxic-ischemic brain damage in neonatal rats. Pediatr. Res. 2003, 53, 770–775. [Google Scholar] [CrossRef]
- Lehnardt, S.; Lachance, C.; Patrizi, S.; Lefebvre, S.; Follett, P.L.; Jensen, F.E.; Rosenberg, P.; Volpe, J.J.; Vartanian, T. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J. Neurosci. 2002, 22, 2478–2486. [Google Scholar] [CrossRef]
- Cai, Z.; Pang, Y.; Lin, S.; Rhodes, P.G. Differential roles of tumor necrosis factor-alpha and interleukin-1 beta in lipopolysaccharide-induced brain injury in the neonatal rat. Brain Res. 2003, 975, 37–47. [Google Scholar] [CrossRef]
- Oskvig, D.B.; Elkahloun, A.G.; Johnson, K.R.; Phillips, T.M.; Herkenham, M. Maternal immune activation by LPS selectively alters specific gene expression profiles of interneuron migration and oxidative stress in the fetus without triggering a fetal immune response. Brain Behav. Immun. 2012, 26, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Manni, G.; Mondanelli, G.; Scalisi, G.; Pallotta, M.T.; Nardi, D.; Padiglioni, E.; Romani, R.; Talesa, V.N.; Puccetti, P.; Fallarino, F.; et al. Pharmacologic Induction of Endotoxin Tolerance in Dendritic Cells by L-Kynurenine. Front. Immunol. 2020, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Salazar, F.; Awuah, D.; Negm, O.H.; Shakib, F.; Ghaemmaghami, A. The role of indoleamine 2,3-dioxygenase-aryl hydrocarbon receptor pathway in the TLR4-induced tolerogenic phenotype in human DCs. Sci. Rep. 2017, 7, srep43337. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, L. The p50-homodimer mechanism in tolerance to LPS. J. Endotoxin Res. 2001, 7, 219–222. [Google Scholar] [CrossRef]
- Chen, X.; El Gazzar, M.; Yoza, B.K.; McCall, C.E. The NF-kappaB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance. J. Biol. Chem. 2009, 284, 27857–27865. [Google Scholar] [CrossRef]
- Xiong, Y.; Medvedev, A.E. Induction of endotoxin tolerance in vivo inhibits activation of IRAK4 and increases negative regulators IRAK-M, SHIP-1, and A20. J. Leukoc. Biol. 2011, 90, 1141–1148. [Google Scholar] [CrossRef]
- Van‘t Veer, C.; van den Pangaart, P.S.; van Zoelen, M.A.; de Kruif, M.; Birjmohun, R.S.; Stroes, E.S. Induction of IRAK-M is associated with lipopolysaccharide tolerance in a human endotoxemia model. J. Immunol. 2007, 179, 7110–7120. [Google Scholar] [CrossRef]
- Wiersinga, W.J.; Veer, C.V.; Pangaart, P.S.V.D.; Dondorp, A.M.; Day, N.P.; Peacock, S.J.; Van Der Poll, T. Immunosuppression associated with interleukin-1R-associated-kinase-M upregulation predicts mortality in Gram-negative sepsis (melioidosis). Crit. Care Med. 2009, 37, 569–576. [Google Scholar] [CrossRef]
- Kobayashi, K.; Hernandez, L.D.; Galán, J.E.; Janeway, C.A.; Medzhitov, R.; Flavell, R.A. IRAK-M Is a negative regulator of toll-like receptor signaling. Cell 2002, 110, 191–202. [Google Scholar] [CrossRef]
- Seeley, J.J.; Ghosh, S. Molecular mechanisms of innate memory and tolerance to LPS. J. Leukoc. Biol. 2016, 101, 107–119. [Google Scholar] [CrossRef]
- Draisma, A.; Pickkers, P.; Bouw, M.P.; van der Hoeven, J.G. Development of endotoxin tolerance in humans in vivo. Crit. Care Med. 2009, 37, 1261–1267. [Google Scholar] [CrossRef]
- Pachot, A.; Lepape, A.; Vey, S.; Bienvenu, J.; Mougin, B.; Monneret, G. Systemic transcriptional analysis in survivor and non-survivor septic shock patients: A preliminary study. Immunol. Lett. 2006, 106, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Sly, L.M.; Rauh, M.J.; Kalesnikoff, J.; Song, C.H.; Krystal, G. LPS-Induced upregulation of SHIP is essential for endotoxin tolerance. Immunity 2004, 21, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Rauh, M.J.; Ho, V.; Pereira, C.; Sham, A.; Sly, L.M.; Lam, V.; Huxham, L.; Minchinton, A.I.; Mui, A.; Krystal, G. SHIP represses the generation of alternatively activated macrophages. Immunity 2005, 23, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Esaba, R.; Sorensen, D.L.; Booth, S.A. MicroRNA-146a: A Dominant, Negative Regulator of the Innate Immune Response. Front. Immunol. 2014, 5, 578. [Google Scholar] [CrossRef]
- Quinn, E.M.; Wang, J.; Redmond, H.P. The emerging role of microRNA in regulation of endotoxin tolerance. J. Leukoc. Biol. 2012, 91, 721–727. [Google Scholar] [CrossRef]
- Nahid, M.A.; Satoh, M.; Chan, E.K.L. Mechanistic role of MicroRNA-146a in endotoxin-induced differential cross-regulation of TLR signaling. J. Immunol. 2010, 186, 1723–1734. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, Q.; Song, Y.; Lai, L.; Wang, J.; Yu, H.; Cao, X.; Wang, Q. MicroRNA-98 negatively regulates IL-10 production and endotoxin tolerance in macrophages after LPS stimulation. FEBS Lett. 2011, 585, 1963–1968. [Google Scholar] [CrossRef]
- El Gazzar, M.; McCall, C.E. MicroRNAs distinguish translational from transcriptional silencing during endotoxin tolerance. J. Biol. Chem. 2010, 285, 20940–20951. [Google Scholar] [CrossRef] [PubMed]
- Androulidaki, A.; Iliopoulos, D.; Arranz, A.; Doxaki, C.; Schworer, S.; Zacharioudaki, V.; Margioris, A.N.; Tsichlis, P.N.; Tsatsanis, C. The Kinase Akt1 Controls Macrophage Response to Lipopolysaccharide by Regulating MicroRNAs. Immunity 2009, 31, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Foster, S.L.; Hargreaves, D.C.; Medzhitov, R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 2007, 447, 972–978. [Google Scholar] [CrossRef] [PubMed]
- El Gazzar, M.; Yoza, B.K.; Hu, J.Y.-Q.; Cousart, S.L.; McCall, C.E. Epigenetic Silencing of tumor necrosis factor α during endotoxin tolerance. J. Biol. Chem. 2007, 282, 26857–26864. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhong, Y.; Fu, X.; Wang, Y.; Ye, P.; Cai, J.; Liu, Y.; Sun, J.; Mei, Z.; Jiang, Y.; et al. H3K4 Methylation Regulates LPS-Induced Proinflammatory Cytokine Expression and Release in Macrophages. Shock 2019, 51, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-L.; Shu, C.-C.; Chen, Y.-M.; Lu, J.-J.; Wu, T.-S.; Lai, W.-F.; Tzeng, C.-M.; Lai, H.-C.; Lu, C.-C. Like Cures Like: Pharmacological Activity of Anti-Inflammatory Lipopolysaccharides from Gut Microbiome. Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- D’Hennezel, E.; Abubucker, S.; Murphy, L.O.; Cullen, T.W. Total Lipopolysaccharide from the Human Gut Microbiome Silences Toll-Like Receptor Signaling. mSystems 2017, 2, e00046-17. [Google Scholar] [CrossRef] [PubMed]
- Coats, S.R.; Do, C.T.; Karimi-Naser, L.M.; Braham, P.H.; Darveau, R.P. Antagonistic lipopolysaccharides block E. coli lipopolysaccharide function at human TLR4 via interaction with the human MD-2 lipopolysaccharide binding site. Cell Microbiol. 2007, 9, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Langridge, W.; Dénes, B.; Fodor, I. Cholera toxin B subunit modulation of mucosal vaccines for infectious and autoimmune diseases. Curr. Opin. Investig. Drugs 2010, 11, 919–928. [Google Scholar]
- Lavelle, E.C.; Ward, R.W. Mucosal vaccines—fortifying the frontiers. Nat. Rev. Immunol. 2021, 22, 236–250. [Google Scholar] [CrossRef]
- Arakawa, T.; Chong, D.K.; Langridge, W.H.R. Efficacy of a food plant-based oral cholera toxin B subunit vaccine. Nat. Biotechnol. 1998, 16, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.E.; Yu, J.; Choi, N.-W.; Hough, J.; Henderson, D.; He, D.; Langridge, W.H.R. Bacterial and plant enterotoxin B subunit–autoantigen fusion proteins suppress diabetes insulitis. Mol. Biotechnol. 2006, 32, 001–016. [Google Scholar] [CrossRef]
- Odumosu, O.; Nicholas, D.; Payne, K.; Langridge, W. Cholera toxin B subunit linked to glutamic acid decarboxylase suppresses dendritic cell maturation and function. Vaccine 2011, 29, 8451–8458. [Google Scholar] [CrossRef]
- Odumosu, O.; Payne, K.; Baez, I.; Jutzy, J.; Wall, N.; Langridge, W. Suppression of dendritic cell activation by diabetes autoantigens linked to the cholera toxin B subunit. Immunobiology 2011, 216, 447–456. [Google Scholar] [CrossRef]
- Atkinson, M.A. Losing a grip on the notion of β-Cell specificity for immune responses in type 1 diabetes: Can we handle the truth? Diabetes 2014, 63, 3572–3574. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef]
- Aspord, C.; Thivolet, C. Nasal administration of CTB-insulin induces active tolerance against autoimmune diabetes in non-obese diabetic (NOD) mice. Clin. Exp. Immunol. 2002, 130, 204–211. [Google Scholar] [CrossRef]
- Dénes, B.; Fodor, I.; Langridge, W.H. Autoantigens plus interleukin-10 suppress diabetes autoimmunity. Diabetes Technol. Ther. 2010, 12, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Ploix, C.; Bergerot, I.; Durand, A.; Czerkinsky, C.; Holmgren, J.; Thivolet, C. Oral administration of cholera toxin B-insulin conjugates protects NOD mice from autoimmune diabetes by inducing CD4+ regulatory T-cells. Diabetes 1999, 48, 2150–2156. [Google Scholar] [CrossRef] [PubMed]
- Ferris, S.T.; Carrero, J.A.; Mohan, J.F.; Calderon, B.; Murphy, K.M.; Unanue, E.R. A minor subset of batf3-dependent antigen-presenting cells in islets of langerhans is essential for the development of autoimmune diabetes. Immunity 2014, 41, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Chilton, P.M.; Rezzoug, F.; Fugier-Vivier, I.; Weeter, L.A.; Xu, H.; Huang, Y.; Ray, M.B.; Ildstad, S.T. Flt3-Ligand treatment prevents diabetes in NOD mice. Diabetes 2004, 53, 1995–2002. [Google Scholar] [CrossRef][Green Version]
- Sun, J.-B.; Czerkinsky, C.; Holmgren, J. Mucosally induced immunological tolerance, regulatory T cells and the adjuvant effect by cholera toxin B subunit. Scand. J. Immunol. 2010, 71, 1–11. [Google Scholar] [CrossRef]
- Shinomiya, M.; Nadano, S.; Shinomiya, H.; Onji, M. In situ characterization of dendritic cells occurring in the islets of nonobese diabetic mice during the development of insulitis. Pancreas 2000, 20, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, J.; Harandi, A.M.; Czerkinsky, C. Mucosal adjuvants and anti-infection and anti-immunopathology vaccines based on cholera toxin, cholera toxin B subunit and CpG DNA. Expert Rev. Vaccines 2003, 2, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Esebanmen, G.E.; Langridge, W.H.R. Mechanism of chimeric vaccine stimulation of indoleamine 2,3-dioxygenase biosynthesis in human dendritic cells is independent of TGF-β signaling. Cell Immunol. 2017, 319, 43–52. [Google Scholar] [CrossRef] [PubMed]
- León, B.; López-Bravo, M.; Ardavín, C. Monocyte-derived dendritic cells. Semin. Immunol. 2005, 17, 313–318. [Google Scholar] [CrossRef]
- León, B.; Ardavín, C. Monocyte-derived dendritic cells in innate and adaptive immunity. Immunol. Cell Biol. 2008, 86, 320–324. [Google Scholar] [CrossRef]
- Ludovini, V.; Bianconi, F.; Siggillino, A.; Vannucci, J.; Baglivo, S.; Berti, V.; Tofanetti, F.R.; Reda, M.S.; Bellezza, G.; Mandarano, M. High PD-L1/IDO-2 and PD-L2/IDO-1 Co-Expression Levels Are Associated with Worse Overall Survival in Resected Non-Small Cell Lung Cancer Patients. Genes 2021, 12, 273. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mbongue, J.C.; Vanterpool, E.; Firek, A.; Langridge, W.H.R. Lipopolysaccharide-Induced Immunological Tolerance in Monocyte-Derived Dendritic Cells. Immuno 2022, 2, 482-500. https://doi.org/10.3390/immuno2030030
Mbongue JC, Vanterpool E, Firek A, Langridge WHR. Lipopolysaccharide-Induced Immunological Tolerance in Monocyte-Derived Dendritic Cells. Immuno. 2022; 2(3):482-500. https://doi.org/10.3390/immuno2030030
Chicago/Turabian StyleMbongue, Jacques C., Elaine Vanterpool, Anthony Firek, and William H. R. Langridge. 2022. "Lipopolysaccharide-Induced Immunological Tolerance in Monocyte-Derived Dendritic Cells" Immuno 2, no. 3: 482-500. https://doi.org/10.3390/immuno2030030
APA StyleMbongue, J. C., Vanterpool, E., Firek, A., & Langridge, W. H. R. (2022). Lipopolysaccharide-Induced Immunological Tolerance in Monocyte-Derived Dendritic Cells. Immuno, 2(3), 482-500. https://doi.org/10.3390/immuno2030030