
Molecular Testing and Treatment Strategies in RET-Rearranged NSCLC Patients: Stay on Target to Look Forward



Abstract
:1. Introduction
2. Molecular Pathway
3. The Available Techniques to Detect RET Rearrangements
4. Improving Patients Care: Mistakes in the Past and Adjustments for the Future

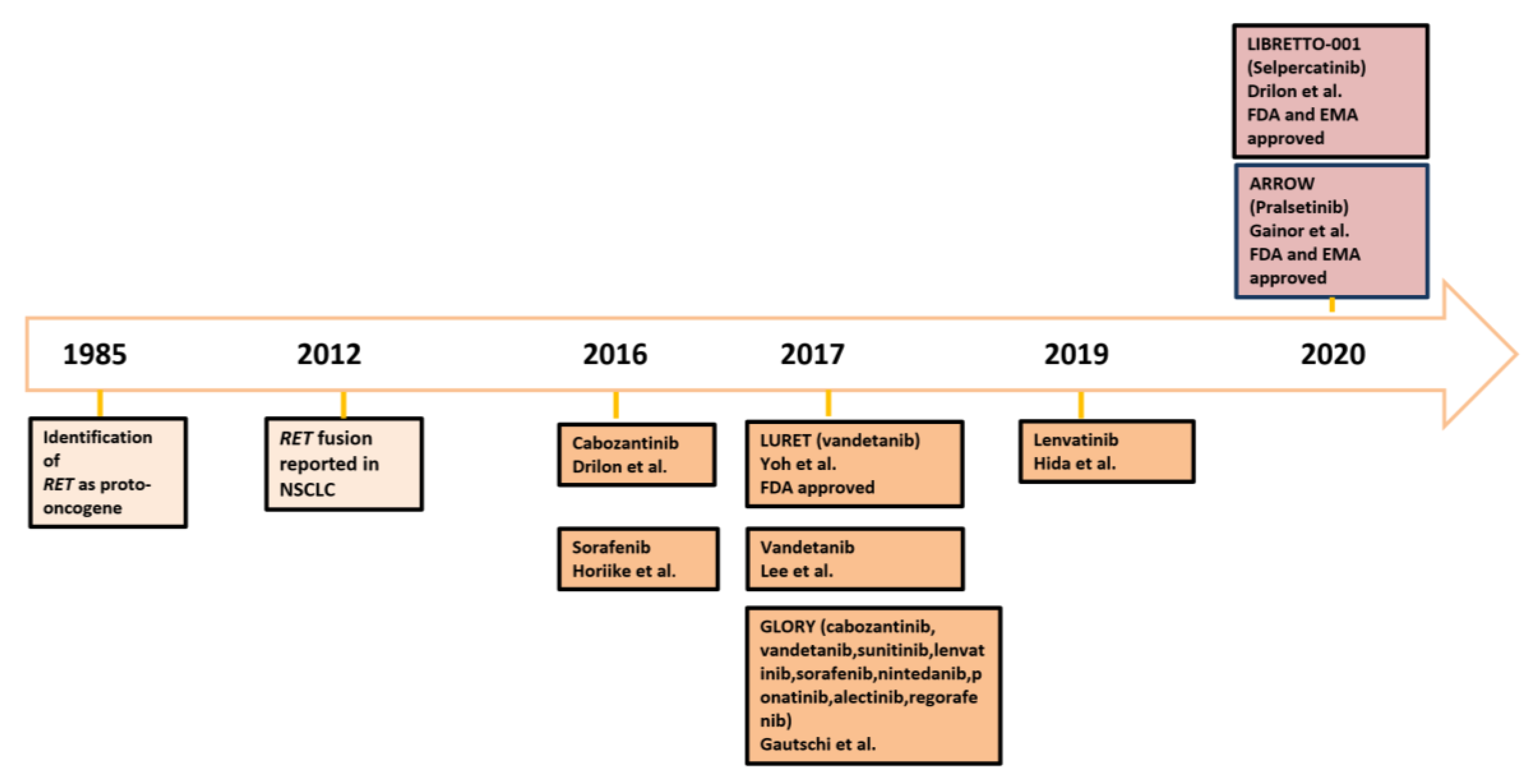
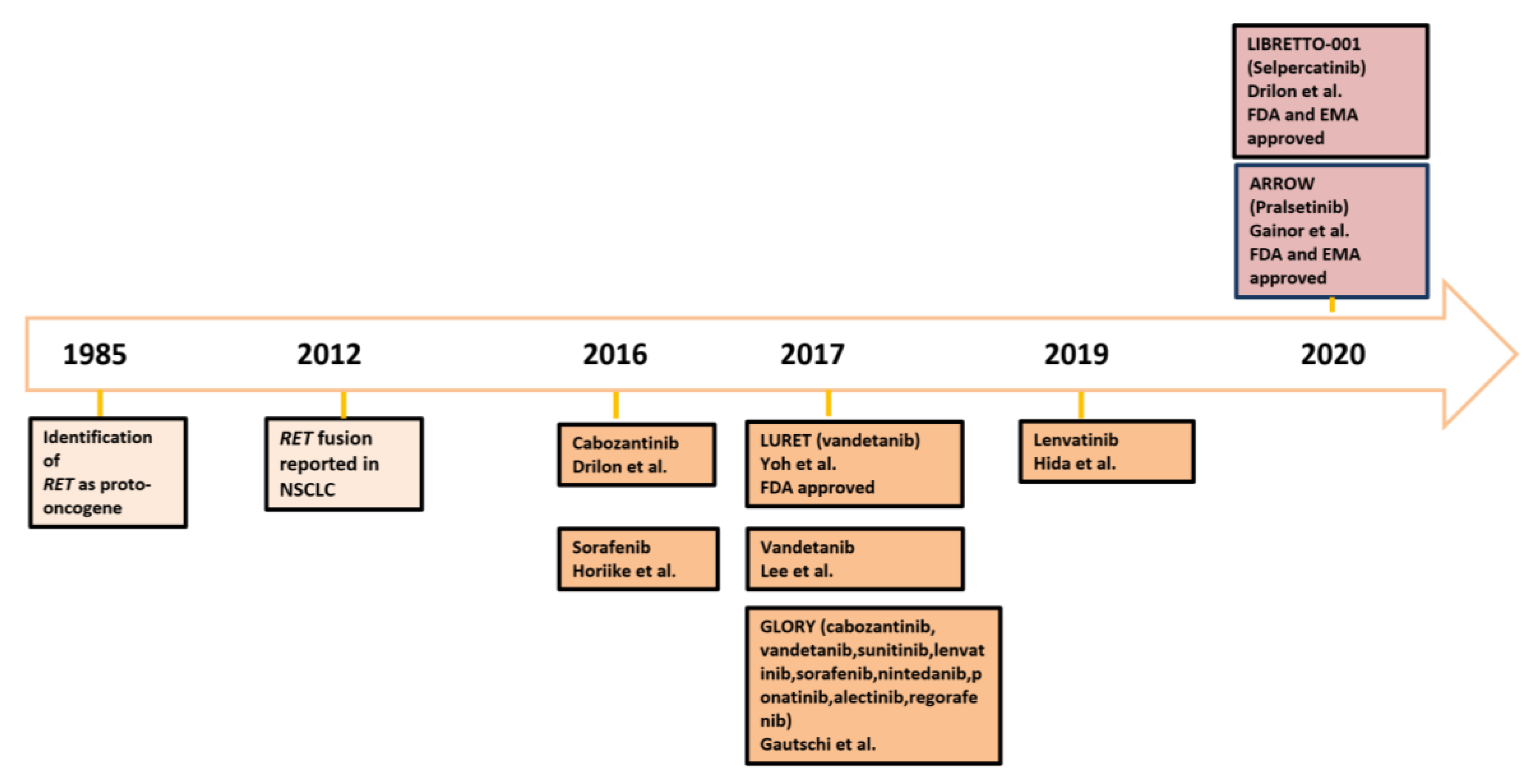{kind=link}
| Drug | First Author, Year | Phase | Number of Patients Enrolled | ORR (%) | Median PFS (Months) | Median OS (Months) | Adverse Events Grade ≥3 |
|---|---|---|---|---|---|---|---|
| Cabozantinib | Drilon, 2016 [45] | II | 26 | 28% (12–49) | 5.5 (3.8–8.4) | 9.9 (8.1–NR) | 47% |
| Gautschi, 2017 [42] | retrospective | 21 | 37% (16.3–61.6) | 3.6 (1.3–7.0) | 4.9 (1.9–14.3) | NA | |
| Lenvatinib | Hida, 2019 [47] | II | 25 | 16% (4.5–36.1) | 7.3 (3.6–10.2) | NA | 92% |
| Gautschi, 2017 [42] | retrospective | 2 | 50% | NA | NA | NA | |
| Vandetanib | Lee, 2017 [50] | II | 18 | 18% | 4.5 | 11.6 | 28% |
| Yoh, 2017 [49] | II | 19 | 53% (28–77) | 4.7 (2.8–8.5) | 11.1 (9.4–NR) | ||
| Gautschi, 2017 [42] | retrospective | 11 | 18% (2.3–51.8) | 2.9 (1.0–6.4) | 10.2 (2.4–NR) | NA | |
| Sorafenib | Horiike, 2016 [43] | II | 3 | 0% | NA | NA | 33% |
| Gautschi, 2017 [42] | retrospective | 2 | 0% | NA | NA | NA | |
| Sunitinib | Gautschi, 2017 [42] | retrospective | 10 | 22% (2.8–60) | 2.2 (0.7–5.0) | 6.8 (1.1–NR) | NA |
| Nintedanib | Gautschi, 2017 [42] | retrospective | 2 | 50% | NA | NA | NA |
| Ponatinib | Gautschi, 2017 [42] | retrospective | 2 | 0% | NA | NA | NA |
| Alectinib | Gautschi, 2017 [42] | retrospective | 2 | 0% | NA | NA | NA |
| Regorafenib | Gautschi, 2017 [42] | retrospective | 1 | 0% | NA | NA | NA |
| Drug | First Author, Year | Phase | Number of Patients | ORR | Intracranial RR | Median Intracranial PFS (Months) | Median DOR (Months) | Median PFS (Months) | Median OS (Months) | Adverse Events Grade ≥3 |
|---|---|---|---|---|---|---|---|---|---|---|
| Selpercatinib | Drilon, 2020 [32] | I/II | 105 pretreated with platinum chemotherapy 39 untreated patients | 64% (54–73) 85% (70–94) | 91% (59–100) (11 patients) | 13.7 (10.9–NE) | 17.5 (12.0–NE) NE (12.0–NE) | 16.5 (13.7–NE) NE (13.8–NE) | NR NR | 28% |
| Pralsetinib | Gainor 2021 [52] | I/II | 92 pretreated with platinum chemotherapy 29 untreated patients | 61% (50–71) 70% (50–86) | 56% (21–86) (9 patients) | NR 9 | 17.1 (8.3–22.1) 9.1 (6.1–13) | NR NR | 48% |
5. Facing Acquired Resistance
6. Future Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Kris, M.G.; Johnson, B.E.; Berry, L.D.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Varella-Garcia, M.; Franklin, W.A.; Aronson, S.L.; Su, P.F.; et al. Using Multiplexed Assays of Oncogenic Drivers in Lung Cancers to Select Targeted Drugs. JAMA 2014, 311, 1998–2006. [Google Scholar] [CrossRef]
- Chu, Q.S. Targeting non-small cell lung cancer: Driver mutation beyond epidermal growth factor mutation and anaplastic lymphoma kinase fusion. Ther. Adv. Med. Oncol. 2020, 12, 1758835919895756. [Google Scholar] [CrossRef] [Green Version]
- Marsh, D.J.; Learoyd, D.L.; Andrew, S.D.; Krishnan, L.; Pojer, R.; Richardson, A.L.; Delbridge, L.; Eng, C.; Robinson, B.G. Somatic mutations in the RET proto-oncogene in sporadic medullary thyroid carcinoma. Clin. Endocrinol. 1996, 44, 249–257. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, K.; Soda, M.; Togashi, Y.; Suzuki, R.; Sakata, S.; Hatano, S.; Asaka, R.; Hamanaka, W.; Ninomiya, H.; Uehara, H.; et al. RET, ROS1 and ALK fusions in lung cancer. Nat. Med. 2012, 18, 378–381. [Google Scholar] [CrossRef]
- Wang, R.; Hu, H.; Pan, Y.; Li, Y.; Ye, T.; Li, C.; Luo, X.; Wang, L.; Li, H.; Zhang, Y.; et al. RET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancer. J. Clin. Oncol. 2012, 30, 4352–4359. [Google Scholar] [CrossRef]
- Song, Z.; Yu, X.; Zhang, Y. Clinicopathologic characteristics, genetic variability and therapeutic options of RET rearrangements patients in lung adenocarcinoma. Lung Cancer 2016, 101, 16–21. [Google Scholar] [CrossRef]
- Klempner, S.J.; Bazhenova, L.A.; Braiteh, F.S.; Nikolinakos, P.G.; Gowen, K.; Cervantes, C.M.; Chmielecki, J.; Greenbowe, J.R.; Ross, J.S.; Stephens, P.J.; et al. Emergence of RET rearrangement co-existing with activated EGFR mutation in EGFR-mutated NSCLC patients who had progressed on first- or second-generation EGFR TKI. Lung Cancer 2015, 89, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Hu, Z.I.; Lai, G.G.Y.; Tan, D.S.W. Targeting RET-driven cancers: Lessons from evolving preclinical and clinical landscapes. Nat. Rev. Clin. Oncol. 2018, 15, 150. [Google Scholar] [CrossRef] [Green Version]
- Plaza-Menacho, I. Structure and function of RET in multiple endocrine neoplasia type 2. Endocr.- Relat. Cancer 2018, 25, T79–T90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, K.; Takahashi, M. Intracellular RET Signaling Pathways Activated by GDNF. Cell Tissue Res. 2020, 382, 113–123. [Google Scholar] [CrossRef]
- Plaza-Menacho, I.; Burzynski, G.M.; de Groot, J.W.; Hofstra, R.M. Current Concepts in RET-Related Genetics, Signaling and Therapeutics. Trends Genet. 2006, 22, 627–636. [Google Scholar] [CrossRef] [Green Version]
- Plaza-Menacho, I.; Mologni, L.; Sala, E.; Gambacorti-Passerini, C.; Magee, A.I.; Links, T.P.; Hofstra, R.M.; Barford, D.; Isacke, C.M. Sorafenib functions to potently suppress RET tyrosine kinase activity by direct enzymatic inhibition and promoting RET lysosomal degradation independent of proteasomal targeting. J. Biol. Chem. 2007, 282, 29230–29240. [Google Scholar] [CrossRef] [Green Version]
- Plaza-Menacho, I.; Morandi, A.; Mologni, L.; Boender, P.; Gambacorti-Passerini, C.; Magee, A.I.; Hofstra, R.M.; Knowles, P.; McDonald, N.Q.; Isacke, C.M. Focal adhesion kinase (FAK) binds RET kinase via its FERM domain, priming a direct and reciprocal RET-FAK transactivation mechanism. J. Biol. Chem. 2011, 286, 17292–17302. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.; Ritz, J.; Cooper, G.M. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell 1985, 42, 581–588. [Google Scholar] [CrossRef]
- Grieco, M.; Santoro, M.; Berlingieri, M.T.; Melillo, R.M.; Donghi, R.; Bongarzone, I.; Pierotti, M.A.; Della Porta, G.; Fusco, A.; Vecchio, G. PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell 1990, 60, 557–563. [Google Scholar] [CrossRef]
- Thein, K.Z.; Velcheti, V.; Mooers, B.H.M.; Wu, J.; Subbiah, V. Precision therapy for RET-altered cancers with RET inhibitors. Trends Cancer 2021, 7, 1074–1088. [Google Scholar] [CrossRef] [PubMed]
- Sipple, J.H. Multiple endocrine neoplasia type 2 syndromes: Historical perspectives. Henry Ford Hosp. Med. J. 1984, 32, 219–221. [Google Scholar]
- Subbiah, V.; Roszik, J. Towards precision oncology in RET-aberrant cancers. Cell Cycle 2017, 16, 813–814. [Google Scholar] [CrossRef] [Green Version]
- Subbiah, V.; Yang, D.; Velcheti, V.; Drilon, A.; Meric-Bernstam, F. State-of-the-Art Strategies for Targeting RET-Dependent Cancers. J. Clin. Oncol. 2020, 38, 1209–1221. [Google Scholar] [CrossRef]
- Wiesner, T.; He, J.; Yelensky, R.; Esteve-Puig, R.; Botton, T.; Yeh, I.; Lipson, D.; Otto, G.; Brennan, K.; Murali, R. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat. Commun. 2014, 5, 3116. [Google Scholar] [CrossRef] [Green Version]
- Ballerini, P.; Struski, S.; Cresson, C.; Prade, N.; Toujani, S.; Deswarte, C.; Dobbelstein, S.; Petit, A.; Lapillonne, H.; Gautier, E.F.; et al. RET fusion genes are associated with chronic myelomonocytic leukemia and enhance monocytic differentiation. Leukemia 2012, 26, 2384–2389. [Google Scholar] [CrossRef] [Green Version]
- Le Rolle, A.F.; Klempner, S.J.; Garrett, C.R.; Seery, T.; Sanford, E.M.; Balasubramanian, S.; Ross, J.S.; Stephens, P.J.; Miller, V.A.; Ali, S.M.; et al. Identification and characterization of RET fusions in advanced colorectal cancer. Oncotarget 2015, 6, 28929–28937. [Google Scholar] [CrossRef] [Green Version]
- Ogino, H.; Yano, S.; Kakiuchi, S.; Yamada, T.; Ikuta, K.; Nakataki, E.; Goto, H.; Hanibuchi, M.; Nishioka, Y.; Ryan, A.; et al. Novel dual targeting strategy with vandetanib induces tumor cell apoptosis and inhibits angiogenesis in malignant pleural mesothelioma cells expressing RET oncogenic rearrangement. Cancer Lett. 2008, 265, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, K. Discovery Stories of RET Fusions in Lung Cancer: A Mini-Review. Front. Physiol. 2019, 10, 216. [Google Scholar] [CrossRef]
- Benayed, R.; Offin, M.; Mullaney, K.; Sukhadia, P.; Rios, K.; Desmeules, P.; Ptashkin, R.; Won, H.; Chang, J.; Halpenny, D.; et al. High Yield of RNA Sequencing for Targetable Kinase Fusions in Lung Adenocarcinomas with No Mitogenic Driver Alteration Detected by DNA Sequencing and Low Tumor Mutation Burden. Clin. Cancer Res. 2019, 25, 4712–4722. [Google Scholar] [CrossRef]
- Tsuta, K.; Kohno, T.; Yoshida, A.; Shimada, Y.; Asamura, H.; Furuta, K.; Kushima, R. RET-rearranged non-small-cell lung carcinoma: A clinicopathological and molecular analysis. Br. J. Cancer 2014, 110, 1571–1578. [Google Scholar] [CrossRef] [Green Version]
- Radonic, T.; Geurts-Giele, W.R.R.; Samsom Kris, G. RET Fluorescence In Situ Hybridization Analysis Is a Sensitive but Highly Unspecific Screening Method for RET Fusions in Lung Cancer. J. Thorac. Oncol. 2021, 16, 798–806. [Google Scholar] [CrossRef]
- Yang, S.R.; Aypar, U.; Rosen, E.Y.; Mata, D.A.; Benayed, R.; Mullaney, K.; Jayakumaran, G.; Zhang, Y.; Frosina, D.; Drilon, A.; et al. A Performance Comparison of Commonly Used Assays to Detect RET Fusions. Clin. Cancer Res. 2021, 27, 1316–1328. [Google Scholar] [CrossRef]
- Go, H.; Jung, Y.J.; Kang, H.W.; Park, I.K.; Kang, C.H.; Lee, J.W.; Ju, Y.S.; Seo, J.S.; Chung, D.H.; Kim, Y.T. Diagnostic method for the detection of KIF5B-RET transformation in lung adenocarcinoma. Lung Cancer 2013, 82, 44–50. [Google Scholar] [CrossRef]
- Yokota, K.; Sasaki, H.; Okuda, K.; Shimizu, S.; Shitara, M.; Hikosaka, Y.; Moriyama, S.; Yano, M.; Fujii, Y. KIF5B/RET fusion gene in surgically-treated adenocarcinoma of the lung. Oncol. Rep. 2012, 28, 1187–1192. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Oxnard, G.R.; Tan, D.S.W.; Loong, H.H.F.; Johnson, M.; Gainor, J.; McCoach, C.E.; Gautschi, O.; Besse, B.; Cho, B.C.; et al. Efficacy of Selpercatinib in RET Fusion-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Das, T.K.; Cagan, R.L. KIF5B-RET Oncoprotein Signals through a Multi-kinase Signaling Hub. Cell Rep. 2017, 20, 2368–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rich, T.A.; Reckamp, K.L.; Chae, Y.K.; Doebele, R.C.; Iams, W.T.; Oh, M.; Raymond, V.M.; Lanman, R.B.; Riess, J.W.; Stinchcombe, T.E.; et al. Analysis of cell-free DNA from 32,989 advanced cancers reveals novel co-occurring activating RET alterations and oncogenic signaling pathway aberrations. Clin. Cancer Res. 2019, 25, 5832–5842. [Google Scholar] [CrossRef] [Green Version]
- Belli, C.; Penault-Llorca, F.; Ladanyi, M.; Normanno, N.; Scoazec, J.Y.; Lacroix, L.; Reis-Filho, J.S.; Subbiah, V.; Gainor, J.F.; Endris, V.; et al. ESMO recommendations on the standard methods to detect RET fusions and mutations in daily practice and clinical research. Ann. Oncol. 2021, 32, 337–350. [Google Scholar] [CrossRef]
- Gainor, J.F.; Curigliano, G.; Kim, D.-W.; Lee, D.H.; Besse, B.; Baik, C.S.; Doebele, R.C.; Cassier, P.A.; Lopes, G.; Tan, D.S.; et al. Registrational Dataset from the Phase I/II ARROW Trial of Pralsetinib (BLU-667) in Patients (Pts) With Advanced RET Fusion+ Non-Small Cell Lung Cancer (NSCLC). J. Clin. Oncol. 2020, 38, 9515–9515. [Google Scholar] [CrossRef]
- Reckamp, K.L.; Patil, T.; Kirtane, K.; Rich, T.A.; Espenschied, C.R.; Weipert, C.M.; Raymond, V.M.; Santana-Davila, R.; Doebele, R.C.; Baik, C.S. Duration of targeted therapy in patients with advanced none-small-cell lung cancer identifed by circulating tumor DNA analysis. Clin. Lung Cancer 2020, 21, 545–552. [Google Scholar] [CrossRef]
- Mack, P.C.; Banks, K.C.; Espenschied, C.R.; Burich, R.A.; Zill, O.A.; Lee, C.E.; Riess, J.W.; Mortimer, S.A.; Talasaz, A.; Lanman, R.B.; et al. Spectrum of driver mutations and clinical impact of circulating tumor DNA analysis in non-small cell lung cancer: Analysis of over 8000 cases. Cancer 2020, 126, 3219–3228. [Google Scholar] [CrossRef]
- Malapelle, U.; Tiseo, M.; Vivancos, A.; Kapp, J.; Serrano, M.J.; Tienamm, M. Liquid Biopsy for Biomarker Testing in Non-Small Cell Lung Cancer: A European Perspective. J. Mol. Pathol. 2021, 2, 22. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Stevens, S.E.; Lin, J.J.; Nagy, R.; Ferris, L.; Shaw, A.T.; Gainor, J.F. Emergence of a RET V804M gatekeeper mutation during treatment with vandetanib in RET-rearranged NSCLC. J. Thorac. Oncol. 2018, 13, e226–e227. [Google Scholar] [CrossRef] [Green Version]
- Hofman, P. What Is New in Biomarker Testing at Diagnosis of Advanced Non-Squamous Non-Small Cell Lung Carcinoma? Implications for Cytology and Liquid Biopsy. J. Mol. Pathol. 2021, 2, 15. [Google Scholar] [CrossRef]
- Gautschi, O.; Milia, J.; Filleron, T.; Wolf, J.; Carbone, D.P.; Owen, D.; Camidge, R.; Narayanan, V.; Doebele, R.C.; Besse, B.; et al. Targeting RET in Patients With RET-Rearranged Lung Cancers: Results From the Global, Multicenter RET Registry. J Clin Oncol. 2017, 35, 1403–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horiike, A.; Takeuchi, K.; Uenami, T.; Kawano, Y.; Tanimoto, A.; Kaburaki, K.; Tambo, Y.; Kudo, K.; Yanagitani, N.; Ohyanagi, F.; et al. Sorafenib treatment for patients with RET fusion-positive non-small cell lung cancer. Lung Cancer. 2016, 93, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a Novel MET and VEGFR2 Inhibitor, Simultaneously Suppresses Metastasis, Angiogenesis, and Tumor Growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drilon, A.; Rekhtman, N.; Arcila, M.; Wang, L.; Ni, A.; Albano, M.; Van Voorthuysen, M.; Somwar, R.; Smith, R.S.; Montecalvo, J.; et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: An open-label, single-centre, phase 2, single-arm trial. Lancet Oncol. 2016, 17, 1653–1660. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, K.; Kodama, K.; Takase, K.; Sugi, N.H.; Yamamoto, Y.; Iwata, M.; Tsuruoka, A. Antitumor activities of the targeted multi-tyrosine kinase inhibitor lenvatinib (E7080) against RET gene fusion-driven tumor models. Cancer Lett. 2013, 340, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hida, T.; Velcheti, V.; Reckamp, K.L.; Nokihara, H.; Sachdev, P.; Kubota, T.; Nakada, T.; Dutcus, C.E.; Ren, M.; Tamura, T. A phase 2 study of lenvatinib in patients with RET fusion-positive lung adenocarcinoma. Lung Cancer. 2019, 138, 124–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlomagno, F.; Vitagliano, D.; Guida, T.; Ciardiello, F.; Tortora, G.; Vecchio, G.; Ryan, A.J.; Fontanini, G.; Fusco, A.; Santoro, M. ZD6474, an Orally Available Inhibitor of KDR Tyrosine Kinase Activity, Efficiently Blocks Oncogenic RET Kinases. Cancer Res. 2002, 62, 7284–7290. [Google Scholar]
- Yoh, K.; Seto, T.; Satouchi, M.; Nishio, M.; Yamamoto, N.; Murakami, H.; Nogami, N.; Nosaki, K.; Kohno, T.; Tsuta, K.; et al. Final survival results for the LURET phase II study of vandetanib in previously treated patients with RET-rearranged advanced non-small cell lung cancer. Lung Cancer 2021, 155, 40–45. [Google Scholar] [CrossRef]
- Lee, S.-H.; Lee, J.-K.; Ahn, M.-J.; Kim, D.W.; Sun, J.M.; Keam, B.; Kim, T.M.; Heo, D.S.; Ahn, J.S.; Choi, Y.L.; et al. Vandetanib in pretreated patients with advanced non-small cell lung cancer-harboring RET rearrangement: A phase II clinical trial. Ann. Oncol. 2017, 28, 292–297. [Google Scholar] [CrossRef]
- Subbiah, V.; Gainor, J.F.; Rahal, R.; Brubaker, J.D.; Kim, J.L.; Maynard, M.; Hu, W.; Cao, Q.; Sheets, M.P.; Wilson, D.; et al. Precision Targeted Therapy with BLU-667 for RET-Driven Cancers. Cancer Discov. 2018, 8, 836–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gainor, J.F.; Curigliano, G.; Kim, D.W.; Lee, D.H.; Besse, B.; Baik, C.S.; Doebele, R.C.; Cassier, P.A.; Lopes, G.; Tan, D.S.W.; et al. Pralsetinib for RET fusion-positive non-small-cell lung cancer (ARROW): A multi-cohort, open-label, phase 1/2 study. Lancet Oncol. 2021, 22, 959–969. [Google Scholar] [CrossRef]
- Besse, B.; Felip, E.; Clifford, C.; Louie-Gao, M.; Green, J.; Turner, C.D.; Popat, S. AcceleRET Lung: A phase III study of first-line pralsetinib in patients (pts) with RET-fusion+ advanced/metastatic non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2020, 38 (Suppl. 15), TPS9633. [Google Scholar] [CrossRef]
- Subbiah, V.; Velcheti, V.; Tuch, B.B.; Ebata, K.; Busaidy, N.L.; Cabanillas, M.E.; Wirth, L.J.; Stock, S.; Smith, S.; Lauriault, V.; et al. Selective RET Kinase Inhibition for Patients with RET-Altered Cancers. Ann. Oncol. 2018, 29, 1869–1876. [Google Scholar] [CrossRef]
- Subbiah, V.; Gainor, J.F.; Oxnard, G.R.; Tan, D.S.W.; Owen, D.H.; Cho, B.C.; Loong, H.H.; McCoach, C.E.; Weiss, J.; Kim, Y.J.; et al. Intracranial Efficacy of Selpercatinib in RET Fusion-Positive Non-Small Cell Lung Cancers on the LIBRETTO-001 Trial. Clin. Cancer Res. 2021, 27, 4160–4167. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Wolf, J.; Elamin, Y. LIBRETTO-431: Selpercatinib in Treatment-Naïve Patients with RET Fusion-Positive Non-Small Cell Lung Cancer (NSCLC). J. Thorac. Oncol. 2021, 16, S228–S229. [Google Scholar] [CrossRef]
- Drilon, A.; Bergagnini, I.; Delasos, L.; Sabari, J.; Woo, K.M.; Plodkowski, A.; Wang, L.; Hellmann, M.D.; Joubert, P.; Sima, C.S.; et al. Clinical outcomes with pemetrexed-based systemic therapies in RET-rearranged lung cancers. Ann. Oncol. 2016, 27, 1286–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.B.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Shaw, A.T. Resisting Resistance: Targeted Therapies in Lung Cancer. Trends Cancer 2016, 2, 350–364. [Google Scholar] [CrossRef] [Green Version]
- Carlomagno, F.; Guida, T.; Anaganti, S.; Vecchio, G.; Fusco, A.; Ryan, A.J.; Billaud, M.; Santoro, M. Disease associated mutations at valine 804 in the RET receptor tyrosine kinase confer resistance to selective kinase inhibitors. Oncogene 2004, 23, 6056–6063. [Google Scholar] [CrossRef] [Green Version]
- Wirth, L.J.; Kohno, T.; Udagawa, H.; Matsumoto, S.; Ishii, G.; Ebata, K.; Tuch, B.B.; Zhu, E.Y.; Nguyen, M.; Smith, S.; et al. Emergence and targeting of acquired and hereditary resistance to multikinase RET inhibition in patients with RET-altered cancer. JCO Precis. Oncol. 2019, 3, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Nakaoku, T.; Kohno, T.; Araki, M.; Niho, S.; Chauhan, R.; Knowles, P.P.; Tsuchihara, K.; Matsumoto, S.; Shimada, Y.; Mimaki, S.; et al. A secondary RET mutation in the activation loop conferring resistance to vandetanib. Nat. Commun. 2018, 9, 625. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Shen, T.; Terzyan, S.S.; Liu, X.; Hu, X.; Patel, K.P.; Hu, M.; Cabanillas, M.; Behrang, A.; Meric-Bernstam, F.; et al. Structural basis of acquired resistance to selpercatinib and pralsetinib mediated by non-gatekeeper RET mutations. Ann. Oncol. 2021, 32, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.J.; Tan, L.; Lin, J.J.; Wong, S.Q.; Hollizeck, S.; Ebata, K.; Tuch, B.B.; Yoda, S.; Gainor, J.F.; Sequist, L.V.; et al. RET Solvent Front Mutations Mediate Acquired Resistance to Selective RET Inhibition in RET-Driven Malignancies. J. Thorac. Oncol. 2020, 15, 541–549. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.J.; Liu, S.V.; McCoach, C.E.; Zhu, V.W.; Tan, A.C.; Yoda, S.; Peterson, J.; Do, A.; Prutisto-Chang, K.; Dagogo-Jack, I.; et al. Mechanisms of resistance to selective RET tyrosine kinase inhibitors in RET fusion-positive non-small-cell lung cancer. Ann. Oncol. 2020, 31, 1725–1733. [Google Scholar] [CrossRef]
- Gainor, J.; Curigliano, G.; Doebele, R.C.; Lin, J.; Ou, S.; Miller, S.; Turner, C.; Subbiah, V. OA05.02 analysis of resistance mechanisms to pralsetinib in patients with RET fusion-positive non-small cell lung cancer (NSCLC) from the ARROW study. J. Thorac. Oncol. 2021, 16, S5. [Google Scholar] [CrossRef]
- Rosen, E.Y.; Johnson, M.L.; Clifford, S.E.; Somwar, R.; Kherani, J.F.; Son, J.; Bertram, A.A.; Davare, M.A.; Gladstone, E.; Ivanova, E.V.; et al. Overcoming MET-Dependent Resistance to Selective RET Inhibition in Patients with RET Fusion-Positive Lung Cancer by Combining Selpercatinib with Crizotinib. Clin. Cancer Res. 2021, 27, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Zhu, V.W.; Zhang, S.S.; Zhang, J.; Swensen, J.; Xiu, J.; Ou, S.I. Acquired Tertiary MET Resistance (MET D1228N and a Novel LSM8-MET Fusion) to Selpercatinib and Capmatinib in a Patient with KIF5B-RET-positive NSCLC With Secondary MET Amplification as Initial Resistance to Selpercatinib. J. Thorac. Oncol. 2021, 16, e51–e54. [Google Scholar] [CrossRef]
- Chang, H.; Sung, J.H.; Moon, S.U.; Kim, H.S.; Kim, J.W.; Lee, J.S. EGF Induced RET Inhibitor Resistance in CCDC6-RET Lung Cancer Cells. Yonsei Med. J. 2017, 58, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Nelson-Taylor, S.K.; Le, A.T.; Yoo, M.; Schubert, L.; Mishall, K.M.; Doak, A.; Varella-Garcia, M.; Tan, A.C.; Doebele, R.C. Resistance to RET-Inhibition in RET-Rearranged NSCLC Is Mediated by Reactivation of RAS/MAPK Signaling. Mol. Cancer Ther. 2017, 16, 1623–1633. [Google Scholar] [CrossRef] [Green Version]
- Schoenfeld, A.J.; Chan, J.M.; Kubota, D.; Sato, H.; Rizvi, H.; Daneshbod, Y.; Chang, J.C.; Paik, P.K.; Offin, M.; Arcila, M.E.; et al. Tumor Analyses Reveal Squamous Transformation and Off-Target Alterations as Early Resistance Mechanisms to First-line Osimertinib in EGFR-Mutant Lung Cancer. Clin. Cancer Res. 2020, 26, 2654–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Rogers, E.; Zhai, D.; Deng, W.; Zhang, X.; Lee, D.; Ung, J.; Whitten, J.; Zhang, H.; Liu, J.; et al. TPX-0046 is a novel and potent RET/SRC inhibitor for RET-driven cancers. Ann. Oncol. 2019, 30 (Suppl. 5), v190–v191. [Google Scholar] [CrossRef]
- Study of TPX-0046, A RET/SRC Inhibitor in Adult Subjects With Advanced Solid Tumors Harboring RET Fusions or Mutations. Available online: https://clinicaltrials.gov/ct2/show/NCT04161391 (accessed on 9 January 2022).
- Schoffski, P.; Chul Cho, B.; Italiano, A.; Loong, H.F.; Massard, C.; Rodriguez, L.M.; Shih, J.; Subbiah, V.; Verlingue, L.; Andreas, K.; et al. BOS172738, a highly potent and selective RET inhibitor, for the treatment of RET-altered tumors including RET-fusion+ NSCLC and RET-mutant MTC: Phase 1 study results. J. Clin. Oncol. 2021, 39 (Suppl. 15), 3008. [Google Scholar] [CrossRef]
- Study of RET Inhibitor TAS0953/HM06 in Patients with Advanced Solid Tumors with RET Gene Abnormalities (MARGARET). Available online: https://clinicaltrials.gov/ct2/show/NCT04683250 (accessed on 9 January 2022).
- Gabrielle, R.; Kolakowski, Anderson, E.D.; Brandhuber, B.J.; Condroski, K.R.; Gomez, E.B.; Irvin, T.C.; Kumar, M.; Patel, N.A.; Watson, F.D. Pre-clinical characterization of potent and selective next-generation RET inhibitors [abstract]. In Proceedings of the American Association for Cancer Research Annual Meeting 2021, Online, 10–15 April and 17–21 May 2021; AACR: Philadelphia, PA, USA, 2021. [Google Scholar]
- Goldman, J.; Besse, B.; Wu, Y.; Yang, J.C.; Paz-Ares, L.; Drilon, A.; Johnson, M.; Xia, M.; Chao, B.H.; Tsuboi, M. P01.01 LIBRETTO-432: A Placebo-Controlled Phase 3 Study of Adjuvant Selpercatinib in Stage IB-IIIA RET Fusion-Positive NSCLC. J. Thorac. Oncol. 2021, 16, S975–S976. [Google Scholar] [CrossRef]
- A Study of Alectinib, Entrectinib, Vemurafenib Plus Cobimetinib, or Pralsetinib in Patients With Resectable Stages II-III Non-Small Cell Lung Cancer With ALK, ROS1, NTRK, BRAF V600, or RET Molecular Alterations. Available online: https://clinicaltrials.gov/ct2/show/NCT04302025 (accessed on 9 January 2022).
| Trial | Experimental Arm | Comparator Arm | Setting | Phase | Primary Endpoint | Status |
|---|---|---|---|---|---|---|
| NCT04161391 | TPX-0046 | -- | N line | 1/2 | DLTs, MTD, ORR | Recruiting |
| NCT04683250 (MARGARET) | TAS0953/HM06 | -- | N line | 1/2 | MTD, RP2D, ORR | Recruiting |
| NCT03037385 (ARROW) | pralsetinib (BLU-667) | -- | 1-N line | 1/2 | MTD, N° of patients with adverse events and serious adverse events, ORR | Recruiting |
| NCT01639508 | Cabozantinib | -- | 1-N line | 2 | ORR | Recruiting |
| NCT03780517 | BOS172738. | -- | N line | 1 | TEAE, MTD, RP2D | Active, not recruiting |
| NCT04268550 (Lung-MAP) | Selpercatinib | -- | N line | 2 | ORR | Recruiting |
| NCT03157128 (LIBRETTO-001) | Selpercatinib | -- | N line | 1/2 | MTD, RP2D, ORR | Recruiting |
| NCT04131543 (CRETA) | Cabozantinib | -- | 2-N line | 2 | RR | Recruiting |
| NCT04194944 | Selpercatinib | Platinum–Pemetrexed with or without Pembrolizumab | 1 line | 3 | PFS | Recruiting |
| NCT04302025 (NAUTIKA1) | SOC chemotherapy + Pralsetinib | -- | Neoadj–Adjuvant | 2 | MPR | Recruiting |
| NCT04222972 | Pralsetinib | Platinum–based chemotherapy with or without pembrolizumab | 1 line | 3 | PFS | Recruiting |
| NCT04819100 (LIBRETTO-432) | Selpercatinib | Placebo | Adjuvant | 3 | EFS | Recruiting |
| NCT02314481 (DARWINII) | Alectinib | -- | N line | 2 | PFS | Recruiting |
| NCT04591431 (ROME) | Alectinib | -- | 2 line | 2 | ORR | Recruiting |
| NCT03178552 (B-FAST) | Alectinib | -- | 1 line | 2/3 | ORR | Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reale, M.L.; Bertaglia, V.; Listì, A.; Novello, S.; Passiglia, F. Molecular Testing and Treatment Strategies in RET-Rearranged NSCLC Patients: Stay on Target to Look Forward. J. Mol. Pathol. 2022, 3, 24-37. https://doi.org/10.3390/jmp3010003
Reale ML, Bertaglia V, Listì A, Novello S, Passiglia F. Molecular Testing and Treatment Strategies in RET-Rearranged NSCLC Patients: Stay on Target to Look Forward. Journal of Molecular Pathology. 2022; 3(1):24-37. https://doi.org/10.3390/jmp3010003
Chicago/Turabian StyleReale, Maria Lucia, Valentina Bertaglia, Angela Listì, Silvia Novello, and Francesco Passiglia. 2022. "Molecular Testing and Treatment Strategies in RET-Rearranged NSCLC Patients: Stay on Target to Look Forward" Journal of Molecular Pathology 3, no. 1: 24-37. https://doi.org/10.3390/jmp3010003
APA StyleReale, M. L., Bertaglia, V., Listì, A., Novello, S., & Passiglia, F. (2022). Molecular Testing and Treatment Strategies in RET-Rearranged NSCLC Patients: Stay on Target to Look Forward. Journal of Molecular Pathology, 3(1), 24-37. https://doi.org/10.3390/jmp3010003